

Summary
Next-generation DNA sequencing (NGS) can be used to reconstruct eco-evolutionary population dynamics and to identify the genetic basis of adaptation in laboratory evolution experiments. Here, we describe how to run the open-source breseq computational pipeline to identify and annotate genetic differences found in whole-genome and whole-population NGS data from haploid microbes where a high-quality reference genome is available. These methods can also be used to analyze mutants isolated in genetic screens and to detect unintended mutations that may occur during strain construction and genome editing.
Keywords: Evolutionary genomics, Genome re-sequencing, Variant caller, Single-nucleotide variant, Structural variant, Insertion sequence, Mobile genetic element, Gene conversion
1. Introduction
Recent developments in next-generation DNA sequencing (NGS) technologies have increasingly made them affordable and accessible to any researcher (1, 2). The NGS platforms that are widely available today – including Illumina, Roche 454, Ion Torrent, Pacific Biosciences, and ABI SOLiD systems – produce many DNA sequencing reads (thousands to millions to billions) of various lengths (~50 to >3000 bases). This data is fundamentally different from classic Sanger sequencing, which generates just one read of 400–900 bases per reaction, in that NGS instruments generate many orders of magnitude greater base coverage, but each individual read sequence is often shorter and of lower quality. NGS technologies can be used for RNA-seq transcriptomics, the de novo assembly of non-model organism genomes, and characterizing epigenetic DNA modifications, among many other types of studies. One of the most common uses of NGS is to “re-sequence” samples from a laboratory organism or population that is very closely related (typically >99.9% nucleotide identity) to a complete, high-quality reference genome to identify the salient genetic differences between them.
There are three main steps in analyzing NGS genome re-sequencing data: 1) mapping each sequencing read to the reference genome, 2) identifying genetic variation present in the sample by searching for discrepancies between aligned reads and the reference genome, and 3) annotating how genes are affected by these sequence differences. Many software tools exist for read mapping, with various trade-offs in speed and sensitivity and algorithmic subtleties that can affect the downstream analysis steps (3). Similarly, variant callers differ a great deal, both in how sophisticated their statistical models are for maximizing sensitivity while minimizing false-positive predictions and in what types of genetic variation they are designed to find (4, 5). Three main categories of genetic variation exist: changes of a single base (Single nucleotide variants, SNVs), insertions and deletions of a few nucleotides (indels), and more complicated chromosomal rearrangements and larger insertions and deletions (structural variants, SVs). The latter types can be considerably more challenging to identify from NGS data. Many research groups and sequencing centers have created custom computational pipelines tailored to their needs by combining any number of read mapping, variant calling, and annotation programs.
Here, we describe how to use breseq, an open-source pipeline that automates all of the NGS genome re-sequencing analysis steps from mapping to annotation. In contrast to workflows developed for analyzing mainly human genomes (4, 5), breseq has been optimized for haploid microbial-sized genomes (<20 Mb). Because breseq is intended for use on laboratory evolution experiments, molecular genetic experiments, and synthetic biology experiments with microbes – where detecting a single key genetic change in a sample can be very important (6–9) – it emphasizes sensitivity over speed and reports evidence for a wider variety of genetic variants than most other tools that are currently available. The breseq pipeline produces output in an annotated HTML format that is accessible to non-experts; in a Genome Diff flat file format for comparing mutations predicted in different samples and for applying mutations to a reference genome; and also in community formats that can be used for visualizing mapped reads and other downstream analyses.
2. Materials
2.1 Computer system and software
Access to a computer system with a command-line prompt in a Unix-like environment, such as Linux or Mac OS X. On Windows machines, it is possible to compile and run breseq under Cygwin. For analysis of a few samples, a personal computer is likely sufficient. If you have many samples to process, you may want to install and use breseq on a computer cluster or in a cloud computing environment.
breseq pipeline (http://barricklab.org/breseq). Download, compile, and install according to the included documentation. Version 0.24 was used to generate the figures and examples for this tutorial.
For editing Genome Diff files, a plain text editor that can be set to not wrap lines of text, such as vi or emacs on a Unix-like system, TextWrangler on a Mac OS X system, or Notepad++ on a Windows system.
For viewing read alignments, Tablet (http://bioinf.scri.ac.uk/tablet/) (10) or the Integrative Genomics Viewer (http://www.broadinstitute.org/igv/) (11).
2.2 Data and tutorial files
NGS read files for clonal or whole-population genomic DNA samples in FASTQ format. These files should be provided by the facility that sequenced your samples. breseq does not require input FASTQ files to use a specific base quality encoding scheme, and it is compatible with most current technologies, except SOLiD color space data.
Reference genome sequence files in GenBank, GFF3, or FASTA format. GenBank or GFF3 files with feature annotations are preferred, as they enable breseq to report the effects of predicted mutations on genes. Suitable reference sequences can be downloaded from GenBank (http://www.ncbi.nlm.nih.gov/genbank) or the European Nucleotide Archive (http://www.ebi.ac.uk/ena) for many organisms. It is generally impractical to use breseq on reference genomes that are >20 Mb in size, and it assumes that the reference genome for clonal samples is haploid.
For this tutorial, archives of example input and results files available from the breseq website (http://barricklab.org/breseq). Example commands in the methods section use these input read and reference sequence files. The archives also contain examples of output files that illustrate specific points about using breseq discussed in the methods section.
3. Methods
Commands to be executed in the shell appear in monospaced font following a prompt ( $), which is not part of the typed command. If there is no new prompt at the beginning of a subsequent printed line, then the full command is wrapped to the following line and should be entered into the shell all at once.
3.1 Predicting mutations in a clonal sample
In a command-line shell, navigate to inside a directory on your computer system containing both the FASTQ read and GenBank reference files. The commands presented here assume that you are inside the Clonal_Sample directory of the downloaded tutorial data. This sample is an Illumina Genome Analyzer NGS dataset of paired-end 36-base reads generated from genomic DNA isolated from a 20,000-generation clone from a long-term laboratory evolution experiment with E. coli (6). The fragment size of the sequenced DNA library was 140 ± 20 bases (s.d.). The reference sequence is the complete genome of the ancestral strain (12). The example commands presented here can be generalized to any similar re-sequencing analysis of your data.
