

Abstract
Genetics have undoubtedly become an integral part of biomedical science and clinical practice, with important implications in deciphering disease pathogenesis and progression, identifying diagnostic and prognostic markers, as well as designing better targeted treatments. The exponential growth of our understanding of different genetic concepts is paralleled by a growing list of genetic terminology that can easily intimidate the unfamiliar reader. Rendering genetics incomprehensible to the clinician however, defeats the very essence of genetic research: its utilization for combating disease and improving quality of life. Herein we attempt to correct this notion by presenting the basic genetic concepts along with their usefulness in the cardiology clinic. Bringing genetics closer to the clinician will enable its harmonious incorporation into clinical care, thus not only restoring our perception of its simple and elegant nature, but importantly ensuring the maximal benefit for our patients.
Basic Principles in Molecular Genetics
The structure of DNA
All inheritable traits of living organisms are determined by their genetic material, the ‘genome’, a long nucleic acid called deoxyribonucleic acid (DNA). The DNA consists of nucleotides. Each nucleotide is made up of a sugar (deoxyribose), a nitrogenous base (adenine (A), guanine (G), cytosine (C) or thymine (T)) and a phosphate group (Fig. 1) [1,2]. The four nitrogenous bases are divided into two groups: purines (including A and G) have two joined heterocyclinc rings and pyrimides (including C and T) have a single heterocyclic ring. Successive sugar and phosphate residues are linked by covalent phosphodiester bonds, forming the backbone of the DNA molecule and a nitrogenous base is attached to each sugar. The stability of DNA is primarily dependent on the strong covalent bonds that connect the constituent atoms of its linear backbone, and also on a number of weak non-covalent bonds that exist. Meanwhile, because of the phosphate group charges present in each nucleotide, DNA is negatively charged and therefore highly soluble in water.
Figure 1. The chemical structure of a nucleotide including the phosphate (yellow), the sugar (deoxyribose in green) and adenine as the nitrogenous base (pink).
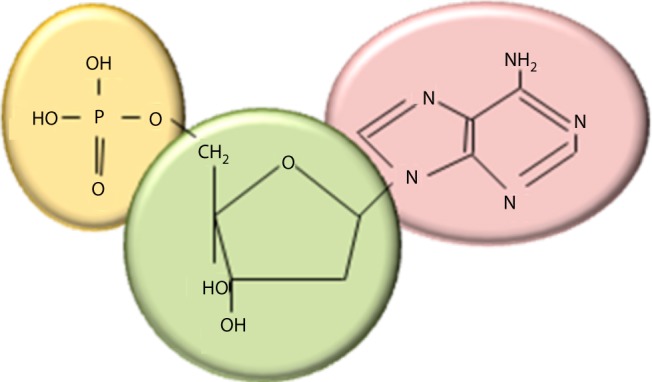
The DNA structure is a double helix, in which two DNA molecules are held together by weak hydrogen bonds [3–7]. Hydrogen bonding occurs between laterally opposed bases, of the two strands according to Watson-Crick rules: A specifically binds to T, and G to C. The two strands are therefore complementary [8]. As the phosphodiester bonds link carbon atoms number 3’ and number 5’ of successive sugar residues, the end of each DNA strand will have a terminal sugar residue where carbon atom number 5 is not linked to a neighboring sugar residue, and is therefore called 5’ end. The other end of the molecule is similarly called 3’ end. The two DNA strands are antiparallel because they always associate (anneal) in such a way that the 5’→3’ direction of one DNA strand is the opposite to that of its partner. To describe a DNA sequence, the sequence of bases of one strand only, are usually provided, and are provided in the 5’→3’ direction. This is the direction of DNA replication as well as transcription.
