

SUMMARY
Caspase-11 is a highly inducible caspase that controls both inflammatory responses and cell death. Caspase-11 controls interleukin 1β (IL-1β) secretion by potentiating caspase-1 activation and induces caspase-1-independent pyroptosis downstream of noncanonical NLRP3 inflammasome activators such as lipopolysaccharide (LPS) and Gram-negative bacteria. However, we still know very little about the downstream mechanism of caspase-11 in regulating inflammation because the known substrates of caspase-11 are only other caspases. Here, we identify the cationic channel subunit transient receptor potential channel 1 (TRPC1) as a substrate of caspase-11. TRPC1 deficiency increases the secretion of IL-1β without modulating caspase-1 cleavage or cell death in cultured macrophages. Consistently, trpc1−/− mice show higher IL-1β secretion in the sepsis model of intraperitoneal LPS injection. Altogether, our data suggest that caspase-11 modulates the cationic channel composition of the cell and thus regulates the unconventional secretion pathway in a manner independent of caspase-1.
INTRODUCTION
Inflammatory responses activated by the innate immunity system constitute a highly efficient barrier in defending against diverse organismal insults. During an innate immune response, early local detection of the insult is mediated by the activation of pattern recognition receptors (PRRs) by pathogen- and damage-associated molecular patterns, which then triggers inflammation by stimulating the production and release of proinflammatory cytokines such as interleukin 1β (IL-1β), as well as pyroptosis, a proinflammatory form of cell death (Keller et al., 2008; Kepp et al., 2010; Martinon et al., 2009). Inflammation, however, must be tightly regulated, as excessive or chronic inflammation may lead to detrimental outcomes, including tissue damage or death as a result of septic shock.
Caspase-11, a member of the caspase family of cysteine proteases, is a critical mediator of inflammation and cell death (Kang et al., 2000; Wang et al., 1998). Casp11−/− mice are highly resistant to lipopolysaccharide (LPS)-induced septic shock, but are more sensitive to infection with Burkholderia, a cytosolic bacterium that escapes the vacuole, demonstrating the inflammatory response as a “double-edged sword” (Aachoui et al., 2013; Wang et al., 1998). Caspase-11 is highly inducible by activators of innate immunity such as LPS, interferon γ (IFN-γ), and IFN-β (Kang et al., 2000, 2002; Lee et al., 2001; Rathinam et al., 2012; Wang et al., 1996). The expression of caspase-11 is required for caspase-1 activation and subsequent IL-1β secretion downstream of a subgroup of stimuli (Wang et al., 1998). Caspase-1 is directly involved in the processing of pro-IL-1β and the secretion of IL-1β. Caspase-11 potentiates caspase-1 activation downstream of noncanonical NLRP3 inflammasome activators, including diverse Gram-negative bacteria such as Escherichia coli (Kayagaki et al., 2011; Wang et al., 1998), but is dispensable for the activation of caspase-1 downstream of canonical NLRP3 inflammasome activators such as extracellular ATP, the lysomotropic agent HLLOMe, and silica crystals, as well as other canonical inflammasome complexes such as the NLRC4 or AIM2 inflammasomes. In addition, caspase-11 triggers caspase-1-independent pyroptosis and subsequent release of proinflammatory factors such as IL-1α and HMGB1 downstream of noncanonical stimuli, including several vacuolar and cytosolic Gram-negative bacteria (Aachoui et al., 2013; Broz et al., 2012; Case et al., 2013; Kayagaki et al., 2011). Finally, caspase-11 modulates actin cytoskeleton dynamics and restricts Legionella pneumophila growth by promoting bacterial vacuole fusion with lysosomes, as well as by regulating cell migration (Akhter et al., 2012; Li et al., 2007). Caspase-11, which was first characterized in the late 1990s (Wang et al., 1996, 1998), has recently received much attention (Broz et al., 2012; Rathinam et al., 2012). However, the downstream molecular effectors of caspase-11 remain to be identified. To the best of our knowledge, caspase-3 and caspase-11 itself are the only reported substrates for caspase-11 (Kang et al., 2000).
Transient receptor potential channel 1 (TRPC1) belongs to the canonical TRP subfamily of proteins with six transmembrane domains that assemble to form cation-permeable pores as homo- or heterotetramers (Clapham, 2003). TRPC1 associates with other TRP channels, such as TRPC4 and TRPC5, as well as scaffolding proteins (Strübing et al., 2001). The composition of these complexes determines the distribution of TRPC1 between the endoplasmic reticulum (ER) and the plasma membrane, as well as the various characteristics and gating mechanisms of the channels in which TRPC1 takes part. Although TRPC1 is widely expressed in a variety of tissues and is known to contribute to various physiological functions, it was not known to be involved in innate immunity.
Here, we identify TRPC1 as a substrate for caspase-11 that regulates inflammatory responses. Upon LPS treatment, caspase-11 induction leads to the degradation of TRPC1 in macrophages. TRPC1-deficient macrophages secrete increased amounts of mature IL-1β in response to NLRP3 activators without affecting caspase-1 cleavage or pyroptosis. In addition, trpc1−/− mice show higher IL-1β secretion in their sera following LPS injection. Our data provide a mechanism whereby caspase-11 promotes IL-1β release by degrading TRPC1 independently of caspase-1, and suggest that TRPC1 plays a role in regulating innate immunity by modulating channel complexes during inflammatory responses.