- For the example data, the following breseq command should be executed from the command prompt and allowed to finish running:
$ breseq -j 4 -o Clonal_Output -r REL606.gbk SRR030257_1.fastq SRR030257_2.fastq
Depending on your computer, it may take several hours to complete this breseq command. The -j option controls how many processes will be used by the pipeline and should be set to the number of CPU cores on your machine for optimal performance. In this example, 4 cores were used. The -o option controls where the analysis files should be directed. In this case, they will be directed to the Clonal_Output directory. If this directory does not exist, it will be created. The -r option identifies the file REL606.gbk as containing the reference genome for comparison to your NGS reads. Finally, all files listed at the end of the line without preceding option flags ( SRR030257_1.fastq and SRR030257_2.fastq) are the read files supplied by the sequencing facility. Typing just breseq with no options will display a help message describing other basic options (Fig. 1). It is important to consider what breseq settings are appropriate for your data. For the input DNA sequencing reads, paired-end information is not currently utilized (see Note 1), and 40- to 100-fold coverage of the reference genome is generally optimal for clonal samples (see Note 2). Reference sequences are expected to be haploid microbial chromosomes or plasmids, and the NGS read dataset for a sample should have nearly complete and even sequencing coverage of each one. Advanced options can relax some of these reference sequence expectations (see Note 3).
After breseq has successfully finished executing, several new directories should exist in the Clonal_Output path. Directories with numbered prefixes 01, 02, 03, and so on, contain intermediate files generated by breseq and can typically be ignored and deleted at this point. They allow breseq to skip steps that are already complete if its execution is interrupted and restarted. The output directory contains files and figures describing mutations predicted in the sample. The data directory contains files that can be used to visualize mapped reads and FASTQ files containing reads that did not match the reference genome sequence. If you are executing breseq on a remote computer, you will need to copy the output and data directories back to your computer to view them locally.
Open the summary.html file located in the output directory in a web browser to bring up a “Summary Statistics” page that displays general information about a breseq run (Fig. 2). The “Read File Information” section reports statistics about the reads and how they aligned to the reference sequence. If the percentage of reads aligned is not >90% in your sample, then you may have a problem with the quality of your sequencing data or it may need further processing before analysis (see Note 4). Alternatively, the reference genome that you provided may be too divergent from your DNA sample to reliably map reads to it and call mutations (see Note 5). The “Reference Sequence Information” section reports the depth of read coverage for each reference sequence. The “coverage” links in this table open image files displaying the coverage across each reference sequence that can be used to detect amplifications or deletions of very large genomic regions.
Open the index.html file located in the s directory in a web browser to display the main “Mutation Predictions” page containing tables listing differences found between the sample and the reference genome (Fig. 2). Breseq utilizes three types of information to predict mutations: read alignment (RA), missing coverage (MC), and new junction (JC) evidence. In the main “Predicted mutations” table at the top of this page, links on the left side of this page display alignments and coverage graphs related to the RA, MC, and JC evidence that supported a particular mutation call. The “Unassigned…” tables contain evidence items that also indicate differences between the sample and the reference, but that could not be fully resolved to describe precise genetic changes. We will discuss how to manually interpret the unassigned evidence in Section 3.2. For now, we concentrate on explaining the three types of evidence and how they are used to predict mutations.
For reasonable input files, nearly all mutations predicted by breseq should be correct. That is, breseq should report only evidence for real sequence differences between the sample and the reference. Every single mutation and evidence item predicted from the Clonal_Sample data is genuine, for example. However, there is always the possibility of some false-positive predictions passing the statistical tests used by breseq, resulting in evidence items appearing in these tables that are not actually well supported upon further examination. As we discuss each type of evidence below, we give advice for recognizing the most common false positives you may encounter. These are further illustrated with breseq output files in the Poor_Evidence_Examples supplement. The opposite problem of false negatives – where breseq does not recover or fully interpret evidence supporting genuine mutations – is discussed in later sections.
Read alignment (RA) evidence is derived from analyzing the columns of bases in reads aligned to each position in a reference sequence. breseq uses a standard Bayesian SNP caller for RA evidence. The “score” reported is the negative log10 of the posterior probability that the base is not the reported base, corrected for multiple testing by multiplying by the total genome size. In this calculation, breseq uses a re-calibrated base error probability model including single-base indels that is fit from the data. RA evidence can support mutations resulting in single-nucleotide substitutions as well as insertions and deletions that are shorter than the read length. Several pieces of adjacent RA evidence may be merged to make a single mutation prediction (e.g., of an insertion of two nucleotides).
You can evaluate RA evidence by clicking on the link to bring up a color-coded alignment of reads overlapping the position in question. The most common causes of false-positive predictions can be readily detected on this page. If the base quality scores supporting the variant are uniformly lower (Fig 3, “RA”, center base in right panel) or almost always associated with reads on one strand (Fig 3, “RA”, right panel, mutated “C” base always maps to the top strand indicated by the arrows to the right of the bases), then this may be a problem sequence context for the NGS technology where there is locally an unexpectedly high rate of errors. Alternatively, false-positive RA evidence may originate from mapping reads from the sample back to the reference genome at the wrong site. This may happen when there is a novel sequence in the sample that is not present in the reference, causing reads derived from this new sequence to be mapped incorrectly to their best match in the reference genome. This situation can often be recognized when a subset of the reads supporting a putative RA evidence item do not match across their entire length or have multiple discrepancies from the reference sequence in common (Fig 4, “Mismapped Reads”). Other false-positive RA evidence may be derived from local misalignment of reads near true examples of short insertions or deletions in the sample. Precautions are taken by breseq to not count “masked” bases on each end of a read (shown in lowercase in the alignment) when these could be aligned incorrectly if they fell within true expansions or contractions of short sequence repeats (Fig 4, “Ambiguous Ends”). However, breseq does not include a full local read realignment step (4), which may be necessary to make the most optimal prediction in cases where there are base errors in reads in close proximity to true indels or point mutations (Fig 4, “Local Misalignment”).