The packing of DNA into chromosomes
The human DNA is estimated to be approximately 2 m long. In order for it to fit in the 10 μm nucleus of human cells it is imperative that it is tightly folded. The DNA double helix is therefore subjected to at least two levels of coiling: the first involving coiling around a central core of eight histone proteins, resulting in units called nucleosomes, which are connected by spacer DNA; and the second involving coiling of this string of nucleosomes into a chromatin fiber [3]. During the different phases of the cell cycle, the DNA varies in the extent of its condensation. For example, during interphase the chromatin fibers are organized into long loops, whereas in metaphase chromosomes, the DNA is compacted to about 1/10,000 of its stretched out length. In humans there are 24 different chromosomes, namely 1–22 autosomes, and sex chromosomes X and Y [9]. Since humans are diploid organisms, our DNA is found in two copies, one inherited from each parent, and is folded into 46 chromosomes. Among the major DNA sequence elements of each chromosome are: the centromeres (constriction site where sister chromatics are joined and chromosomes link to the mitotic spindle), the telomeres (structures capping the ends of chromosomes) and the origins of replication (where DNA replication begins). Chromatin is encountered in extended (euchromatin) or highly condensed (heterochromatin) states, which in turn affect the transcriptional status of the corresponding DNA regions (being active or inactive, respectively) [10]. Under the light microscope, these regions appear as light and dark bands of metaphase chromosomes (Fig. 2).
Figure 2. Diagrammatical representation of the human karyotype of haploid chromosome set with X and Y as the sex chromosome complement. The alternating light and dark bands are characteristic of each chromosome in standard G-banding karyotype, and they represent euchromatic and heterochromatic regions, respectively.

Genomic DNA contains coding as well as non-coding regions. The non-coding regions are involved in DNA folding, chromosome formation, chromatin organization within the nucleus, regulation of transcription and more [11–14]. The coding regions are responsible for the transcription of RNA molecules and ultimately protein synthesis.
The composition and function of genes
The genes are stretches of DNA that code for polypeptides. Specifically, genes contain regulatory and coding regions, which regulate their transcription or code for the polypeptide product, respectively. A key regulatory region is the promoter, where the transcription machinery binds for transcription to be initiated. Other possible regulatory regions include enhancers, which regulate gene expression in different tissues or cells, and can be found upstream or downstream of the coding region, as far as several thousand bases. The coding regions are represented by exons, whose size and number varies among different genes. Interspersed between gene exons, there are non-coding sequences named introns, which tend to make up the largest percentage of a gene. The human genome is estimated to contain approximately 20,000 different genes [15]. Interestingly, it is estimated that 80% of the human genome is expressed, yet only 2% is coding for proteins [16].
The central dogma of molecular biology
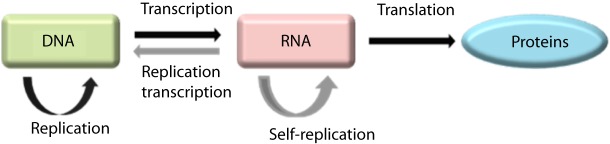
The central dogma of molecular biology was first stated in 1958 and re-stated in 1970 by Francis Crick [17,18]. According to this dogma there are three major classes of biopolymers: DNA, RNA and protein, and three classes of direct transfer of information that can occur between these biopolymers: general transfers, special transfers and unknown transfers. Of these, only the general transfers are believed to occur normally in most cells and they involve DNA replication to DNA, DNA transcription to mRNA and mRNA translation to proteins (Fig. 3).
Figure 3. The central dogma of molecular biology, as it currently applies in most cells (general transfers = black), or under specific conditions (in some viruses and in vitro: special transfers = grey).
