

Abstract
We have previously shown that auditory Pavlovian fear conditioning is associated with an increase in DNA methyltransferase (DNMT) expression in the lateral amygdala (LA) and that intra-LA infusion or bath application of an inhibitor of DNMT activity impairs the consolidation of an auditory fear memory and long-term potentiation (LTP) at thalamic and cortical inputs to the LA, in vitro. In the present study, we use awake behaving neurophysiological techniques to examine the role of DNMT activity in memory-related neurophysiological changes accompanying fear memory consolidation and reconsolidation in the LA, in vivo. We show that auditory fear conditioning results in a training-related enhancement in the amplitude of short-latency auditory-evoked field potentials (AEFPs) in the LA. Intra-LA infusion of a DNMT inhibitor impairs both fear memory consolidation and, in parallel, the consolidation of training-related neural plasticity in the LA; that is, short-term memory (STM) and short-term training-related increases in AEFP amplitude in the LA are intact, while long-term memory (LTM) and long-term retention of training-related increases in AEFP amplitudes are impaired. In separate experiments, we show that intra-LA infusion of a DNMT inhibitor following retrieval of an auditory fear memory has no effect on post-retrieval STM or short-term retention of training-related changes in AEFP amplitude in the LA, but significantly impairs both post-retrieval LTM and long-term retention of AEFP amplitude changes in the LA. These findings are the first to demonstrate the necessity of DNMT activity in the consolidation and reconsolidation of memory-associated neural plasticity, in vivo.
Keywords: DNA methytransferase, epigenetics, reconsolidation, fear conditioning, neural plasticity
1. Introduction
Epigenetic mechanisms, including modifications in chromatin structure and DNA methylation, have been widely implicated in synaptic plasticity and memory formation (Barrett & Wood, 2008; Jiang, et al., 2008; Levenson & Sweatt, 2005, 2006; Monsey, et al., 2011). Chromatin modifications, the most commonly studied epigenetic modification, are typically associated with the acetylation of histones by histone acetyltransferases (HATs), a process which results in the relaxation of chromatin and contributes to enhanced transcription of memory-related genes (Levenson & Sweatt, 2005; Barrett & Wood, 2008). Conversely, the methylation of cytosine residues on DNA via DNA methyltransferases (DNMTs) is typically thought to negatively regulate gene transcription via preventing the binding of transcription factors (Levenson, et al., 2006; Levenson & Sweatt, 2005; Miller, et al., 2010; Miller & Sweatt, 2007; Miranda & Jones, 2007). In development, the methylation of DNA has been associated with gene silencing and cellular differentiation in actively dividing cells, and has traditionally been viewed as a long-lasting, static process (Levenson & Sweatt, 2005; Miranda & Jones, 2007). Post-mitotic neurons, however, are known to express high levels of DNMT mRNA into adulthood (Brown et al., 2008; Feng et al., 2010), and emerging evidence has suggested that dynamic regulation of DNA methylation in adult neurons may be critical for regulating the transcription of genes involved in creating and maintaining stable memories (Miller, et al., 2010; Miller & Sweatt, 2007).
Recent studies have implicated alterations in DNA methylation in both hippocampal-and amygdala-dependent memory formation. Contextual fear conditioning has been observed to increase the expression of DNMT3A/B mRNA in hippocampal area CA1 and to regulate the methylation of the plasticity-related genes PP1, reelin, and BDNF (Lubin, Roth, & Sweatt, 2008; Miller & Sweatt, 2007). Further, intra-hippocampal infusion of an inhibitor of DNMT activity has been observed to impair the consolidation of a contextual fear memory (Miller, et al., 2008; Miller & Sweatt, 2007). Similarly, our lab has recently observed that DNMT3A protein is regulated in an associative manner within the lateral nucleus of the amygdala (LA) following auditory fear conditioning (Monsey, et al., 2011), and that intra-LA infusion of an inhibitor of DNMT activity shortly following auditory fear conditioning or retrieval of an auditory fear memory impairs the consolidation and reconsolidation of an auditory fear memory, respectively (Maddox & Schafe, 2011b; Monsey, et al., 2011).
In parallel to behavioral studies showing the involvement of DNMT activity in memory consolidation and reconsolidation processes, a number of studies have shown that DNMT activity is required for synaptic plasticity, in vitro. Bath application of an inhibitor of DNMT activity has been observed to impair the induction of long-term potentiation (LTP) in hippocampal area CA1 while having no effect on baseline synaptic transmission (Levenson, et al., 2006; Miller, et al., 2008). Further, our lab has recently observed that bath application of an inhibitor of DNMT activity to slices containing the amygdala impairs LTP at both thalamic and cortical input pathways to the LA (Monsey, et al., 2011). While these studies have revealed a role for DNMT activity in in vitro models of synaptic plasticity in both the hippocampus and amygdala, no study has to date examined the role of DNMT activity in neural plasticity that accompanies naturally-occurring memory formation, in vivo. In the present study, we used awake-behaving neurophysiology techniques to examine the role of DNMT activity in the consolidation of neural plasticity in the LA associated with initial fear memory consolidation and in the retention of training-related neural plasticity in the LA following retrieval of a fear memory.
2. Materials & Methods
2.1. Subjects
Adult-male Sprague-Dawley rats (Harlan), weighing 300–350g, were housed individually in plastic cages and maintained on a 12:12 hr light/dark cycle with food and water provided ad libitum.