Missing coverage (MC) evidence is derived from finding places in the genome where no reads align and then extending these intervals in both directions and through repeat regions. Extension of the MC interval is stopped when uniquely mapped read coverage exceeds a threshold that is automatically set by fitting the distribution of read-depth coverage found across all normal sites in the current reference sequence. Deletion mutations with precise endpoints are predicted from MC evidence in conjunction with JC evidence, or from MC evidence alone when the ends of the deletion are in similarly-oriented copies of the same repeat region (Fig. 5). Otherwise, the MC evidence is left unassigned.
You can evaluate MC evidence by clicking on the link to bring up a graph of read depth coverage at this location. The MC positions have a white background, and the graph is expanded on each end to show the context of surrounding regions with a gray-shaded background. It is important to understand the difference between “unique” and “repeat” coverage lines in this graph. The former indicates that reads mapped to that location only mapped to one place in the entire reference genome. The latter means reads that mapped there also mapped to other locations in the genome. So even if this example of the repeat was deleted in a sample, you would still find reads that matched this location in the reference. (The contribution to the repeat coverage graph at this location is normalized: if a read mapped four places equally well, it contributes 0.25 to the repeat coverage at each site.) The most common cause of false-positive MC predictions is low sequencing coverage in a sample leading to some regions of the genome not being sampled by chance. This usually gives rise to MC evidence where coverage gradually decreases to zero on each side (Fig. 3, “MC”, right panel), rather than the sharp cliffs on each end expected for real deletions in unique regions (Fig. 3, “MC”, left panel). You can display the alignment of reads that overlap each edge of the predicted MC evidence through the additional asterisk links shown after selecting it.
New junction (JC) evidence is predicted from split-read matches where read sequences start matching one location in the reference and then “jump” to matching another distant site. breseq uses the depth and evenness with which reads are tiled across a putative junction to judge support for it. A bona fide junction should resemble any other site in the genome as far as having an even tiling of reads with many different starting points that map across it (Fig. 3, “JC”, left panel). This characteristic is measured in the reported skew, which is the negative log10 probability of the hypothesis that this tiling is unusual. Junction predictions are rejected when they have a high skew. Predicting junctions is computationally expensive and can be disabled using the --no-junction-prediction flag if you are not interested in mutations that generate structural variants. JC evidence is used to predict insertions of new copies of mobile genetic elements that create two new junctions between the target site and existing, usually multicopy, sequences corresponding to the element elsewhere in the genome. Note that precise prediction of mobile element insertions requires these elements to be annotated as repeat_region features in the input reference files. As mentioned before, JC and MC evidence are used together to predict deletions with precise endpoints as long as at least one side of the new junction maps to a unique site in the genome (Fig. 5).
You can evaluate JC evidence by clicking on the link to bring up an alignment of reads matching across the putative junction better than they match to any position in the reference sequence. False-positive JC predictions can be detected from uneven tiling of reads across the sequence junction, especially when many reads supporting the junction have unaligned portions at their ends that may indicate that they are being mismapped in the junction context (Fig. 3, “JC”, right panel). Additional links from this page display each of the two sites in the original genome connected by the new junction. Usually, these graphs should show mostly one side of a read mapping up to the junction and then the match stopping because the rest of the read supports the new junction and jumps to the distant site in the genome. But in some cases, such as for tandem amplifications, both the old and new junctions may be present, so some reads may align well to the original genome sequence and some may align to the junction. If a side of the JC evidence is highlighted in orange, this indicates that side falls in a sequence repeat and could also have matched other coordinates in the genome in addition to the one that is displayed. This situation is common for mobile element insertions and for unassigned evidence that requires human intervention to resolve.
Of the forty-five mutations separating this evolved clone from the ancestor (6), thirty-nine are completely predicted by breseq on the first pass. Four of the remaining mutations are not completely predicted by breseq because they involve repeat sequences, but they can be resolved from unassigned evidence items, as detailed in the next section. The final two remaining mutations are a large chromosomal inversion mediated by two oppositely oriented copies of an IS1 transposable element and a base substitution in one of the 28 original copies of IS1 in the reference genome. They were identified either by Southern blotting (13) or by Sanger sequencing, rather than by analyzing NGS data. Though relatively rare in most samples (2/45 mutations in this one), it is important to be aware of these potential “holes” in a breseq analysis. Some types of mutations are difficult or simply impossible to predict from short-read NGS data (see Note 6).
Fig. 1.

Basic breseq command line help.
Fig. 2.
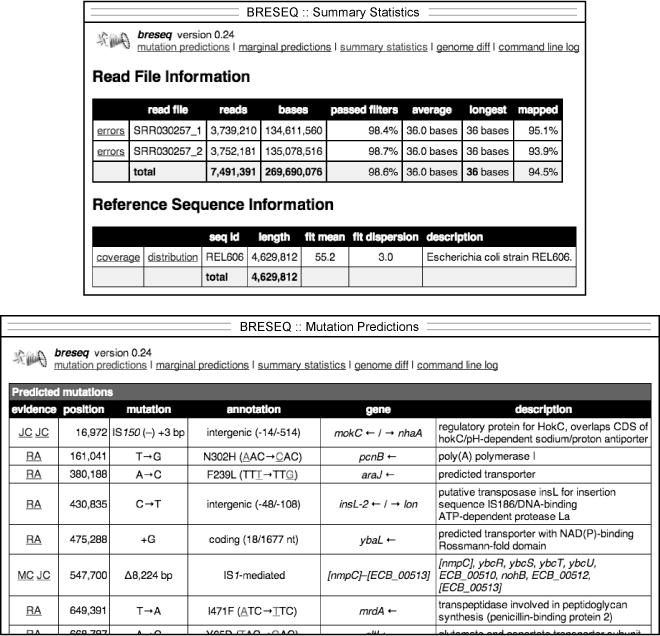
Example of breseq output. The upper panel shows a portion of the summary.html file which displays general information about the read data sets, reference sequence, and run parameters. The lower panel shows part of the main index.html page reporting predicted mutations.