RESULTS
TRPC1 Is a Substrate for Caspase-11
A yeast two-hybrid screen identified TRPC1 as an interacting protein of inflammatory caspases (data not shown). When coexpressed in human embryonic kidney 293T (HEK293T) cells, the larger isoform of pro-caspase-11 (p43) coimmunoprecipitated with hemagglutinin-TRPC1 (HA-TRPC1; Figure 1A). This interaction of TRPC1 with caspase-11 is selective because, among all TRPC channels, only TRPC1 and (with a lower affinity) TRPC2 could be immunoprecipitated with caspase-11 (Figure S1). Subunits p10 and p20 of caspase-11, but not caspase recruitment domain (CARD) alone, could be coimmunoprecipitated with TRPC1 (Figure 1B). TRPC1 contains six transmembrane domains in its central part and two large N- and C-terminal cytosolic domains (Beech, 2005). FLAG-caspase-11 was able to coimmunoprecipitate independently with HA-TRPC1 N-terminal (HA-TRPC1-N) alone, the N-terminal domain with the transmembrane domain (HA-TRPC1-ΔC), and the transmembrane domain with the C-terminal domain (HA-TRPC1-ΔN), but not with the central transmembrane domain alone (HA-TRPC1-TM; Figure 1C).
Figure 1. Caspase-11 Interacts with TRPC1.
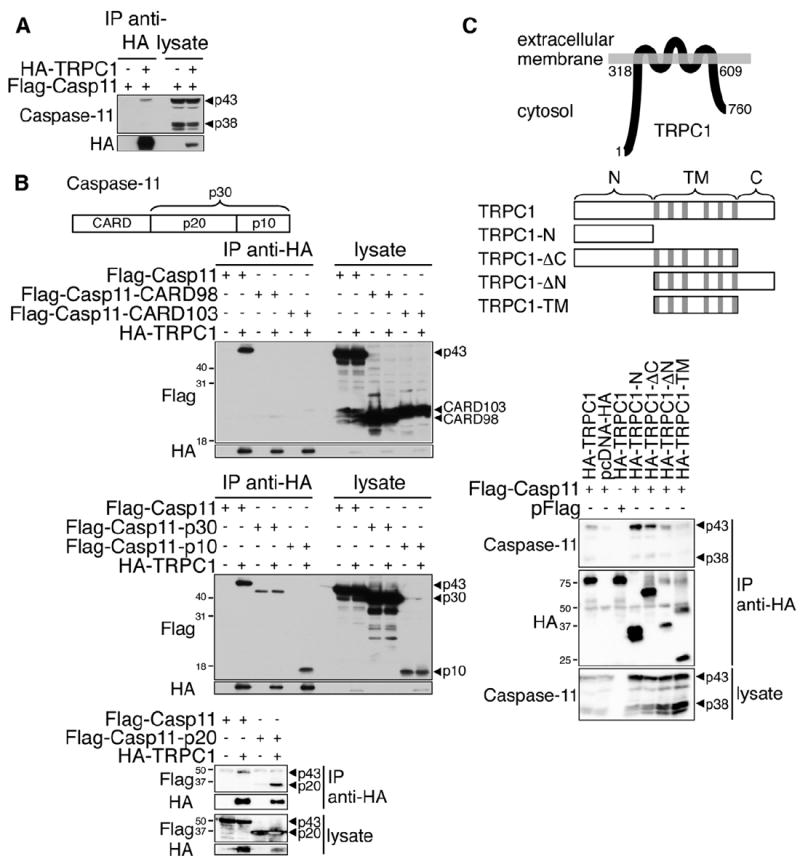
(A) Lysates of HEK293T cells expressing HA-TRPC1 and FLAG-Casp11 were subjected to anti-HA immunoprecipitation. The western blots were analyzed using anti-caspase-11 and anti-HA.
(B) Lysates of HEK293T cells expressing HA-TRPC1 and FLAG-Casp11, FLAG-Casp11-CARD98, FLAG-Casp11-CARD103, FLAG-Casp11-p30, FLAG-Casp11-p20, or FLAG-Casp11-p10 were subjected to anti-HA immunoprecipitation. The western blots were analyzed using anti-FLAG and anti-HA.
(C) Lysates of HEK293T cells expressing FLAG-Casp11 and HA-TRPC1, HA-TRPC1-N, HA-TRPC1-ΔC, HA-TRPC1-ΔN, or HA-TRPC1-TM were subjected to anti-HA immunoprecipitation. Diagrams of TRPC1 association with membrane and truncation expression constructs of TRPC1 are shown; gray sections represent transmembrane spans. The western blots were analyzed using anti-caspase-11 and anti-HA.
See also Figure S1.
Since the catalytic domain (p20+p10) of caspase-11 interacts with the TRPC1 channel, we hypothesized that TRPC1 may be a substrate for caspase-11. Indeed, coexpression of caspase-11 and TRPC1 in HEK293T cells greatly decreased the levels of TRPC1 protein, which were restored upon addition of the caspase inhibitor z-VAD.fmk (Figure 2A). Notably coexpression of the proteolytically inactive mutant caspase-11C255G did not have any impact on the levels of TRPC1. Furthermore, incubation of purified recombinant caspase-11 p30 with S35-labeled TRPC1 in vitro led to the appearance of multiple TRPC1 fragments and a reduction of full-length TRPC1, which was inhibited by z-VAD.fmk (Figure 2B). On the other hand, recombinant caspase-1 did not cleave TRPC1 in vitro, although it effectively cleaved pro-IL-1β (Figures 2C and S2A). Coexpression of TRPC1 with caspase-1 did not lead to a reduction of TRPC1, contrary to that observed with caspase-11 (Figure S2B), although overexpressed caspase-1 could be coimmunoprecipitated with overexpressed TRPC1 (Figure S2C). Thus, TRPC1 is a substrate for caspase-11, but not caspase-1.