2.2. Electrode implantation procedures
Rats were anesthetized with i.p. administration of Ketamine (100 mg/kg) and Xylazine (6.0 mg/kg) and implanted in the left LA with a tungsten recording electrode adhered to a 26-gauge guide cannula (AP: −3.2 mm; ML: ±5.2; DV: −7.4). A second 26-gauge guide cannula was implanted in the right LA. A low-impedance copper wire was connected to a stainless steel bone screw drilled into the skull contralateral to the side of the recording electrode ~1 mm posterior to bregma to serve as the reference for recording purposes. Another stainless steel screw attached to a copper wire was drilled into the skull ~3 mm posterior to lambda and served as the ground electrode. Dental cement was used to anchor the electrodes and connecting device to the skull. Buprenex (0.2 mg/kg) was administered as an analgesic, and rats were provided with at least five days of post-operative recovery time. All surgical procedures were conducted under the guidelines provided in the National Institutes of Health Guide for the Care and Use of Experimental Rats and were approved by the Yale University Institutional Animal Care and Use Committee.
2.3. Drugs
The DNA methyltransferase inhibitors 5-AZA-2`- deoxycytidine (5-AZA; Sigma Cat# A3656) and N-Phthalyl-L-Tryptophan (RG108; Sigma Cat# R8279) were dissolved in 100% DMSO to a 2 μg/μl stock solution. Both drugs were then diluted 1:1 in ACSF to a final 1μg/μl solution. All vehicle solutions consisted of 50% DMSO in ACSF.
2.4. Electrophysiological recordings
Awake-behaving electrophysiology took place in a custom-made electromagnetic shielded recording chamber designed for delivery of auditory stimuli and recording. The chamber was kept within a ventilated and temperature-regulated acoustic isolation room. Stimulus delivery and data acquisition were controlled by SciWorks Experimenter Real-time 7.0 (DataWave). During recording sessions, rats were exposed to a series of 20 tone ‘pips’ as a conditioned stimulus (CS). Each pip consisted of a 50 ms, 75 dB, 1 kHz tone, delivered at 1 Hz from a speaker mounted on the ceiling of the recording chamber. The tone pip series CS was triggered by TTL signals generated by SciWorks. The TTL signals were converted (Coulbourn, H91-24, 5V TTL to 24 V converter) and sent to a tone generator (Coulbourn, H12-07, Seven-Tone Audio Cue). During recordings, the implanted electrodes were connected to a Micro-Miniature Headstage (DataWave). Neural signals were picked up (Legacy PCI data acquisition bundles, Model: DT3010), amplified (16-channel A-M Systems microelectrode amplifier, Model: AM-3600) and saved for off-line analysis.
For the memory consolidation experiments, rats were handled and habituated to the recording chamber and cable connection for 15 min on Day 1. On days 2 and 3, baseline auditory-evoked field potentials (AEFPs) elicited by 3 presentations of the 20 tone pip CS series were recorded (ITI between series = 2 mins) from the LA, for a total of 60 tone pip presentations. On day 4, rats received three trials of fear conditioning consisting each of a series of 20 tone pip CS presentations that co-terminated with a 1s, 1.0 mA footshock administered through the grid floor. AEFPs were not recorded during fear conditioning. One hour following training, rats received intra-LA infusion of either vehicle (0.5 μl/side), RG108 (500 ng/side; 0.5 μl) or 5-AZA (500 ng/side; 0.5 μl). This time point was chosen based on previous behavioral experiments from our lab (Maddox & Schafe, 2011b; Monsey, et al., 2011) and is consistent with the relatively late peak of DNMT3A protein expression (at 90 mins) observed after auditory fear conditioning (Monsey, et al., 2011). Infusions were made over 4 min and the infusion cannulas were left in place for at least 2 min following infusion to facilitate diffusion of the drug throughout the LA. Three hours after drug infusions rats were placed into a modified chamber which included a flat black peppermint scented plastic floor for short-term memory (STM) testing and recordings of AEFPs. The STM test consisted of 3 presentations of the tone pip CS series (ITI = 2 mins), for a total of 60 tone pip presentations (identical to baseline recordings). The following day (~24 hrs later), rats were placed back in the modified testing chamber and tested for long-term memory (LTM) with 9 tone pip CS presentations (ITI = 2 mins) while AEFPs were recorded from the LA.
For the memory reconsolidation experiments, rats underwent habituation, baseline recording sessions, and fear conditioning as in the consolidation experiments. The next day (Day 5), rats were placed in the modified testing chamber (black peppermint scented plastic floor) and received a single tone pip series CS presentation (20 pips) to serve as a memory ‘reactivation’ trial. Additional groups of rats, designated as ‘no-reactivation’ controls, were placed in the testing chamber but did not receive a tone pip series CS presentation. One hour following the memory reactivation (or no-reactivation) session, rats were infused with either vehicle (0.5 μl/side), RG108 (500 ng/side; 0.5 μl) or 5-AZA (500 ng/side; 0.5 μl). Three hours after infusions, rats were tested for post-reactivation (PR)-STM consisting of 3 presentations of the tone pip CS series while AEFPs were recorded from the LA. Twenty-four hr later (Day 6) rats were tested for PR-LTM consisting of 9 presentations of the tone pip CS series while AEFPs were recorded from the LA.
Rats’ freezing behavior was recording during each test session for off-line scoring. Following the completion of testing, all rats were rapidly and deeply anesthetized and perfused transcardially with 0.9% saline followed by 10% formalin. Brains were extracted from the skull, sectioned at 50 μM thickness on a cryostat, and stained for Nissl for cannula and electrode placement analyses. Only those rats with electrode and cannula placements confined to the borders of the LA were included in the subsequent data analyses.