Fig. 3.
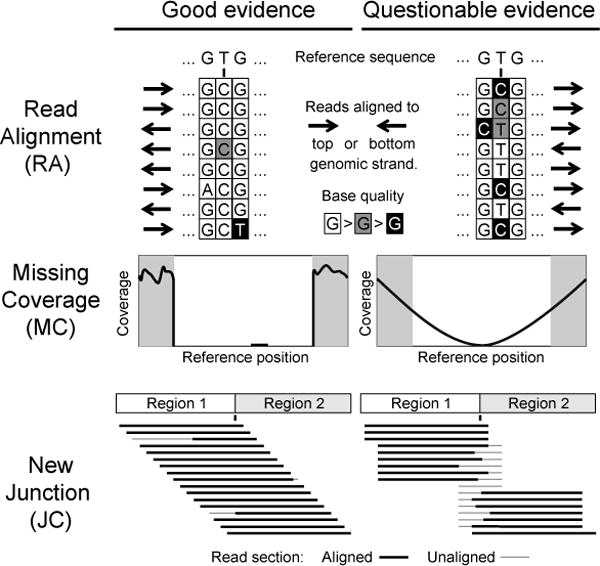
Evaluating evidence supporting predicted mutations. Characteristics of high-quality (left column) and low-quality (right column) evidence items that you may encounter in breseq output are shown as discussed in the text.
Fig. 4.
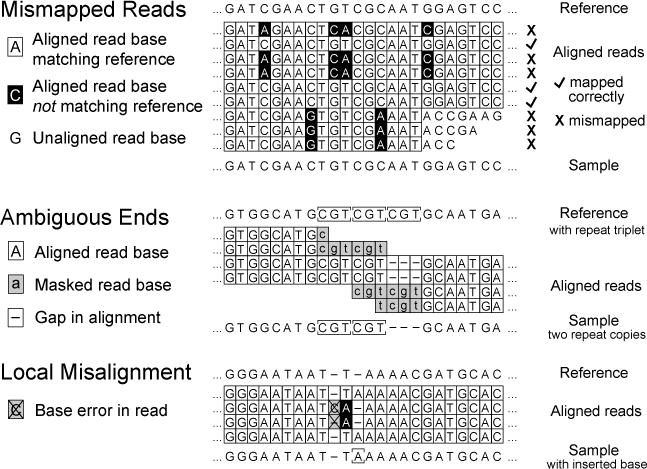
Possible causes of spurious or low-quality read alignment (RA) evidence. As described in the text, mismapping of reads to an incorrect reference genome site or local misalignment of bases in correctly mapped reads containing base errors can degrade accuracy and sensitivity when predicting micro-indels and single nucleotide variants.
Fig. 5.
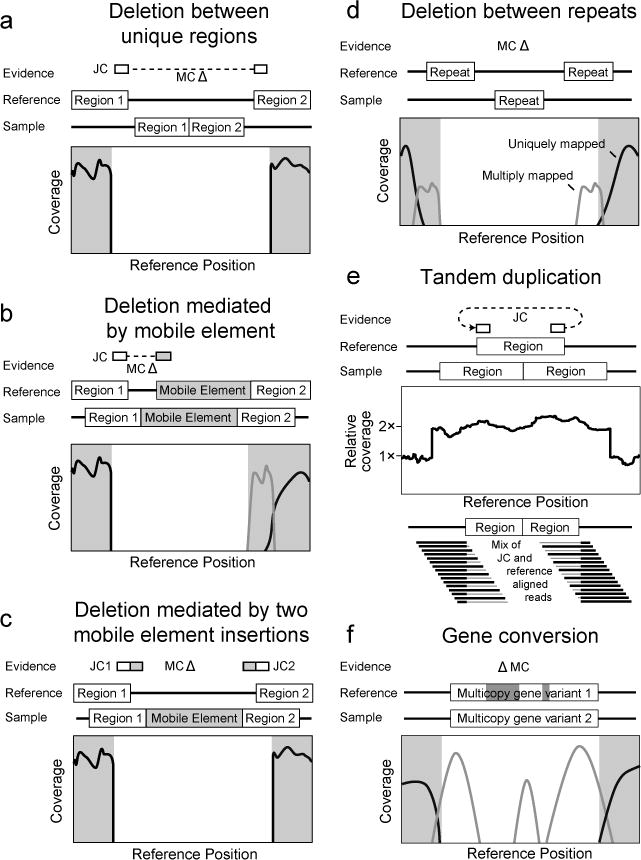
Evidence supporting complex mutations. In each case schematics of the reference genome and the genome of a sequenced clone are shown. Evidence items that would support the genetic difference between the two genomes are shown above. Relevant graphs of read-depth coverage or read alignments are shown below. See the discussion in the text for more details.
3.2 Resolving unassigned evidence and mutating a reference sequence
Using a plain text editor, open the file output.gd located in the output directory of the breseq results from running the commands in Section 3.1. (Alternatively, download the Clonal_Output archive containing the output and data result directories.) This output file is a “Genome Diff” that describes mutational differences in a sample with respect to the reference genome. The purpose of this breseq-specific file is related to, but slightly different from, the Variant Call Format (VCF) file format (14) (see Note 7). Genome Diffs are tab-delimited text files where each line describes a mutation or a piece of evidence which could support a mutation. A full description of the file format can be found in an appendix of the documentation included with the breseq source code. The gdtools command installed as part of breseq can be used to perform several types of operations on Genome Diff files (see Note 8).
In addition to false-positive predictions that are possible for the reasons discussed in Section 3.1, breseq may also sometimes incorrectly rule out a piece of evidence that actually supports a real mutation. These false-negative predictions may sometimes be caught in the “Marginal Predictions” output in the file marginal.html, which shows the next-best pieces of evidence that fall below score thresholds for further manual inspection. In the example, none of these evidence items support real mutations, but they provide many examples of poor evidence (Fig. 3). False-negative predictions can occur for MC evidence if there is cross-contamination of reads between several samples, some with a deletion and some without it, such that there are enough spurious reads that coverage does not reach zero within a true deletion in a sample.