Transcription
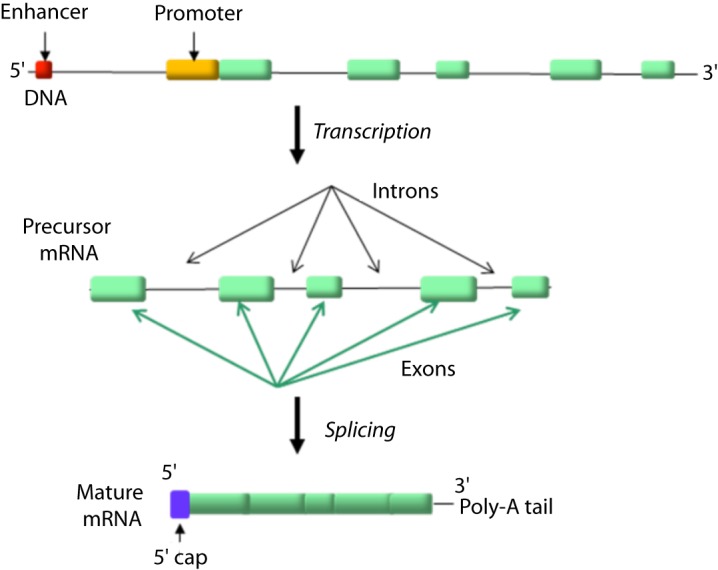
The transfer of information from the DNA to the protein level is achieved step-wise and starts with the transcription of a gene to mRNA. Specifically, the transcription machinery (including RNA polymerase and a variety of transcription factors) binds to the gene promoter, the double helix opens in that location and a single strand primary mRNA molecule (hn-RNA), complementary to that gene sequence, is synthesized base by base (Fig. 4) [19]. RNA as opposed to DNA, is a single strand nucleic acid containing ribose instead of deoxyribose and uracil instead of thymine. The heterogenous (hn)-RNA molecules go through a series of processing steps including a 5’ cap, a poly-A (50–250 adenine molecules and a 70kDa protein) tail at the 3’ end and splicing, to remove the intronic sequences (Fig. 5) [20–23]. Alternative splicing can also occur, which removes certain exons and contributes to the diversity of proteins any single gene can produce [24].
Figure 4. During transcription the transcription machinery (including RNA polymerase and a variety of transcription factors) binds to the gene promoter, the double helix opens in that location and a single strand primary mRNA molecule (hn-RNA), complementary to that gene sequence, is synthesized base by base.
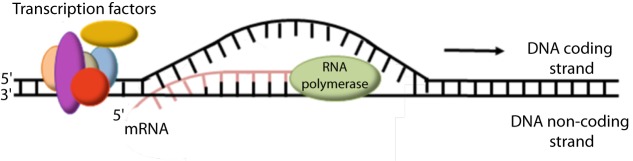
Figure 5. The promoter and enhancer elements of each gene are involved in gene transcription to precursor mRNA (hn-RNA) molecules which are then appropriately processed to give mature mRNA.
Translation
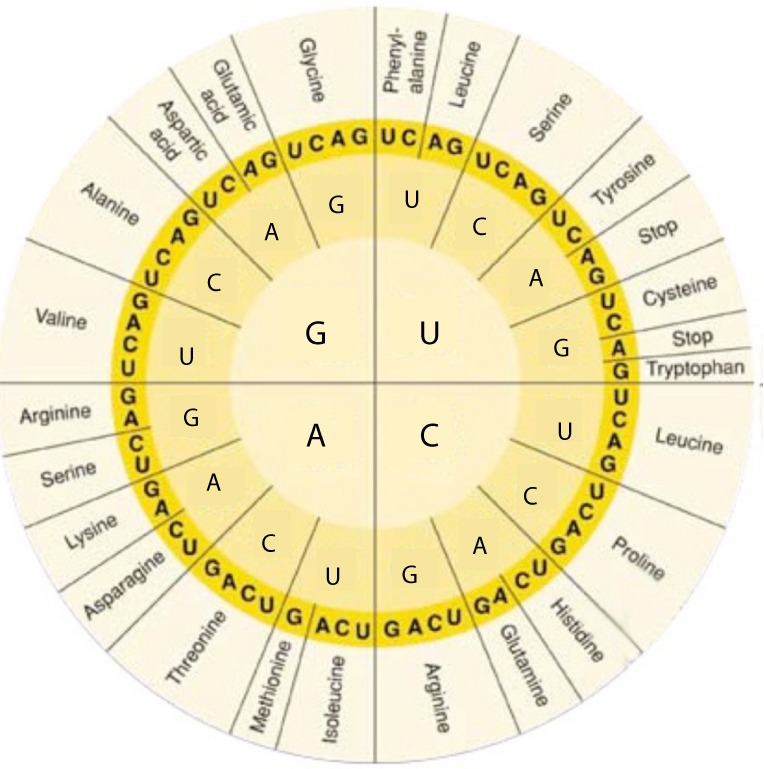
The mature mRNA molecules can be translated to proteins [25]. This process takes place in the cytoplasm with the aid of ribosomes, which are complexes of RNAs and proteins called ribonucleoproteins. The ribosomes are divided into two subunits: the smaller subunit binds to the mRNA, while the larger subunit binds to the tRNA which carries the amino acids. When a ribosome finishes reading a mRNA, these two subunits split apart. In particular, ribosomes bind mRNA and “read” through it as triplet codons, usually starting with an AUG triplet (initiation codon) downstream of the ribosome binding site. For each codon, the ribosome, with the aid of initiation and elongation factors, recruits a complementary tRNA molecule, which in turn carries a specific amino acid. Each codon codes for a specific amino acid as shown in Fig. 6. As the amino acids are linked into the growing peptide chain, they begin folding into the correct conformation. The translation process ends with the stop codons UAA, UGA or UAG. The nascent polypeptide chain is then released from the ribosome as a mature protein [25]. In some cases the new polypeptide chain requires additional processing to make a mature protein. Mature proteins in turn can be subjected to a range of post-translational modifications. Their ultimate roles in cell physiology can be highly variable including cytoarchitecture, enzymatic activity, intracellular signalling, transportation, communication etc.