Figure 2. TRPC1 Is a Substrate of Caspase-11 and Degraded following LPS Treatment.
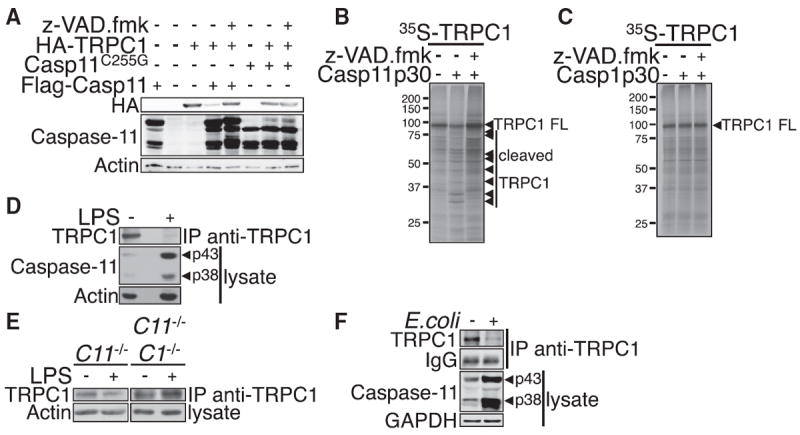
(A) HEK293T cells were cotransfected with expression vectors of HA-TRPC1 and FLAG-Casp11 or Casp11C255G catalytic mutant, and cultured for 48 hr. z-VAD.fmk (20 μM) was added as indicated for the last 24 hr of culture. The levels of HA-TRPC1 protein were assessed by western blotting using anti-HA, anti-caspase-11, and anti-actin (as a control).
(B) In vitro cleavage assay of 35S-labeled TRPC1 by purified recombinant caspase-11 p30.
(C) TRPC1 is not a substrate of caspase-1. In vitro cleavage assay of 35S-labeled TRPC1 by recombinant caspase-1 p30.
(D–F) Macrophages from WT (D) or casp11−/− or casp1−/− casp11−/− mice (E) were treated with LPS (100 ng/ml) for 16 hr. Macrophages from WT mice were infectedwith E. coli (J53strain, moi20) for 16hr (F). The expression levels of endogenous TRPC1 were assessed by anti-TRPC1 immunoprecipitation followed by anti-TRPC1 western blotting.
See also Figure S2.
Given that the expression of caspase-11 is highly inducible by LPS (Wang et al., 1996), we next assessed the cleavage and levels of TRPC1 in LPS-treated cells. Ectopic HA-TRPC1 protein levels were substantially reduced 8 hr following LPS treatment, but not in the presence of the pan caspase inhibitor z-VAD.fmk or IDUN-6556, suggesting that TRPC1 is degraded by inflammatory caspases (Figure S2D). Consistently, the levels of endogenous TRPC1 protein decreased in primary macrophages upon LPS treatment, which induced the expression of caspase-11 (Figure 2D). Strikingly, TRPC1 remained stable in casp1−/− casp11−/−, and casp11−/− macrophages treated with LPS (Figure 2E). On the other hand, the amounts of trpc1 mRNA remained unchanged following LPS treatment of both wild-type (WT) and casp11−/− macrophages, excluding the possibility that TRPC1 is regulated at the transcriptional level (Figure S2E). Furthermore, we found that stimulation of primary macrophages with E. coli (J53 strain), which induced the expression of caspase-11, also led to a reduction in the levels of endogenous TRPC1 protein (Figure 2F). Notably, TRPC1 levels remained stable in WT macrophages treated with low-dose LPS (1 ng/ml) for 3.5 hr followed by ATP, which was sufficient to lead to the secretion of caspase-1, but not sufficient to induce the expression of caspase-11 (Figure S2F). On the other hand, TRPC1 was degraded in WT, but not casp11−/−, macrophages treated with high-dose LPS (100 ng/ml) for 16 hr followed by ATP, which was sufficient to lead to expression of caspase-11 (Figure S2F). Thus, caspase-1 activation in the absence of caspase-11 expression does not lead to TRPC1 degradation. Taken together, these results suggest that TRPC1 is a physiologically relevant substrate of caspase-11 and is degraded in a caspase-11-dependent manner in macrophages upon induction of caspase-11 after the activation of TLR signaling.
Caspase-11 Controls the TRPC1-Dependent Decrease in Cytosolic Ca2+ following LPS Treatment
Since TRPC1 is a membrane protein that can form channels permeable to Ca2+, we next assessed the effect of TRPC1 and caspase-11 on macrophages’ cytosolic Ca2+ concentration in response to LPS stimulation. We found that during continuous LPS treatment, cytosolic Ca2+ concentration demonstrated a transient increase at 2 hr followed with a sustained decrease for up to 10 hr (Figure 3A). Interestingly, this sustained decrease of Ca2+ concentration is dependent on caspase-11 and TRPC1, as no reduction of cytosolic Ca2+ concentration was observed in either trpc1−/− or casp11−/− macrophages at 10 hr after LPS treatment (Figure 3B). Thus, we conclude that TRPC1 cleavage by caspase-11 has a functional impact on Ca2+ flux after LPS stimulation.
Figure 3. LPS Triggers a Caspase-11- and TRPC1-Dependent Cytosolic Ca2+ Decrease.