There is a special table titled “Marginal mixed read alignment evidence…” on the “Marginal Predictions” page that reports places in the genome where there is sufficient statistical support for RA evidence supporting that the sample consisted of a mixture of bases at a single reference location, rather than one consensus base. Most of the time (as in this example), these items of evidence are spurious, resulting from poor quality reads or misalignment, but it is possible that they may represent true mutations. For example, mixed RA evidence with a frequency of around 50% when sequencing a haploid genome, may indicate that a reference region was duplicated and then one of the two copies sustained a point mutation.
While on the topic of examining questionable RA evidence, it is important to point out that the files in the data directory produced by a breseq run can be used to further examine how reads are mapped to the reference genome. This can be accomplished by using the breseq bam2aln subcommand, which operates similarly to bam2cov (discussed below) but generates an HTML pileup of read alignments for the specified region instead of a coverage graph. Alternatively the reference.bam, reference.fasta, and reference.gff3 files in the data directory can be loaded into Tablet (10) or the Integrative Genomics Viewer (11) to interactively explore how breseq mapped reads to the reference genome.
In the example, each of the pieces of “Unassigned…” evidence on the main “Mutation Predictions” page supports a real mutation. In the next steps, we will explain how to examine the breseq output to manually figure out these more complicated types of mutations (Fig. 5). Then, we will code them into the Genome Diff file so that we have a complete manually curated description of all the mutations in the sample. After that, we can apply the Genome Diff to generate a mutated reference sequence and re-run the NGS data against it to verify that we have found (and correctly coded) all of the variation it revealed between the sample and reference genome.
For the second unassigned piece of MC evidence that overlaps 23 genes, the endpoints could not be accurately ascertained by breseq because they fall in two nearly identical genes (manB and cpsG). This deletion resulted from a homologous recombination event between equivalent positions in these genes that deleted the intervening sequences and left one hybrid gene copy behind (Fig. 5d). Notice that the ends of this MC evidence item are listed as ranges of positions because they fall in sequence repeats.
- The breseq bam2cov subcommand can generate graphs and tables of coverage over a user-specified region of the genome. This command requires several files as input, but by default it will use the *.bam and *.fasta files located in a directory named data within the current working directory. So, if you run this command from within the main results directory of a breseq run you can type:
$ breseq bam2cov -t -p 0 REL606:2031650-2055600
The output REL606/2031650-2055600.tab file is tab-delimited and can be opened in a spreadsheet program such as Excel. The 3rd through 6th columns can be used to determine where the deletion starts and stops based on the breakpoints in the coverage of uniquely mapped reads. - When dealing with repeat regions like this, it can also be very helpful to use BLAST (on the NCBI website or locally) to identify all of the other equivalent or near-equivalent copies of the repeat of interest in the genome. In this case, you might BLAST the sequence from the left side of the junction (obtained by clicking on the left-most star of the first MC item):
> TGCGCCAGTTTGCTGTTGATCTCACCGCTTGCCG
If you query this against the appropriate organism (Escherichia coli B str. REL606) on the NCBI website version of BLAST, then you get two perfect matches to coordinates 2031650–2031683 and 2054943–2054976. Note that the deletion can be described in multiple ways to yield the exact same change in the nucleotide sequence of the reference genome. For example, as a deletion of bases 2031650–2054942 or 2031684–2054976. The first MC evidence item and the first JC evidence item describe a deletion between the edge of an existing IS1 mobile element and a sequence within the ECB_00513 gene. The coordinates connected by the new junction agree with the ends of the missing coverage and indicate that 8224 bases corresponding to coordinates 547700–555923 were deleted. This mutation was not fully predicted by breseq because there was uncertainty in assigning the ends of the junction. Notice the orange highlighting of the IS1 side, as expected for a repetitive element, but also that the ECB_00513 side is orange. In fact, the latter junction side maps equally well to the reference starting at position 1604671 and continuing on the reverse genomic strand, explaining why breseq did not have the confidence to initially pair these two evidence items. This type of deletion may be mediated by an insertion of a new copy of a mobile element followed by rapid recombination with an existing mobile element copy to delete one copy and the sequence between them (Fig. 5b).
The second and fifth pieces of unassigned new junction evidence listed correspond to an IS150 mobile element insertion into a copy of IS1 that already exists in the genome. breseq does not fully predict this mutation because the insertion could be in any IS1 copy – the read length is too short to disambiguate which of the 28 ancestral copies – and it arbitrarily predicts junctions to different copies. Annotating this event is therefore best done by looking at the position within the IS1 feature listed (specifically, 437/768 and 435/768 nt). The true coordinates within one representative copy of IS1 of each junction end can be confirmed using BLAST. In this case, if we mapped both junctions onto the IS1 element at 241257–242024 in the genome, the first junction would match coordinates upward through 241693 and the other junction would match coordinates 241691 and above. To determine the size of the target site duplication, count how many bases are on both sides of the insertion. Specifically, the 435th, 436th, and 437th bases of the IS1 ( GTA it its direction) are now repeated on both sides of the new IS, for a target site duplication of three bases. The relative orientation of the inserted IS relative to the genome is determined by identifying how the edges of the IS150 element map within the IS1. In this case, the first base of the IS150 element (1/1443 nt) is connected to the coordinate 241691 and upward side and the last base of the IS150 element (1443/1443 nt) is connected to the up-to-coordinate 241693 side, showing that it inserted in the negative orientation within this IS1 copy (which is itself in the positive genomic direction).