2.5. Data analysis
For data analysis during STM/PR-STM sessions, all 60 AEFPs were averaged into a single waveform. Data analysis for the LTM/PR-LTM sessions was conducted based on the average waveform from the last 60 AEFPs (the last 3 trials of the LTM/PR-LTM test). Spike2 software (Cambridge Electronics Design, Cambridge, UK) was used to measure the amplitude of the short-latency negative-going component of the AEFP from the initial point of deflection to its maximal negativity, which occurs ~12–16 ms from the onset of the pip (Doyère, et al., 2007; Maddox, Watts, et al., 2013; Maddox, et al., 2013; Quirk, et al., 1995; Rogan, et al., 1997; Schafe, et al., 2005). The amplitude of AEFPs recorded from STM/PR-STM and LTM/PR-LTM sessions was expressed as a percentage of the averaged AEFP amplitude on the second day of baseline recording (Day 3). Behavioral and neurophysiological data were analyzed using Analysis of Variance (ANOVA) and Duncan’s post-hoc t-tests. Differences were only considered significant if p < 0.05.
3. Results
3.1. Intra-LA infusion of an inhibitor of DNMT activity following fear conditioning impairs fear memory consolidation and the consolidation of training-related neural plasticity in the LA
In our first series of experiments, we examined the effect of intra-LA infusion of two pharmacologically distinct DNMT inhibitors on fear memory consolidation and the consolidation of training-related changes in auditory-evoked field potentials (AEFPs) in the LA. Rats were fear conditioned with 3 tone pip series (CS)-shock (US) pairings, followed 1 hr later by intra-LA infusion of vehicle (0.5 μl/side), RG108 (500 ng/side; 0.5 μl) or 5-AZA (500 ng/side; 0.5 μl). All rats then received tests of STM (3 hrs later) and LTM (~24 hrs later) while AEFPs were recorded from the LA (Figure 1a). During training, we observed no differences in post-shock freezing between the three groups (not shown). An ANOVA (group by trial) revealed a main effect of trial [F(3,45) = 698.7, p < 0.05] but not of group [F(2,15) = 0.22, p > 0.05]. Similarly, we found no significant difference in freezing between vehicle, RG108 and 5-AZA groups during the STM test [F(2,15) = 0.2, p > 0.05; Figure 1b]. However, the following day the RG108 and 5-AZA-treated rats exhibited impaired LTM relative to the vehicle group [F(2,15) = 31.8, p < 0.01; Figure 1b]. Duncan’s post-hoc t-tests revealed that the vehicle group was significantly different from both the RG108 and 5-AZA groups [p < 0.05], which were not found to differ significantly different from each another [p > 0.05].
Figure 1. Intra-LA infusion of an inhibitor of DNMT activity impairs fear memory consolidation and training-related neural plasticity in the LA.

(a) Rats were habituated to handling and given two days of baseline recording sessions in which AEFPs were recorded from the LA. On the 4th day, rats were fear conditioned with 3 tone pip series (CS)-shock (US) pairings followed 1 hr later by intra-LA infusion of either vehicle (n = 6), RG108 (500 ng/side; n = 6) or 5-AZA (500 ng/side; n = 6). Rats in each group were then tested for STM and LTM 3 and 21 hrs later, respectively, while AEFPs were recorded from the LA. (b) Mean (± SEM) percent freezing during the STM and LTM tests in vehicle, RG108 and 5-AZA infused groups. *p<0.05 relative to vehicle-infused controls. (c) Mean (± SEM) percent of change in AEFP amplitude during the STM and LTM tests in vehicle, RG108 and 5-AZA-infused rats, relative to baseline. *p<0.05 relative to vehicle-infused controls. (d) Correlation between freezing scores and AEFP amplitudes in RG108- and 5-AZA-treated rats during the LTM test, each expressed as a percentage of freezing and AEFP amplitude change during the STM test. (e) Representative AEFPs recorded from the LA for each group during baseline (light gray trace), STM and LTM sessions (darker traces). Scale bar =10 μV, 5ms.
Analysis of the neurophysiology during the baseline and STM sessions revealed that fear conditioning led to a significant enhancement of the amplitude of the short-latency (~12–15 ms) component of the AEFP in the LA relative to baseline in each of the three groups (Figure 1c). An ANOVA (group by session) revealed a main effect of session [baseline vs. STM; F(1,15) = 39.06, p < 0.05] but not of group [F(2,15) = 0.16, p > 0.05]. Further analysis determined that there was no difference in the amplitude change of AEFPs during the STM test across the 3 groups [F(2,15) = 0.01, p > 0.05; Figure 1c], suggesting that all 3 groups exhibited equivalent training-related AEFP enhancement. However, during the LTM test the RG108 and 5-AZA-treated rats exhibited significantly less AEFP amplitude change relative to vehicle-infused controls [F(2,15) = 19.7, p < 0.05; Figure 1c]. Duncan’s post-hoc t-tests determined that the vehicle group was significantly different from both the RG108 and 5-AZA groups [p < 0.05], which were not found to be significantly different from one another [p > 0.05]. To further examine this effect, we performed a regression analysis within the RG108- and 5-AZA-treated groups between freezing scores and percentage of change in AEFP amplitudes in the LA. For this analysis, we expressed each of these measures during the LTM test as a percentage of freezing and AEFP amplitude change during the STM test for each rat. The analysis revealed a highly significant correlation in the RG108 (r(6) = 0.88, p < 0.05) and 5-AZA groups (r(6) = 0.77, p < 0.05; Figure 1d), indicating that the more effective RG108 and 5-AZA were at impairing the consolidation of fear memory, the more effective these drugs were at impairing the consolidation of training-related neural plasticity in the LA. Thus, intra-LA infusion of a DNMT inhibitor shortly following training can significantly impair not only the consolidation of a fear memory but also the consolidation of memory- associated neural plasticity in the LA.