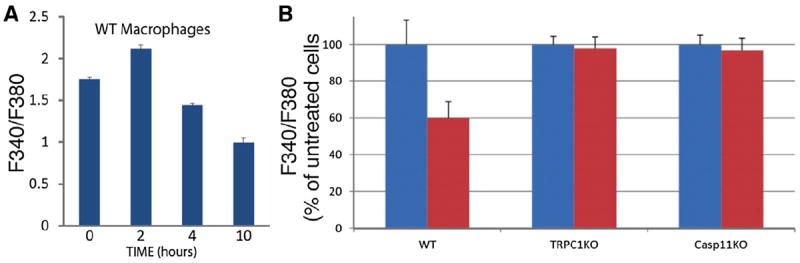
(A) Macrophages from WT mice were treated with LPS (100 ng/ml) for the indicated times. Intracellular Ca2+ was measured through ratiometric imaging of Fura-2AM (F340/F380). Error bars indicate SE from the mean.
(B) WT, trpc1−/−, and casp11−/− macrophages were treated with LPS (100 ng/ml, red bars) for 10 hr or left untreated (blue bars). Intracellular Ca2+ was measured through ratiometric imaging of Fura-2AM (F340/F380). Error bars indicate SE.
TRPC1 Deficiency Promotes IL-1β Release but Has No Effect on Caspase-1 Activation or Pyroptosis
Next, we investigated the implication of TRPC1 in the inflammatory response regulated by caspase-11. Caspase-11 is a critical mediator of LPS-induced septic shock (Wang et al., 1996, 1998); therefore, we first assessed the response of trpc1−/− mice to intraperitoneally injected LPS. Indeed, trpc1−/− mice showed significantly higher levels of IL-1β in serum following LPS injection as compared with WT mice, indicating that TRPC1 modulates inflammation in vivo by inhibiting IL-1β secretion (Figure 4A). On the other hand, trpc1−/− mice did not exhibit higher constitutive levels of circulating IL-1β in basal conditions, suggesting that the absence of TRPC1 alone is not sufficient to elicit an inflammatory response (Figure S3A). At the cellular level, caspase-11 is required for caspase-1 activation, IL-1β and IL-18 secretion, and pyroptosis in macrophages infected with E. coli (Kayagaki et al., 2011). Consistently, trpc1−/− macrophages showed higher secretion of mature IL-1β and IL-18 in response to E. coli (Figures 4B–4D). Since TRPC1 deficiency did not have any effect on caspase-1 cleavage, as indicated by the normal appearance of the caspase-1 subunit p20, we conclude that the cleavage of TRPC1 likely impacts events downstream of caspase-1 activation, including the unconventional secretion pathway that is responsible for mature IL-1β release. Interestingly, trpc1−/− macrophages also showed increased mature IL-1β release triggered by canonical NLRP3 inflammasome activators such as ATP, HLLOMe, and silica, which is independent of caspase-11 (Kayagaki et al., 2011), but showed no difference in caspase-1 cleavage (Figures 4E and 4F). We thus conclude that LPS priming modulates the TRPC1 complexes by both caspase-11-dependent and -independent, redundant mechanisms. Notably, TRPC1 deficiency did not lead to any increase in the levels of pro-IL-1β, or secretion of TNF-α following LPS priming, ruling out a role of TRPC1 in TLR4 signaling (Figures 4C, 4F, and S3B). In addition, TRPC1-deficiency did not sensitize cells to caspase-11-dependent pyroptosis following infection by E. coli, or to caspase-1-dependent pyroptosis induced by canonical stimuli, including ATP and HLLOMe (Figures S3C–S3F), supporting the notion that pyroptosis occurs independently of TRPC1 degradation.
Figure 4. TRPC1 Inhibits Mature IL-1β Release.
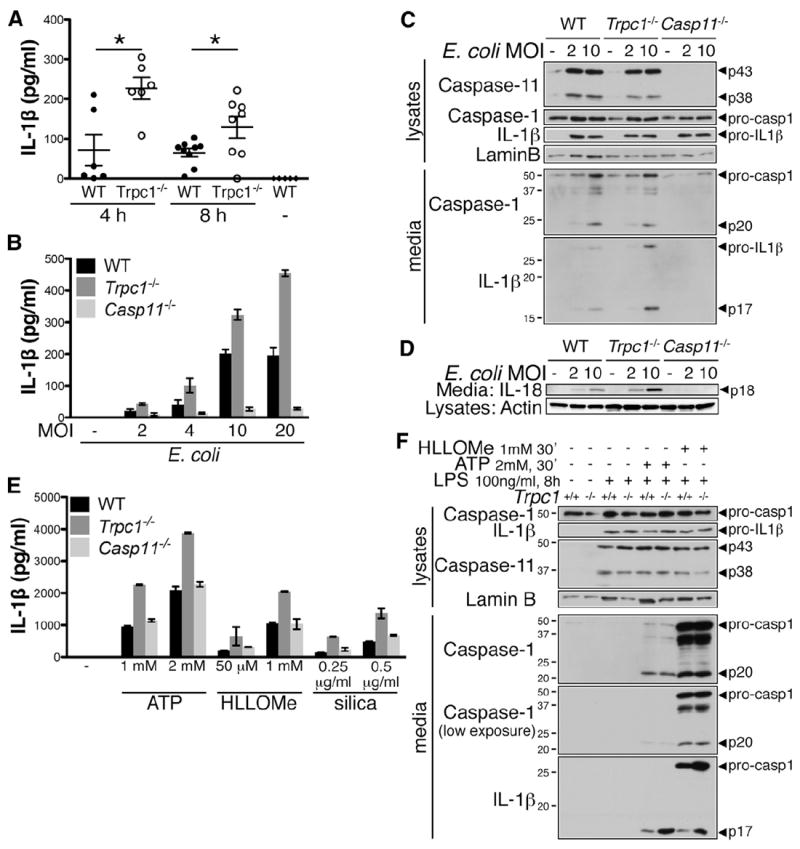
(A) Trpc1−/− mice secrete more IL-1β than WT mice in the serum following LPS challenge. WT and trpc1−/− mice were injected intraperitoneally with LPS (4 mg/kg). The sera were harvested after 4 hr (n = 6) or 8 hr (n = 8–9). IL-1β secretion in sera was assessed by ELISA. Error bars represent SE. Untreated mice were used as negative control (−). t test, *p < 0.05.