The third and fourth unassigned new junctions can also be paired together by running BLAST on each sequence against the REL606 genome. The ldrC sequence ( CCGGATAATTCCGGCTTGGTGTGGATACTACTTCTC) has a single perfect mapping at 1270660–1270692 while the ldrD sequence ( CCGGTGAGGCGCAATGTTGCGGGGGCTTTATCCCTGG) has five perfect matches including one at positions 1270628–1270663. These two junctions should be paired together based on their proximity to each other (within 3 bases of one of the alternative best matches of ldrD) and that each is paired with the opposite end of an IS150 insertion. The insertion orientation is determined by identifying what edge of the IS150 element corresponds to left side of the non-insertion element (up to 1270663 in this case). As in the previous case, this corresponds to the end of the IS150 element (1977/1977 nt) and the other junction matches the beginning of the element (1/1977 nt) for another negatively oriented insertion. Bases 1270660–1270663 now exist on each side of the newly inserted IS copy, meaning that there was a target site duplication of 4 bases.
- In order to verify that our interpretation of the unassigned evidence agrees with the NGS data, we can apply our mutations to the reference genome and re-query the data against the mutated version. The following four lines of text should be added to the output.gd file to describe the four mutations that we just manually predicted:
DEL_10000_._REL606_547700_8224_mediated=IS1 DEL_10001_._REL606_2031684_23293 MOB_10002_._REL606_241691_IS150_-1_3_ambiguous=1 MOB_10003_._REL606_1270660_IS150_-1_4
Each item within a line must be separated by tab characters, rather than the underscores shown here for the sake of clarity. - Once the additional mutations are added, the gdtools APPLY command can be run to generate a mutated reference file in GFF3 format:
$ gdtools APPLY -r REL606.gbk -f GFF3 -o mutated_reference.gff3 Clonal_Output/output/output.gd
- The new GFF3 file can then be used as a reference file for rerunning breseq with the read files to verify that all mutations have been correctly predicted:
$ breseq -j 4 -r mutated_reference.gff3 -o mutated_output SRR030257_1.fastq SRR030257_2.fastq
Looking at the resulting index.html file should show a “Mutation Predictions” page with no mutations (see Note 9).
3.3 Additional examples of complex mutations
There are several other types of mutations that can be reconstructed from unassigned evidence that you may encounter (Fig. 5). The Mutation_Examples supplement contains breseq output generated from sequencing other clonal samples derived from the same E. coli ancestor to illustrate these cases.
The first type of mutation is a compound event where there has been a mobile element insertion, then later another mobile element insertion in the same orientation nearby followed by recombination to generate a deletion between them (Fig. 5c). This results in two JC items at the ends to copies of the same the mobile element with missing coverage between. It can be coded in the GenomeDiff as two separate events: one mobile element insertion and then a mobile element-mediated deletion. See the 2X_Mobile_Element_&_Deletion files for an example of this type of mutation, which is more common in longer evolution experiments.
The second additional type of mutation is a tandem duplication or amplification (Fig. 5e). This event can be detected as unassigned JC evidence between two unique regions where the original junctions remain present. To confirm that there is the expected increase in coverage of the reference genome region “looped” by this junction and to get a better estimate of its new copy number, use the breseq bam2cov subcommand as covered in Section 3.2. See the Amplification supplementary files for an example of this type of evidence.
The third type of mutation that may explain some of the unassigned evidence is a gene conversion event (Fig. 5f). This mutation occurs when a portion of one copy of a near-identical repeat element in a genome is “repaired” to have the same sequence as another repeat copy by recombination. It shows up as MC evidence because reads that used to uniquely map to parts of the repeat that differed by only a few point mutations from the other copies are no longer present. Do not be confused and believe that this missing coverage supports a deletion. The reads that map to this location now map to one of the other copies, so it is true that this sequence is no longer present in the genome. But, it has been replaced at this location with the homologous sequence, rather than deleted. Gene conversions in microbial genomes commonly occur between slightly diverged ribosomal RNA copies. See the supplementary archive Gene_Conversion files for an example.
3.4 Identifying and comparing variants in whole-population samples
Using the shell prompt, navigate to the Population_Sample directory in the supplementary archive. The FASTQ files in this directory are for genomic DNA isolated from whole-population samples of E. coli after 2,000, 5,000, 10,000, 15,000, and 20,000 generations of a long-term evolution experiment (7). They consist of 36-base single-end reads generated by an Illumina Genome Analyzer II instrument. The GenBank reference file is the clone that was used to begin the evolution experiment (12). Genetic diversity changed over time in this population as new genetic variants arose, competed, and replaced their ancestors. Therefore, each DNA read may be from an individual with a different set of mutations relative to the ancestor (i.e., these samples are “metagenomic”).
- To identify mutations that may be present in only a fraction of a mixed-population sample, add the -p option to the breseq command line to switch from consensus mode (the default) to polymorphism prediction mode. The following commands should be run to analyze the population samples for all time points:
$ breseq -p -j 4 -o 2K -r REL606.gbk SRR032370.fastq $ breseq -p -j 4 -o 5K -r REL606.gbk SRR032371.fastq $ breseq -p -j 4 -o 10K -r REL606.gbk SRR032372.fastq $ breseq -p -j 4 -o 15K -r REL606.gbk SRR032373.fastq $ breseq -p -j 4 -o 20K -r REL606.gbk SRR032374.fastq
In consensus mode, breseq assumes mutations are present in 100% of a clonal sample. Switching to polymorphism mode enables breseq to identify mutations present at intermediate frequencies in the population. Mutations present in 100% of the mixed sample are still identified, but polymorphic variation is also reported if the evidence for it is statistically significant versus the null hypothesis that the sample was not a mixture. This null hypothesis holds that discrepancies between the reads and the reference genome are adequately explained by sequencing errors. Polymorphism mode output from breseq is nearly identical to that for a clonal sample as discussed in Section 3.1. The primary addition is a “freq” column, representing the estimated frequency at which each mutation was detected within the population, to the predicted mutations table located on the main index.html output page.
Predictions of polymorphisms can be bedeviled by various types of non-ideality in the input data where certain sequence contexts or locations are hotspots for sequencing errors at a much greater rate than expected. To reduce the false-positive rate for these predictions breseq employs several filters, particularly for polymorphic point mutations. In some cases, it may be desirable to adjust the stringency of these filters (see Note 10).