3.2. Intra-LA infusion of an inhibitor of DNMT activity following fear memory retrieval impairs fear memory reconsolidation and the retention of memory-related neural plasticity in the LA
In our second series of experiments, we examined the effect of intra-LA infusion of a DNMT inhibitor following fear memory retrieval on fear memory reconsolidation and the retention of memory-associated AEFPs in the LA. Rats were trained with 3 tone pip series (CS)-shock (US) presentations, followed 24 h later by a memory reactivation session consisting of a single tone pip series (CS) presentation. One hr following the memory reactivation session, rats received intra-LA infusion of either vehicle (0.5 μl/side), RG108 (500 ng/side; 0.5 μl) or 5-AZA (500 ng/side; 0.5 μl) followed 3 and 21h by tests of PR-STM and PR-LTM while AEFPs were recorded from the LA (Figure 2a). Analysis of the reactivation session revealed that all three groups exhibited significant and equivalent memory recall during the reactivation session; the ANOVA (group by trial) revealed a significant effect of trial [pre-CS vs. CS; F(1,16) = 3573.02, p < 0.01], but not of group [F(2,16) = 0.11, p > 0.05; Figure 2b]. Similarly, we found no significant difference in freezing between vehicle, RG108 and 5-AZA groups during the PR-STM test [F(2,16) = 0.49, p > 0.05; Figure 2c]. However, the following day the RG108 and 5-AZA-treated rats exhibited impaired PR-LTM relative to the vehicle group [F(2,16) = 68.9, p < 0.01; Figure 2c]. Duncan’s post-hoc t-tests revealed that the vehicle group was significantly different from both the RG108 and 5-AZA groups [p < 0.05], which were not found to be significantly different from one another [p > 0.05].
Figure 2. Intra-LA infusion of an inhibitor of DNMT activity impairs fear memory reconsolidation and retention of memory-associated neural plasticity in the LA.

(a) Rats were habituated and given two days of baseline AEFP recording sessions, followed by fear conditioning with 3 tone pip series-(CS) shock (US) pairings. Twenty four hrs following training rats were given a memory reactivation session consisting of a single tone pip series CS presentation followed 1 hr later by intra-LA infusion of vehicle (n = 6), RG108 (500 ng/side; n = 6) or 5-AZA (500 ng/side; n = 7). Rats in each group were then tested for PR-STM and PR-LTM 3 and 21 hrs later, respectively, while AEFPs were recorded from the LA. (b) Memory retrieval data during the reactivation session for the vehicle, RG108 and 5-AZA-infused groups. *p<0.05 relative to the pre-CS period. (c) Mean (± SEM) percent freezing during the PR-STM and PR-LTM tests in vehicle, RG108 and 5-AZA-infused groups. *p<0.05 relative to vehicle-infused controls. (d) Mean (± SEM) percent change in AEFP amplitude during the PR-STM and PR-LTM tests in vehicle, RG108 and 5-AZA-infused rats, relative to baseline. *p<0.05 relative to vehicle-infused controls. (e) Correlation between freezing scores and AEFP amplitudes in RG108- and 5-AZA-treated rats during the PR-LTM test, each expressed as a percentage of freezing and AEFP amplitude change during the PR-STM.test. (f) Representative AEFPs recorded from the LA for each group during baseline (light gray trace), PR-STM and PR-LTM sessions (darker traces). Scale bar =10 μV, 5ms.
Analysis of the neurophysiology revealed significant retention of training-related enhancements in the amplitude of the AEFP in the LA during the PR-STM test relative to baseline in each of the three groups (Figure 2d). An ANOVA (group by session) revealed a main effect of session [baseline vs. PR-STM; F(1,16) = 15.99, p < 0.05] but not of group [F(2,16) = 0.11, p > 0.05]. Further analysis determined that there was no difference in the training-related retention of AEFPs during PR-STM between the 3 groups [F(2,16) = 0.02, p > 0.05; Figure 2d], suggesting that all 3 groups exhibited equivalent retention of the training-related AEFP enhancement. However, during the PR- LTM test the RG108 and 5-AZA-treated rats exhibited significantly less AEFP amplitude change relative to vehicle-infused controls [F(2,16) = 7.18, p < 0.05; Figure 2d]. Duncan’s post-hoc t-tests determined that the vehicle group was significantly different from both the RG108 and 5-AZA groups [p < 0.05], which were not found to be significantly different from one another [p > 0.05]. As before, we performed a regression analysis within the RG108- and 5-AZA-treated groups between freezing scores and percentage of change in AEFP amplitudes during the PR-LTM test, both expressed as a percentage each of these measures during the PR-STM test. The analysis revealed a highly significant correlation in the RG108 (r(6) = 0.77, p < 0.05) and 5-AZA groups (r(7) = 0.77, p < 0.05; Figure 2e), indicating that the more effective RG108 and 5-AZA were at impairing the reconsolidation of fear memory, the more effective these drugs were at impairing the retention of training-related neural plasticity in the LA. Thus, intra-LA inhibition of DNMT activity shortly following fear memory retrieval significantly impairs the reconsolidation of a fear memory and, in parallel, leads to a reversal in training-related enhancements in tone-evoked neural activity in the LA.
3.3. Intra-LA infusion of an inhibitor of DNMT activity in the absence of fear memory retrieval has no effect on fear memory reconsolidation or memory-associated neural plasticity in the LA
In our final experiment, we asked whether the reconsolidation impairment induced by intra-LA infusion of an inhibitor of DNMT activity is specific to a fear memory that has undergone retrieval. Rats were trained with 3 tone pip series (CS)-shock (US) presentations as before, followed 24 h later by a ‘no-reactivation’ session in which they were placed in the testing chamber without a tone pip series (CS) presentation. One hr following the ‘no-reactivation’ session, rats received intra-LA infusion of either vehicle (0.5 μl/side), RG108 (500 ng/side; 0.5 μl) or 5-AZA (500 ng/side; 0.5 μl) followed 3 and 21h by tests of ‘PR’-STM and ‘PR’-LTM (Figure 3a). Thus, rats in this experiment were treated identically to those in the reconsolidation experiment, with the exception that their fear memory was not reactivated prior to drug treatment. Analysis of the no reactivation session data revealed that all 3 groups exhibited equivalently low levels of freezing during the ‘pre-CS’ period and during the 20 sec period when the tone pip series would have been presented during the reactivation session (Figure 3b). An ANOVA (group by trial) revealed no significant effect of group [F(2,11) = 0.27, p > 0.05] or trial [F(1,11) = 0.11, p > 0.05]. Further, all three groups exhibited equivalently high levels of freezing during the ‘PR’-STM test [F(2,11) = 1.34, p > 0.05; Figure 3c] and during the ‘PR’-LTM test [F(2,11) = 1.65, p > 0.05; Figure 3c].