(B) Macrophages from WT, trpc1−/−, and casp11−/− mice were infected with E. coli for 16 hr, and secretion of mature IL-1β in the culture supernatant was assessed by ELISA. Error bars indicate SD from the mean.
(C and D) Caspase-11, caspase-1, IL-1β, IL-18 expression level, cleavage, and secretion were assessed by western blotting of cell lysates and TCA-precipitated culture media.
(E) Macrophages from WT, trpc1−/−, and casp11−/− mice were treated with LPS (100 ng/ml, 8 hr) followed by ATP (30 min), HLLOMe (30 min), or silica (2 hr) at the indicated concentrations. Mature IL-1β in the culture supernatant was assessed by ELISA. Error bars indicate SD.
(F) Caspase-11, caspase-1, IL-1β expression level, cleavage, and secretion were assessed by western blot on cell lysates and TCA-precipitated culture media.
See also Figure S3.
DISCUSSION
In this study, we describe the involvement of the cationic channel subunit TRPC1 in promoting mature IL-1β release upon inactivation by caspase-11-mediated cleavage in response to Gram-negative bacteria E. coli and LPS. Our identification of TRPC1 as a substrate for caspase-11, and evidence for its degradation upon LPS-induced caspase-11 expression, suggest that the TRPC1 complex is remodeled at the onset of inflammation and plays a role in regulating innate immunity. Consistently, trpc1−/− macrophages show increased secretion of mature IL-1β upon infection by E. coli or stimulation with LPS together with extracellular ATP or lysosome disruption, but show no defect in caspase-1 cleavage or pyroptosis, suggesting that remodeling of the TRPC1 complex regulates the unconventional secretion pathway independently of caspase-1 activation. Thus, our results demonstrate a role for caspase-11 in regulating IL-1β secretion through the degradation of TRPC1 and the remodeling of associated channel complexes independently of caspase-1.
As we reported previously (Kang et al., 2000), casp1−/− mice, which were independently generated by two separate groups, do not express caspase-11 and accordingly should be referred to as casp1−/− casp11−/− mice. Casp1−/− Casp11Tg mice that are deficient for caspase-1 and express caspase-11 were generated recently and helped to clarify the distinct functional roles of caspase-1 and caspase-11 in innate immunity (Kayagaki et al., 2011). Importantly, caspase-11, but not caspase-1, is critical for sensitivity to LPS-induced septic shock and confers resistance against two pathogenic species of Gram-negative Burkholderia; therefore, caspase-11 is an important regulator of inflammation (Kayagaki et al., 2011; Rathinam et al., 2012; Wang et al., 1996, 1998). Caspase-11 is unique among caspases in that it is highly regulated at levels of transcription upon the activation of inflammatory responses (Wang et al., 1996, 1998). Pro-caspase-11 is inducible upon stimulation of multiple ligands involved in innate immunity, including LPS, IFNγ, and type I IFN (Broz et al., 2012; Gurung et al., 2012; Hur et al., 2001; Lee et al., 2001; Lin et al., 2000; Rathinam et al., 2012; Schauvliege et al., 2002; Wang et al., 1998; Yen and Ganea, 2009). Whether expression of caspase-11 is sufficient for its autoactivation is the subject of debate (Broz et al., 2012; Kang et al., 2000, 2004; Rathinam et al., 2012; Wang et al., 1998). Indeed, although pro-caspase-11 induction by extracellular LPS or IFNγ is sufficient for its autoprocessing in the p30 form, an additional signal that was recently identified to be cytosolic LPS is required to trigger caspase-11-dependent pyroptosis and caspase-1 activity (Broz et al., 2012; Hagar et al., 2013; Kayagaki et al., 2013; Rathinam et al., 2012). However, due to the lack of a specific caspase-11 activity assay and identification of a caspase-11-specific substrate, caspase-11 activity per se was not assessed in previous studies. Therefore, cytosolic LPS may also control pyroptosis and caspase-1 activation by acting downstream of caspase-11 activation. Detection of autoprocessed p30 demonstrates that caspase-11 is active in macrophages treated with extracellular LPS (Rathinam et al., 2012). In the present study, we show that TRPC1 is degraded by caspase-11 in macrophages treated with extracellular LPS, demonstrating that caspase-11 can be activated without triggering pyroptosis.