- Commonly, one may want to compare the results of sequencing many related samples side-by-side to see what mutations are in common between clones or to examine how the frequency of a genetic variant changed over time in a population. Once the five breseq commands have finished running, the following commands should be run in the Population_Sample directory to copy and rename the output Genome Diff files:
$ cp 2K/output/output.gd 2K.gd $ cp 5K/output/output.gd 5K.gd $ cp 10K/output/output.gd 10K.gd $ cp 15K/output/output.gd 15K.gd $ cp 20K/output/output.gd 20K.gd
To generate an HTML file with a table comparing all of the mixed population samples in this example, use the the gdtools COMPARE subcommand as follows:$ gdtools COMPARE -o compare.html -r REL606.gbk 2K.gd 5K.gd 10K.gd 15K.gd 20K.gd
In the resulting comparison table in file compare.html, rows represent specific mutations and columns represent different samples (Fig. 6). In the portion of the table shown, one can see that the mrdA mutation arose and fixed between 2,000 and 5,000 generations. In contrast, it took some time for the later araJ mutation to sweep to 100% frequency.
Fig. 6.
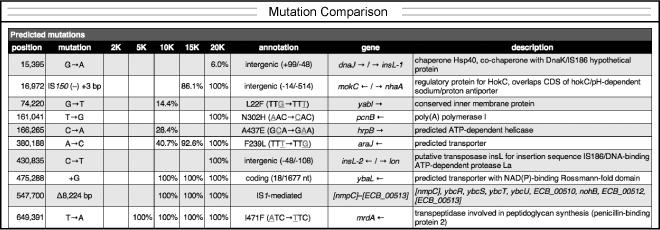
Example time course of mutation frequencies in an evolving population. A portion of the comparison file generated from the results of analyzing several whole-population samples is shown. Each column (e.g. 2K) is for a sample from a different time point (e.g., 2000 generations).
Acknowledgments
DED was supported by a University of Texas at Austin CPRIT Cancer Research Traineeship. Development of breseq has been supported by an NSF Postdoctoral Research Fellowship in Biological Informatics (DBI-0630687) and by grants from the NSF BEACON Center for the Study of Evolution in Action (DBI-0939454), NIH (R00-GM087550), and CPRIT (RP130124) to JEB. Additional programmers and users who have provided valuable feedback and bug reports are thanked in the breseq documentation.
Footnotes
For mapping to the reference genome, breseq currently treats all input reads as if they are single-end data generated from a DNA fragment library. That is, it does not use paired-end or mate-paired constraints on the orientation and distance between reads. In theory, this information can be used to better predict certain structural variants and mutations in repetitive regions when the distance between read pairs is great enough (see Note 6). We find that breseq’s analysis of just split-read alignments, where the two halves of one sequencing read match sites that are distant from one another in the reference genome, reliably predicts most new sequence junctions resulting from structural variation. This has also been the conclusion of the developers of other tools such as TopHat-fusion (5).
In our experience, read-depth coverage of the reference genome of ≥40-fold gives very accurate mutation predictions for Illumina data. Greater coverage beyond this usually does not result in any improvements and can make the pipeline take substantially longer to run. So, it may be desirable to truncate overly large FASTQ input files to an estimated ~100-fold coverage of the reference genome to decrease runtime in some cases. This can be easily accomplished by using Unix commands like head to extract a subset of lines from a large FASTQ file. The exception to this rule is for whole-population samples (Section 3.4) where read-depth can limit the discovery of rare variants and as much read data as possible should generally be used.
If you performed some sort of enrichment for DNA from specific genomic regions in preparing your sample (e.g., you are sequencing PCR amplicons), add the -t flag to the breseq command to activate targeted sequencing mode. This relaxes the assumption that there will be equal coverage over the entire reference sequence and prevents analysis steps that only apply when this is true (e.g., predicting deletions from missing coverage). If your sample has foreign DNA in it (such as transposons, viruses, or plasmids) and your only interest in mutations involving these sequences is in their potential insertion into the host chromosome (for example, you sequenced a transposon insertion mutant library and have the sequence of a suicide plasmid that carried the transposon), reference files for each of the foreign genomes should be supplied using the junction-only reference ( -s) option in addition to the main reference of interest which is supplied using the typical -r option. This usage provides three main benefits: SNPs, indels, and other mutations affecting only the foreign DNA are ignored, insertions into the genome of interest will be correctly identified (e.g., where the transposon from the suicide plasmid integrated), and it prevents reads originating from the foreign DNA from accidentally mismapping to the genome of interest where they could possibly lead to spurious mutation calls.
It is not unusual for 5–10% of total reads in any NGS data set to not align to the reference genome because they pass quality filters but contain many incorrect base calls. A higher percentage of reads not mapping may indicate a poorly constructed DNA fragment library containing many adapter dimers, for example. Another common problem is not removing barcode sequences at the ends of reads after de-multiplexing samples. This can result in sub-optimal mapping of reads and spurious mutation predictions in breseq. We suggest checking the quality of your input FASTQ file using a tool such as FastQC that can detect these and other concerns (15). Tools like FLEXBAR (16) can be used to trim adaptor and barcode sequences before they are used as input for breseq.
The re-sequencing strategy employed by breseq breaks down when there is sufficient sequence divergence between the sample and the reference genome to lead to mismapping of reads, such as when there is a high level of local nucleotide divergence or there are novel sequences in the sample that are not in the reference genome. In these cases, it may be valuable to use de novo assembly tools, such as Velvet (17) or ALLPATHS-LG (18), on either the unmapped reads after a breseq run (located at data/unmatched.*) or all reads, and then compare assemblies to the reference genome or to one another using tools for comparing whole genome sequences, such as MUMmer (19).