Figure 3. Intra-LA infusion of an inhibitor of DNMT activity in the absence of fear memory retrieval has no effect on fear memory reconsolidation or the retention of memory-associated neural plasticity in the LA.

(a) Rats were given two days of baseline AEFP recording sessions, followed by fear conditioning with three tone pip series (CS)-shock (US) pairings. Twenty four hrs following training rats were given a ‘no reactivation’ session in which they were placed in the testing context but not presented with a tone pip series. One hour following the ‘no reactivation’ session, rats received infusion of vehicle (n = 5), RG108 (500 ng/side; n = 5) or 5-AZA (500 ng/side; n = 4). Rats in each group were then tested for ‘PR’-STM and ‘PR’-LTM 3 and 21 hrs later, respectively, while AEFPs were recorded from the LA. (b) Memory retrieval data for the vehicle, RG108 and 5-AZA-infused groups during the ‘no reactivation’ session. (c) Mean (± SEM) percent freezing during the ‘PR’-STM and ‘PR’-LTM tests in vehicle, RG108 and 5-AZA-infused groups. (d) Mean (± SEM) percent change in AEFP amplitude during the ‘PR’-STM and ‘PR’-LTM tests in vehicle, RG108 and 5-AZA-infused rats, relative to baseline. (e) Representative AEFPs recorded from the LA for each group during baseline (light gray trace), ‘PR’-STM and ‘PR’-LTM sessions (darker traces). Scale bar =10 μV, 5ms.
Analysis of the neurophysiology revealed significant retention of training-related enhancements in the amplitude of the AEFP in the LA during the ‘PR’-STM test relative to baseline in each of the three groups. An ANOVA (group by session) revealed a main effect of session [baseline vs. ‘PR’-STM; F(1,11) = 39.94, p < 0.05] but not of group [F(2,11) = 0.96, p > 0.05]. Further analysis determined that there was no difference in the training-related retention of AEFPs during ‘PR’-STM between the 3 groups [F(2,16) = 0.05, p > 0.05; Figure 3d], suggesting that all 3 groups exhibited equivalent retention of the training-related AEFP enhancement. Further, we observed no differences in AEFP amplitude between the 3 groups during the ‘PR’-LTM test [F(2,11) = 0.01, p > 0.05; Figure 3d]. Thus, intra-LA inhibition of DNMT activity is only effective at impairing fear memory reconsolidation and retention of memory-associated neural plasticity in the LA when it is administered around the time of active memory recall.
4. Discussion
Much progress has been made in defining the cellular and molecular mechanisms underlying fear memory consolidation and reconsolidation processes in the LA (Alberini, 2005; Rodrigues, et al., 2004; Schafe, et al., 2001; Tronson & Taylor, 2007), including the role of NMDA-receptor driven activation of protein kinase signaling cascades (Ben Mamou, et al., 2006; Duvarci, et al., 2005; S.M. Rodrigues, et al., 2001; Schafe, et al., 2000; Tronson, et al., 2006), transcription factors (Josselyn, et al., 2001; Kida, et al., 2002), de novo mRNA and protein synthesis (Bailey, et al., 1999; Duvarci, et al., 2008; Nader, et al., 2000; Schafe & LeDoux, 2000), and the involvement of immediate early genes (Lee, et al., 2005; Maddox, et al., 2011; Maddox & Schafe, 2011a; Ploski, et al., 2008). More recent work has suggested that an additional level of transcriptional control, including alterations in chromatin structure and DNA methylation, also plays a critical role in learning and memory and synaptic plasticity (Barrett & Wood, 2008; Jiang, et al., 2008; Levenson & Sweatt, 2005). In the fear memory system, our lab has recently shown that both histone acetylation and DNA methylation are critical for LTP in the LA as well as the consolidation and reconsolidation of a fear memory (Maddox & Schafe, 2011b; Maddox, et al., 2013a; Maddox, et al., 2013b; Monsey, et al., 2011). In the present study, we sought to further characterize the role of DNMT activity in fear memory consolidation and reconsolidation processes by examining the role DNMT activity in the formation and retention of memory-related neural plasticity in the LA, in vivo, that accompanies fear memory consolidation and reconsolidation.
Previous studies have shown that DNMTs are dynamically regulated in the hippocampus and amygdala by either contextual or auditory fear conditioning, respectively (Miller & Sweatt, 2007; Monsey et al, 2011). Further, local infusion or bath application of inhibitors of DNMT activity in the hippocampus impair contextual fear memory and LTP in hippocampal area CA1 (Miller & Sweatt, 2007; Miller et al., 2008), and intra-LA infusion or bath application of an inhibitor of DNMT activity has been shown to impair the consolidation of an auditory fear memory and LTP at thalamic and cortical inputs to the LA (Monsey et al, 2011). In agreement with these findings, we show that intra-LA infusion of two pharmacologically distinct inhibitors of DNMT activity shortly following auditory fear conditioning have no effect on STM, but significantly impair LTM. In parallel, we show that inhibition of DNMT activity in the LA has no effect on short-term enhancements in tone-evoked neural activity in the LA, but significantly impairs long-term retention of training-related neural plasticity in LA neurons. Collectively, this pattern of findings suggests that DNMT activity in the LA is critical not only for the consolidation of a fear memory at the behavioral level, but also for the consolidation of memory-associated neural plasticity in the LA.