Despite the high interest in caspase-11, very few of its substrates have been identified. Caspase-11 can control cell mobility and intracellular vesicular trafficking by modulating actin cytoskeleton independently of its protease activity (Akhter et al., 2012; Li et al., 2007). On the other hand, caspase-11 is capable of autoprocessing (Kang et al., 2000). In addition, direct cleavage of caspase-3 by caspase-11 was shown to contribute to deleterious cell death in several animal models of neurodegenerative disease (Hisahara et al., 2001; Kang et al., 2000; Shibata et al., 2000). However, the requirement for caspase-3 in caspase-11-dependent pyroptosis has not yet been investigated (Kang et al., 2002; Kayagaki et al., 2011). Lastly, the molecular mechanism of caspase-11-dependent caspase-1 activation downstream of noncanonical NLRP3 activators is unknown. Although caspase-11 associates with caspase-1, caspase-1 is not a direct substrate for caspase-11 (Wang et al., 1998). Thus, TRPC1 constitutes the only noncaspase substrate for caspase-11 identified so far.
TRPC1 assembles with other TRPCs to form heterotetrameric cation-permeable channels with current characteristics different from those of homotetramers (Strübing et al., 2001). In particular, TRPC1 reduces the Ca2+ permeability of the respective channels in homodimers while participating in the channel pore of at least TRPC3, TRPC4, and TRPC5. TRPC1 also inhibits TRPV6 expression at the plasma membrane (Schindl et al., 2012; Strübing et al., 2001). In neurons, TRPC1 forms a complex with TRPC5 that localizes at the plasma membrane (Strübing et al., 2001). However, TRPC1 cannot be detected at the plasma membrane in macrophages where TRPC5 is not expressed (Kunert-Keil et al., 2006). Given the complexity of these functional interactions, TRPC1 degradation during an inflammatory response may alter the gating properties of TRPC1-containing channels in the plasma or intracellular membranes to promote unconventional protein secretion.
TRPC1 deficiency increases the secretion of mature IL-1β and IL-18 by macrophages without affecting pyroptosis or the cleavage of caspase-1. Thus, caspase-11 regulates IL-1β and IL-18 secretion both at the cytokine maturation level through caspase-1 and at the extracellular release level through TRPC1. Notably, TRPC1 deficiency potentiates not only caspase-11-dependent IL-1β secretion but also caspase-11-independent IL-1 secretion, suggesting a potentially wider role of TRPC1 in regulating secretion. Since TRPC1 degradation induced by LPS strictly depends on caspase-11, LPS priming may modulate the channelosome through redundant TRPC1- and caspase-11-independent mechanisms, and bypass the caspase-11 requirement in unconventional protein secretion in response to canonical NLRP3 stimulation. It would be interesting to investigate the roles of caspase-11 and caspase-1 in modulating other TRP channel activities in other inflammatory responses.
Taken together, our data point to a role of cationic channels in the downstream process of unconventional secretion of IL-1β and IL-18, and suggest that in addition to its already described regulatory functions in the cytoskeleton, cell death, and caspase-1 activation, caspase-11 controls IL-1β and IL-18 release by modulating the cationic channel composition of the cell. Our results also suggest a possible role for the TRPC1-associated channelosome in regulating unconventional secretion in general, which needs to be investigated in future studies.
EXPERIMENTAL PROCEDURES
Mice
C57B6, casp11−/− (Wang et al., 1998), casp1−/− mice deficient for both caspase-1 and caspase-11 (casp1−/− casp11−/− mice) (Kang et al., 2000; Kuida et al., 1995), and trpc1−/− (Liu et al., 2007) mice were housed at the Warren Alpert Animal Facility (Harvard Medical School) and Children’s Hospital Animal Facility under specific pathogen-free conditions. Experiments were performed in accordance with federal and institutional guidelines. The mice received food and water ad libitum. Inflammatory peritoneal macrophages were elicited by intraperitoneal injection of 0.75 ml thioglycolate broth and harvested by peritoneal wash 4 days later.
Cell-Growth Conditions
HEK293T and LN-18 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) and DMEM/F12 (GIBCO), respectively, supplemented with 1× penicillin/streptomycin (PS; GIBCO) and 10% fetal bovine serum (FBS; GIBCO). Inflammatory peritoneal macrophages were cultured at 0.5–1 × 106 cells/ml in DMEM (GIBCO) supplemented with 1× PS and 10% FBS. For all E. coli infection experiments, cells were grown in the absence of PS. For trichloroacetic acid (TCA) precipitation of the media, cells were rinsed and treated in DMEM+PS without serum.
Activation of the Inflammasome
E. coli (strain J53) were grown in lysogeny broth (LB) to log expansion phase, resuspended in PBS, and added to macrophages. For IL-1β secretion assay, gentamycin (50 μg/ml) was added 1.5 hr after infection and supernatant was collected after a total of 16 hr following infection.
Constructs
Constructs coding for FLAG-caspase-11, FLAG-caspase-11CARD98 (aa 1–98), FLAG-caspase-11CARD103 (aa 1–103), FLAG-caspase-11p30, FLAG-caspase-11p10, pcDNA-IL-1β, and pcDNA-Casp11C255G were previously described (Li et al., 2008). FLAG-caspase-11p20 (aa 79–285) was cloned in pFLAG3X. HA-TRPC1, HA-TRPC1DC (deleted of the C-terminal domain, aa 1–649), HA-TRPC2, HA-TRPC3, HA-TRPC4, HA-TRPC5, and HA-TRPC6 were kind gifts from P.F. Worley and J. Yuan (Yuan et al., 2003). HA-TRPC1-N (N-terminal domain, aa 1–315), HA-TRPC1ΔN (deleted of the N-terminal domain, aa 306–760), HA-TRPC1-TM (transmembrane domain, aa 306–613) were cloned following PCR amplification.