Due to inherent limitations in the information present in short read data and the algorithms used by breseq, certain kinds of mutations will never be predicted. These are generally related to sequence repeats in the reference genome that are longer than the read length, such as multi-copy mobile genetic elements, ribosomal RNA operons, and recently duplicated genes. It may be impossible to span these repeats with a single read to anchor the two unique sides relative to each other. This may result in an inability to detect the new junctions formed by large chromosomal inversions, deletions, or tandem amplifications that occur through equivalent sites of the same repeat. Deletions between these elements can be found by looking for missing coverage (as detailed for the manB–cpsG deletion). Duplications or amplifications should result in higher read coverage of the amplified region between repeats. This type of copy number variation is not currently automatically predicted by breseq, but it can be detected by generating and manually examining read-coverage graphs that tile the genome at a high enough resolution (see options for breseq bam2cov). Chromosomal inversions through these repeats will not result in a change in coverage, and therefore cannot be predicted. An inability to map reads uniquely to multi-copy sequence repeats can also make it difficult to detect a new point mutation in any one copy of the repeat element. Theoretically, these mutations could be detected by looking for polymorphisms within the “population” of the repeat sequence within a single clonal genome, but breseq does not currently attempt this complicated analysis.
Files in the community Variant Call Format (VCF) describe sequence variation between a sample and a reference genome (14). Genome Diff files output by breseq list mutational events separating two genomes. This is a subtle but important difference. Currently, Genome Diff files can encode additional information regarding evolutionary processes and mechanisms that does not fit naturally in a VCF file. For example, imagine a complex mutation that resulted from a new copy of a mobile element inserting in a genome and then later recombination between this element and an existing copy of the mobile element resulting in a large deletion. A VCF file describing this sequence variation would record the large deletion only. A Genome Diff file could include entries for each of the two separate mutations, so that they could be counted separately or used to construct a better phylogenetic tree relating multiple samples. Another example where information would be lost in a VCF file is when a point mutation occurs within a region that is later deleted. It is possible that future versions of VCF will have ways to precisely describe these situations. For now, GenomeDiff files can be converted to standard VCF files for display in NGS browsers such as the Integrated Genomic Viewer (IGV) using the gdtools GD2CVF subcommand.
In addition to the subcommands described in the text for applying and comparing Genome Diff files, gdtools provides basic support for set operations ( UNION, INTERSECTION, SUBTRACT), format conversion ( GD2VCF), analyzing the overall characteristics of mutations ( COUNT), or drawing images of genomes showing mutations ( GD2CIRCOS). Be aware that commands that rely on outside software or formats are not always up-to-date or as stable as core breseq operations.
If any evidence has been discarded as a false positive, it will likely still be listed again after the breseq run on the mutated genome. Any new mutations or evidence found as a result of re-querying the NGS data against the updated reference will have their locations relative to the mutated genome and not to the original genome. So, be sure that any additional updates to the original Genome Diff file that you make are based on the original genome coordinates and not the updated genome.
breseq employs several filters to attempt to catch the bulk of false-positive predictions of polymorphisms that are not ruled out by the underlying error model (7). First, many spurious polymorphism predictions can be recognized because all of the reads supporting the variant correspond to only one of the two genomic strands, rather than occurring evenly on both strands as would be expected for a real mutation. breseq uses Fisher’s exact test to judge the significance of this bias. Second, the bases supporting a variant may have uniformly lower quality scores than those supporting the consensus. breseq uses a Kolmogorov-Smirnov test to detect this bias. The --polymorphism-bias-cutoff option sets the p-value cutoff for both of these tests. Lower values of this parameter will reject fewer polymorphism predictions. The option --polymorphism-minimum-coverage-each-strand can also be used to add a hard requirement that a certain number of reads on each strand must support a variant for it to be reported. Indels are rare in Illumina data and very common in Roche 454 data. Each situation can lead to over-estimating the significance of this type of variation, so there are options to specifically not predict polymorphisms in repeats of a single base that exceed a certain length ( --polymorphism-reject-homopolymer-length) or to not predict any indel polymorphisms ( --polymorphism-no-indels). Finally, polymorphisms with lower frequencies are more prone to misprediction, so you can apply a simple frequency cutoff criterion to all predictions using the --polymorphism-frequency-cutoff option. Predicting polymorphisms is inherently noisier than predicting consensus mutations. You may need to optimize these parameters for characteristics of your particular samples to achieve the best results.
References
- 1.Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genom Human Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 2.Eid J, Fehr A, Gray J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 3.Trapnell C, Salzberg SL. How to map billions of short reads onto genomes. Nat Biotechnol. 2009;27:455–457. doi: 10.1038/nbt0509-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrick JE, Yu DS, Yoon SH, et al. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature. 2009;461:1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- 7.Barrick JE, Lenski RE. Genome-wide mutational diversity in an evolving population of Escherichia coli. Cold Spring Harbor Symp Quant Biol. 2009;74:119–129. doi: 10.1101/sqb.2009.74.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woods RJ, Barrick JE, Cooper TF, et al. Second-order selection for evolvability in a large Escherichia coli population. Science. 2011;331:1433–1436. doi: 10.1126/science.1198914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blount ZD, Barrick JE, Davidson CJ, Lenski RE. Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature. 2012;489:513–518. doi: 10.1038/nature11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milne I, Stephen G, Bayer M, et al. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform. 2013;14:193–202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 11.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeong H, Barbe V, Lee CH, et al. Genome sequences of Escherichia coli B strains REL606 and BL21(DE3) J Mol Biol. 2009;394:644–652. doi: 10.1016/j.jmb.2009.09.052. [DOI] [PubMed] [Google Scholar]
- 13.Schneider D, Duperchy E, Coursange E, et al. Long-term experimental evolution in Escherichia coli. IX. Characterization of insertion sequence-mediated mutations and rearrangements. Genetics. 2000;156:477–488. doi: 10.1093/genetics/156.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrews S. FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- 16.Dodt M, Roehr J, Ahmed R, Dieterich C. FLEXBAR—Flexible barcode and adapter processing for next-generation sequencing platforms. Biology. 2012;1:895–905. doi: 10.3390/biology1030895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ribeiro FJ, Przybylski D, Yin S, et al. Finished bacterial genomes from shotgun sequence data. Genome Res. 2012;22:2270–2277. doi: 10.1101/gr.141515.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]