In another recent study, we have shown that post-retrieval infusion of an inhibitor of DNMT activity into the LA impairs the reconsolidation of auditory fear memory in a time-limited and retrieval-dependent manner (Maddox & Schafe, 2011b). In that study, we showed that inhibition of DNMT activity in the LA at the time of memory retrieval, but not 6 hrs following memory retrieval, significantly impairs the reconsolidation of a fear memory (Maddox & Schafe, 2011b). Further, we showed that fear memories that are lost due to inhibition of DNMT activity in the LA are impaired in an enduring manner; they do not recover with the passage of time, when the animals are exposed to a reminder shock (reinstatement), or when animals are tested in a new context (renewal; Maddox & Schafe, 2011b). In the present study, we examined the effect of DNMT inhibition in the LA shortly after memory recall on the retention of training-related enhancements in tone-evoked activity in the LA. In parallel to our behavioral findings, we show that post-retrieval inhibition of DNMT activity in the LA leaves the expression of training-related enhancements in tone-evoked activity intact during the PR-STM test, and leads to a reversal of the training-related enhancements in tone-evoked neural activity in the LA during the PR-LTM test. Further, at both the behavioral and neurophysiological levels, we show that the memory impairment induced by DNMT inhibition in the LA shortly after retrieval is dependent on active memory recall; it is not observed when DNMT inhibition occurs in the absence of memory retrieval. To our knowledge, this is the first demonstration that post-retrieval manipulation of DNMT activity impairs, in parallel, not only the reconsolidation of fear a fear memory but also the reconsolidation of memory-related neural plasticity in the LA. Collectively, our findings suggest that dynamic regulation of DNMT activity in the LA at the time of fear memory recall is critical for the reconsolidation process and both the behavioral and neurophysiological levels.
While DNA methylation has largely been considered to be transcriptionally repressive, emerging evidence suggests that this view may be too simplistic. In agreement with previous findings examining the role of DNMT activity in hippocampal-dependent memory, we report memory deficits in both the consolidation and reconsolidation of a fear memory accompanying post-training or post-retrieval inhibition of DNMT activity in the LA (Maddox & Schafe, 2011b; Miller, et al., 2008; Monsey, et al., 2011). These findings are at first glance counterintuitive given that traditional views of DNA methylation would predict that inhibition of DNMT activity should result in enhanced transcription and thus an enhancement of both memory consolidation and associated neural plasticity. However, several hypotheses exist to reconcile our findings. First, emerging evidence suggests that a complex interaction exists between DNA methylation and histone acetylation during fear memory consolidation and reconsolidation processes. We have previously shown, for example, that inhibition of DNMT activity in the LA significantly interferes with training and retrieval-related changes in histone acetylation (Maddox & Schafe, 2011b; Miller, et al., 2008; Monsey, et al., 2011), suggesting that DNMT inhibition may promote impairments in memory consolidation and synaptic plasticity via inhibition of the training and retrieval-related regulation of histone acetylation in the LA. A second hypothesis involves recent work suggesting that alterations in DNA methylation accompanying learning may be preferential to memory suppressing genes, such as protein phosphatase 1 (PP1), rather than to memory promoting genes, such as reelin (Miller & Sweatt, 2007). In the hippocampus, for example, contextual fear conditioning has been shown to regulate the methylation of the PP1 gene, leading to a decrease in PP1 gene expression. In the presence of DNMT inhibitors, however, methylation of the PP1 gene is significantly reduced, with a corresponding increase in PP1 gene expression (Miller & Sweatt, 2007). Thus, one mechanism by which DNMT inhibitors impair synaptic plasticity and memory in the LA may be to promote the expression of memory suppressor genes that would otherwise be suppressed by fear learning. Finally, a third hypothesis is that DNA methylation may not be exclusively repressive of transcription. Recent studies have suggested that, under certain circumstances, DNA methylation can promote, rather than inhibit, transcription, particularly when the gene in question contains a cAMP-response element (CRE) sequence in its promoter region (Chahrour, et al., 2008; Gupta, et al., 2010). Given that many of the genes known to be regulated by auditory fear memory conditioning and retrieval of a fear memory, including Arc/Arg3.1 and Egr-1 (Maddox, Monsey, & Schafe, 2010; Maddox & Schafe, 2011a; Ploski, et al., 2008), contain CRE-response elements, it is possible that inhibition of DNMT activity in the LA may result in reduced expression of these genes following fear conditioning or fear memory retrieval and thereby contribute to the observed behavioral and neurophysiological deficits in the present study. Future work is needed to more closely examine the interaction between DNA methylation and histone acetylation during memory consolidation and reconsolidation as well as to examine in depth how the methylation patterns of genes such as Egr-1 and Arc/Arg3.1 are altered in the LA as a result of auditory fear conditioning or fear memory retrieval.
While our findings of impaired fear memory consolidation, reconsolidation, and memory-related synaptic plasticity following treatment with 5-AZA are consistent with those observed in previous studies that have used this compound to examine the role of DNA methylation in hippocampal- and amygdala-dependent learning paradigms (Maddox and Schafe 2011; Miller and Sweatt 2007; Lubin et al. 2008; Miller et al. 2008, 2010; Monsey et al. 2011), it is worth noting that, outside of the CNS, 5-AZA is considered an S-phase-specific nucleoside analogue that inhibits DNA methylation during DNA replication where it is incorporated into DNA to trap DNMTs. Thus, the precise mechanism by which 5-AZA works in post-mitotic cells of the CNS is presently unknown. However in line with the potential for active DNA demethylation within neurons, recent evidence has suggested that cytosine analogs can be incorporated into the genome of non-dividing cells in vitro (Yamagata et al., 2012) and several studies have shown that 5-AZA can effectively modulate DNA methylation in the hippocampus (Miller and Sweatt 2007) and prefrontal cortex (Miller et al. 2010). Importantly, in our experiments we showed that the pharmacologically distinct non-nucleoside DNMT inhibitor RG108 results in very similar deficits in auditory fear memory consolidation, reconsolidation and memory-related synaptic plasticity LA. The use of two pharmacologically distinct inhibitors of DNMT activity thus makes it highly unlikely that our pharmacological manipulations are producing memory and plasticity deficits by some non-specific means. However, additional experiments will be required to determine how both 5-AZA and RG108 are affecting the methylation of genes relevant to synaptic plasticity in the LA following fear conditioning and fear memory retrieval.