Figure 6. The RNA codon table.
Genetic variation
Genetic variation refers to genetic difference between individuals within or between different populations. This variation is what renders each individual unique in its phenotypic characteristics. Genetic variation occurs on many different scales, ranging from gross alterations in the human karyotype to single nucleotide changes. These variations can be divided in polymorphisms and mutations.
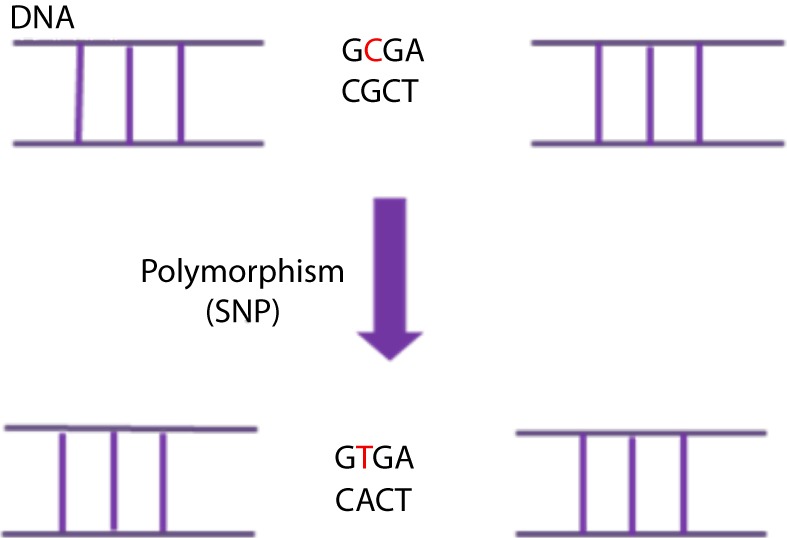
Polymorphisms are defined as variants found in >1% of the general population [26]. Due to their high frequency they are considered unlikely to be causative of genetic disease. They can however, together with other genetic and environmental factors, affect disease predisposition, disease progression or response to treatments (e.g. [27]). Three common types of polymorphisms are the single nucleotide polymorphisms (SNPs), small insertions/deletions (indels) and the large-scale copy number polymorphisms (CNPs or CNVs). SNPs are single base changes that occur on average about every 1000 bases in the genome. Their distribution is not homogenous and they occur more frequently in non-coding regions where there is less selective pressure (Fig. 7) [28,29]. Most SNPs are neutral; yet 3–5% are thought to have a functional role, i.e. affect the phenotype of the individual carrying them. Depending on their effect at the protein level, SNPs can be characterized as synonymous (coding for the same amino acid as the wild type DNA sequence) or non-synonymous (coding for a different amino acid than the wild type DNA sequence) [29]. Indels are small insertions or deletions ranging from 1 to 10,000 bp in length, although the majority involves only a few nucleotides [30,31]. They are considered the second most common form of variation in the human genome following SNPs, with over 3 million short indels listed in public databases. CNVs are variations in the number of copies of DNA regions. They can involve loss of one or both copies of a region of DNA, or the presence of more than two copies of this region. They can arise from DNA deletions, amplifications, inversions or insertions and their size can range from 1 kb (1,000 bases) to several megabases [32]. SNPs, indels and CNVs can either be inherited or arise de novo.