Reagents
The following reagents were used: LPS from E. coli 0111:B4, ATP, and nigericin (Sigma-Aldrich); silica (MIN-U-SIL 5; U.S. Silica); HLLOMe (Chem-Impex International); z-VAD-fmk (Alexis); and IDUN-6556 (Linton et al., 2005).
Immunoprecipitation
For coimmunoprecipitation experiments, HEK293T cells were lysed in lysis buffer (Tris HCl, pH 7.6 50 mM, NaCl 400 mM, NP-40 1%, PMSF 1 mM, and protease inhibitors). HA-TRPC1 and FLAG-caspase-11 were immunoprecipitated with anti-HA and anti-FLAG M2 agarose beads (Sigma-Aldrich), respectively, for 4 hr at 4°C. Immunoprecipitations were washed in lysis buffer and proteins were eluted in Laemmli sample buffer 2×. For anti-TRPC1 immunoprecipitation, 5 × 106 macrophages were lysed in RIPA buffer. TRPC1 was immunoprecipitated using protein A/G UltraLink resin (Thermo Scientific) and anti-TRPC1 antibody (Millipore), and eluted in Laemmli sample buffer.
Transfection
HEK293T cells were transfected using the CaCl2 procedure. LN-18 cells were transfected using transIT-LT1 transfection reagent (Mirus Bio).
Western Blot Analysis
Samples were analyzed by SDS-PAGE, transferred to PVDF membranes, blocked, and probed according to standard procedures. The following antibodies were used: anti-caspase-11 (Wang et al., 1998), anti-caspase-1 (Wang et al., 1998), anti-HA (Santa Cruz biotechnology), anti-FLAG M2 (Sigma-Aldrich), anti-TRPC1 (Millipore), anti-actin (Millipore), anti-IL-1β (R&D Systems), anti-IL-18 (MBL), and anti-laminB (Santa Cruz).
In Vitro Caspase Cleavage Assay
In vitro production of 35S-labeled TRPC1 and IL-1β was performed using TNT-coupled transcription/translation kits (Promega). Purification of recombinant Caspase-11 p30 and in vitro cleavage assay by recombinant Caspase-11 p30 or active human Caspase-1 (p30; Chemicon International) were performed as described previously (Kang et al., 2000).
ELISA
In vitro secretion of IL-1β and TNF-α in macrophage culture supernatants was measured using mouse IL-1β/IL-1F2 and TNF-α DuoSet kits (R&D Systems). In vivo secretion of IL-1β in the serum was assessed using the mouse IL-1β/IL-1F2 Quantikine kit (R&D).
Intracellular Ca2+ Measurement
Cells were loaded with the ratiometric Ca2+ indicator Fura-2/AM in Opti-MEM (at 37°C, 5% CO2) for 30 min, washed, and incubated for an additional 15 min prior to imaging. Ca2+ imaging was performed in Ringer’s solution containing 2 mM Ca2+. Excitation was carried out at 340 nm and 380 nm using a Lambda DG4 Ultra High Speed Wavelength Switcher (Sutter Instrument), and emission was collected at 510 ± 10 nm using a Hamamatsu Orca R2 CCD camera and Slidebook data acquisition software.
Cytotoxicity Assay
Cell death was measured as the percentage of lactate dehydrogenase (LDH) release according to standard protocols. DMEM+10% FBS was used as the blank control, and supernatants of control cells treated with Triton X-100 0.01% were used to measure total LDH release. Cell death was measured by propidium iodide (PI) incorporation. PI (1 μg/ml) was added to the cell culture media, and cells were observed by live imaging using a Nikon Ti inverted microscope equipped with a 10× Plan Apo NA 0.3 objective lens. Images were acquired with a Hamamatsu ORCA-R2 cooled CCD camera controlled with MetaMorph 7 software (Molecular Devices).
Supplementary Material
Acknowledgments
We thank M. Freichel (Heidelberg University) and V. Flockerzi (Saarland University) for the gift of trpc1−/− mice, P.F. Worley (Johns Hopkins University School of Medicine) for sharing the HA-TRPC1 and HA-TRPC1ΔC constructs, and J. Yuan (University of North Texas) for sharing the HA-TRPC2, HA-TRPC3, HA-TRPC4, HA-TRPC5, and HA-TRPC6 constructs. This work was supported in part by the National Institute on Aging (R37 AG012859 to J.Y.), the French National Research Agency (ANR-13-JSV3-0002-01 to B.P.), and the European Research Council (ERC-2012-StG_20111109 to T.H.).
Footnotes
AUTHOR CONTRIBUTIONS
B.F.P. and J.Y. conceived the experiments. B.F.P., B.N.D., M.J., A.P., M.M.L., M.K., and H.Z. performed the experiments. B.F.P., B.N.D., D.E.C., and J.Y. analyzed the results. A.D. and T.H. contributed reagents. B.F.P. and J.Y. wrote the manuscript.
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2014.02.015.