In summary, the present findings strongly suggest that the consolidation and post-retrieval retention of conditioning-related changes in tone-evoked neural activity in the LA are regulated by DNMT activity. These findings are the first to demonstrate that DNMT activity is required for memory-related neural plasticity in vivo and further contribute to our understanding of the epigenetic mechanisms that are required for fear memory consolidation and reconsolidation.
Figure 4. Electrode placements in each experiment.
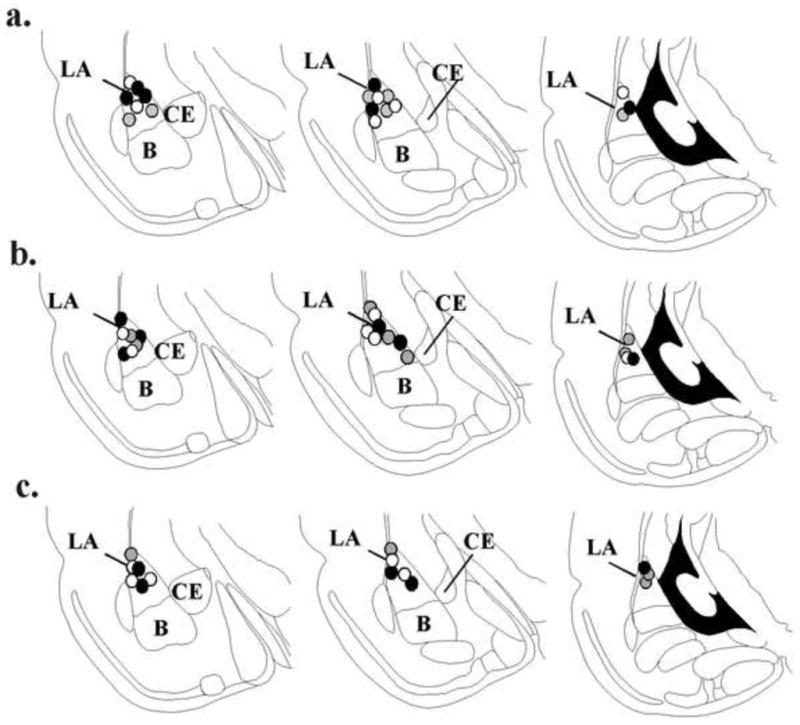
(a) Histological location of recording electrode placements for rats infused with vehicle (black circles), 5-AZA (gray circles) or RG108 (white circles) in the consolidation experiment (Figure 1). (b) Histological location of recording electrode placements for rats infused with vehicle (black circles), 5-AZA (gray circles) or RG108 (white circles) in the reconsolidation experiment (Figure 2). (c) Histological location of recording electrode placements for rats infused with vehicle (black circles), 5-AZA (gray circles) or RG108 (white circles) in the non-reactivated experiment (Figure 3). All panels adapted from Paxinos and Watson (1998). LA = lateral amygdala; CE = central nucleus of amygdala; B = basal nucleus of the amygdala
Highlights.
DNMT inhibition impairs fear memory consolidation
DNMT inhibition impairs the consolidation of neural plasticity in the LA
DNMT inhibition impairs fear memory reconsolidation
DNMT inhibition impairs the reconsolidation of neural plasticity in the LA
Acknowledgments
This research was supported by National Institutes of Health Grant MH 073949 (to G.E.S.) and by Yale University. This research was made with Government support under and awarded by DoD, Air Force Office of Scientific Research, National Defense Science and Engineering Graduate (NDSEG) Fellowship, 32 CRF 168a, awarded to S.A.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alberini CM. Mechanisms of memory stabilization: are consolidation and reconsolidation similar or distinct processes? Trends Neurosci. 2005;28(1):51–56. doi: 10.1016/j.tins.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Bailey DJ, Kim JJ, Sun W, Thompson RF, Helmstetter FJ. Acquisition of fear conditioning in rats requires the synthesis of mRNA in the amygdala. Behav Neurosci. 1999;113(2):276–282. doi: 10.1037//0735-7044.113.2.276. [DOI] [PubMed] [Google Scholar]
- Barrett RM, Wood MA. Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem. 2008;15(7):460–467. doi: 10.1101/lm.917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Mamou C, Gamache K, Nader K. NMDA receptors are critical for unleashing consolidated auditory fear memories. Nat Neurosci. 2006;9(10):1237–1239. doi: 10.1038/nn1778. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyère V, Debiec J, Monfils MH, Schafe GE, LeDoux JE. Synapse-specific reconsolidation of distinct fear memories in the lateral amygdala. Nat Neurosci. 2007;10(4):414–416. doi: 10.1038/nn1871. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Nader K, LeDoux JE. Activation of extracellular signal-regulated kinase- mitogen-activated protein kinase cascade in the amygdala is required for memory reconsolidation of auditory fear conditioning. Eur J Neurosci. 2005;21(1):283–289. doi: 10.1111/j.1460-9568.2004.03824.x. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Nader K, LeDoux JE. De novo mRNA synthesis is required for both consolidation and reconsolidation of fear memories in the amygdala. Learn Mem. 2008;15(10):747–755. doi: 10.1101/lm.1027208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, et al. Histone methylation regulates memory formation. J Neurosci. 2010;30(10):3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Langley B, Lubin FD, Renthal W, Wood MA, Yasui DH, et al. Epigenetics in the nervous system. J Neurosci. 2008;28(46):11753–11759. doi: 10.1523/JNEUROSCI.3797-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA, Jr, Neve RL, Nestler EJ, Davis M. Long-Term Memory Is Facilitated by cAMP Response Element-Binding Protein Overexpression in the Amygdala. J Neurosci. 2001;21(7):2404–2412. doi: 10.1523/JNEUROSCI.21-07-02404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, et al. CREB required for the stability of new and reactivated fear memories. Nat Neurosci. 2002;5(4):348–355. doi: 10.1038/nn819. [DOI] [PubMed] [Google Scholar]
- Lee JL, Di Ciano P, Thomas KL, Everitt BJ. Disrupting reconsolidation of drug memories reduces cocaine-seeking behavior. Neuron. 2005;47(6):795–801. doi: 10.1016/j.neuron.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, et al. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281(23):15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6(2):108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms: a common theme in vertebrate and invertebrate memory formation. Cell Mol Life Sci. 2006;63(9):1009–1016. doi: 10.1007/s00018-006-6026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28(42):10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox SA, Monsey MS, Schafe GE. Early growth response gene 1 (Egr-1) is required for new and reactivated fear memories in the lateral amygdala. Learn Mem. 2010;18(1):24–38. doi: 10.1101/lm.1980211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox SA, Monsey MS, Schafe GE. Early growth response gene 1 (Egr-1) is required for new and reactivated fear memories in the lateral amygdala. Learn Mem. 2011;18(1):24–38. doi: 10.1101/lm.1980211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox SA, Schafe GE. The activity-regulated cytoskeletal-associated protein (Arc/Arg3.1) is required for reconsolidation of a Pavlovian fear memory. J Neurosci. 2011;31(19):7073–7082. doi: 10.1523/JNEUROSCI.1120-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox SA, Schafe GE. Epigenetic alterations in the lateral amygdala are required for reconsolidation of a Pavlovian fear memory. Learn Mem. 2011;18(9):579–593. doi: 10.1101/lm.2243411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox SA, Watts CS, Doyère V, Schafe GE. A naturally-occurring histone acetyltransferase inhibitor derived from Garcinia indica impairs newly acquired and reactivated fear memories. PLoS One. 2013;8(1):e54463. doi: 10.1371/journal.pone.0054463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox SA, Watts CS, Schafe GE. p300/CBP histone acetyltransferase activity is required for newly acquired and reactivated fear memories in the lateral amygdala. Learn Mem. 2013;20(2):109–119. doi: 10.1101/lm.029157.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Campbell SL, Sweatt JD. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem. 2008;89(4):599–603. doi: 10.1016/j.nlm.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Gavin CF, White JA, Parrish RR, Honasoge A, Yancey CR, et al. Cortical DNA methylation maintains remote memory. Nat Neurosci. 2010;13(6):664–666. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53(6):857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. 2007;213(2):384–390. doi: 10.1002/jcp.21224. [DOI] [PubMed] [Google Scholar]
- Monsey MS, Ota KT, Akingbade IF, Hong ES, Schafe GE. Epigenetic alterations are critical for fear memory consolidation and synaptic plasticity in the lateral amygdala. PLoS One. 2011;6(5):e19958. doi: 10.1371/journal.pone.0019958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406(6797):722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Ploski JE, Pierre VJ, Smucny J, Park K, Monsey MS, Overeem KA, et al. The activity-regulated cytoskeletal-associated protein (Arc/Arg3.1) is required for memory consolidation of pavlovian fear conditioning in the lateral amygdala. J Neurosci. 2008;28(47):12383–12395. doi: 10.1523/JNEUROSCI.1662-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Repa C, LeDoux JE. Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: parallel recordings in the freely behaving rat. Neuron. 1995;15(5):1029–1039. doi: 10.1016/0896-6273(95)90092-6. [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE. Intraamygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J Neuroscience. 2001;21(17):6889–6896. doi: 10.1523/JNEUROSCI.21-17-06889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004;44(1):75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390(6660):604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J Neurosci. 2000;20(21):8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Doyère V, LeDoux JE. Tracking the fear engram: the lateral amygdala is an essential locus of fear memory storage. J Neurosci. 2005;25(43):10010–10014. doi: 10.1523/JNEUROSCI.3307-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE. Memory Consolidation of Auditory Pavlovian Fear Conditioning Requires Protein Synthesis and Protein Kinase A in the Amygdala. J Neurosci. 2000;20(18):RC96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Nader K, Blair HT, LeDoux JE. Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci. 2001;24(9):540–546. doi: 10.1016/s0166-2236(00)01969-x. [DOI] [PubMed] [Google Scholar]
- Tronson NC, Taylor JR. Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci. 2007;8(4):262–275. doi: 10.1038/nrn2090. [DOI] [PubMed] [Google Scholar]
- Tronson NC, Wiseman SL, Olausson P, Taylor JR. Bidirectional behavioral plasticity of memory reconsolidation depends on amygdalar protein kinase A. Nat Neurosci. 2006;9(2):167–169. doi: 10.1038/nn1628. [DOI] [PubMed] [Google Scholar]
- Yamagata Y, Szabo P, Szuts D, Bacquet C, Aryanyi T, Paldi A. Rapid turnover of DNA methylation in human cells. Epigenetics: Official Journal of the DNA Methylation Society. 2012;7:141–145. doi: 10.4161/epi.7.2.18906. [DOI] [PMC free article] [PubMed] [Google Scholar]