References
- Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, Miao EA. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339:975–978. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DH, Voss OH, Doseff AI, Hassan H, Azad AK, et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity. 2012;37:35–47. doi: 10.1016/j.immuni.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ. TRPC1: store-operated channel and more. Pflugers Arch. 2005;451:53–60. doi: 10.1007/s00424-005-1441-3. [DOI] [PubMed] [Google Scholar]
- Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. 2012;490:288–291. doi: 10.1038/nature11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, Zamboni DS, Roy CR. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci USA. 2013;110:1851–1856. doi: 10.1073/pnas.1211521110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Gurung P, Malireddi RK, Anand PK, Demon D, Vande Walle L, Liu Z, Vogel P, Lamkanfi M, Kanneganti TD. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-β (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem. 2012;287:34474–34483. doi: 10.1074/jbc.M112.401406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisahara S, Yuan J, Momoi T, Okano H, Miura M. Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J Exp Med. 2001;193:111–122. doi: 10.1084/jem.193.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur J, Kim SY, Kim H, Cha S, Lee MS, Suk K. Induction of caspase-11 by inflammatory stimuli in rat astrocytes: lipopolysaccharide induction through p38 mitogen-activated protein kinase pathway. FEBS Lett. 2001;507:157–162. doi: 10.1016/s0014-5793(01)02975-1. [DOI] [PubMed] [Google Scholar]
- Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, Huang Z, Srinivasan A, Tomaselli KJ, Thornberry NA, et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol. 2000;149:613–622. doi: 10.1083/jcb.149.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SJ, Wang S, Kuida K, Yuan J. Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ. 2002;9:1115–1125. doi: 10.1038/sj.cdd.4401087. [DOI] [PubMed] [Google Scholar]
- Kang SJ, Popat R, Bragdon C, Odonnell K, Phelan S, Yuan J, Sonis ST. Caspase-11 is not necessary for chemotherapy-induced intestinal mucositis. DNA Cell Biol. 2004;23:490–495. doi: 10.1089/1044549041562302. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszyński A, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- Keller M, Rüegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- Kepp O, Galluzzi L, Zitvogel L, Kroemer G. Pyroptosis - a cell death modality of its kind? Eur J Immunol. 2010;40:627–630. doi: 10.1002/eji.200940160. [DOI] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Kunert-Keil C, Bisping F, Krüger J, Brinkmeier H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genomics. 2006;7:159. doi: 10.1186/1471-2164-7-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Hur J, Lee P, Kim JY, Cho N, Kim SY, Kim H, Lee MS, Suk K. Dual role of inflammatory stimuli in activation-induced cell death of mouse microglial cells. Initiation of two separate apoptotic pathways via induction of interferon regulatory factor-1 and caspase-11. J Biol Chem. 2001;276:32956–32965. doi: 10.1074/jbc.M104700200. [DOI] [PubMed] [Google Scholar]
- Li J, Brieher WM, Scimone ML, Kang SJ, Zhu H, Yin H, von Andrian UH, Mitchison T, Yuan J. Caspase-11 regulates cell migration by promoting Aip1-Cofilin-mediated actin depolymerization. Nat Cell Biol. 2007;9:276–286. doi: 10.1038/ncb1541. [DOI] [PubMed] [Google Scholar]
- Li J, Yin HL, Yuan J. Flightless-I regulates proinflammatory caspases by selectively modulating intracellular localization and caspase activity. J Cell Biol. 2008;181:321–333. doi: 10.1083/jcb.200711082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin XY, Choi MS, Porter AG. Expression analysis of the human caspase-1 subfamily reveals specific regulation of the CASP5 gene by lipopolysaccharide and interferon-gamma. J Biol Chem. 2000;275:39920–39926. doi: 10.1074/jbc.M007255200. [DOI] [PubMed] [Google Scholar]
- Linton SD, Aja T, Armstrong RA, Bai X, Chen LS, Chen N, Ching B, Contreras P, Diaz JL, Fisher CD, et al. First-in-class pan caspase inhibitor developed for the treatment of liver disease. J Med Chem. 2005;48:6779–6782. doi: 10.1021/jm050307e. [DOI] [PubMed] [Google Scholar]
- Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, Swaim WD, Beech D, Yildrim E, Singh BB, et al. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(-/-) mice. Proc Natl Acad Sci USA. 2007;104:17542–17547. doi: 10.1073/pnas.0701254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauvliege R, Vanrobaeys J, Schotte P, Beyaert R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J Biol Chem. 2002;277:41624–41630. doi: 10.1074/jbc.M207852200. [DOI] [PubMed] [Google Scholar]
- Schindl R, Fritsch R, Jardin I, Frischauf I, Kahr H, Muik M, Riedl MC, Groschner K, Romanin C. Canonical transient receptor potential (TRPC) 1 acts as a negative regulator for vanilloid TRPV6-mediated Ca2+ influx. J Biol Chem. 2012;287:35612–35620. doi: 10.1074/jbc.M112.400952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Hisahara S, Hara H, Yamawaki T, Fukuuchi Y, Yuan J, Okano H, Miura M. Caspases determine the vulnerability of oligodendrocytes in the ischemic brain. J Clin Invest. 2000;106:643–653. doi: 10.1172/JCI10203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Wang S, Miura M, Jung Yk, Zhu H, Gagliardini V, Shi L, Greenberg AH, Yuan J. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem. 1996;271:20580–20587. doi: 10.1074/jbc.271.34.20580. [DOI] [PubMed] [Google Scholar]
- Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92:501–509. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- Yen JH, Ganea D. Interferon beta induces mature dendritic cell apoptosis through caspase-11/caspase-3 activation. Blood. 2009;114:1344–1354. doi: 10.1182/blood-2008-12-196592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JP, Kiselyov K, Shin DM, Chen J, Shcheynikov N, Kang SH, Dehoff MH, Schwarz MK, Seeburg PH, Muallem S, Worley PF. Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell. 2003;114:777–789. doi: 10.1016/s0092-8674(03)00716-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.