Figure 7. A Single-Nucleotide DNA Polymorphism (SNP) is defined as a single DNA variation detected when a single nucleotide in the genome (or other common sequence) is different between species or paired chromosomes in an individual. In this case there is a substitution of a C (Cytosine) in a T (Tymine) which causes the change of a G (Guanine) in a A (Adenine) in the complementary DNA strand.
Mutations on the other hand, are rare (by some defined as variations with <1% frequency in the general population, although there are many exceptions to this rule) changes in the DNA sequence that can change the resulting protein, impair or inhibit the expression of the gene, or leave both the gene function and protein levels/structure unaffected. Although a variety of definitions have been considered over the years, for most scientists mutation has become synonymous with disease. They can arise during DNA replication or as a result of DNA damage through environmental agents including sunlight, cigarette smoke and radiation. A variety of different types of mutations exist and the terminology used to describe them is based on their effect either on DNA structure, on protein product function, or on the fitness of the individual carrying them.
In terms of DNA structure modification, mutations can be categorized as:
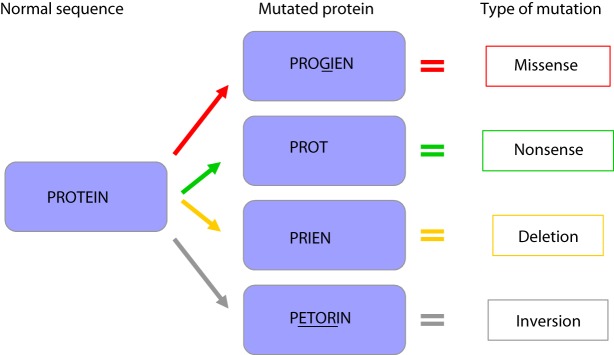
A) point mutations in which a single nucleotide is changed for a different one (Fig. 8). These are divided into missense mutations (meaning that when translated this DNA sequence leads to the incorporation of a different amino acid into the produced protein, with possible implications in the protein function), nonsense mutations (where the new nucleotide changes the sequence so that a “stop” codon is formed earlier than in the normal sequence and therefore the produced protein is truncated), silent mutations (where the nucleotide change does not affect the amino acid in the corresponding position of the produced protein, and therefore the final protein product remains unaltered), and splice-site mutations (which affects the splice site invariant donor or acceptor dinucleotides (5’GT or 3’AG).
Figure 8. Different types of mutation and possible conseuquence on protein function: A) missense mutation; B) nonsense mutation; C) deletion; D) inversion.
B) insertions in which one or more nucleotides are inserted in the normal DNA sequence, therefore disrupting it. This can have a moderate or severe effect on the corresponding mutant protein product. For example it can affect the splicing or the reading frame ( frame-shift mutations), therefore leading an incorrect “reading” of all the downstream nucleotide triplets and consequently their translation to a significantly different and/or truncated amino acid sequence.
C) deletions in which one or more nucleotides are deleted from the normal DNA sequence (Fig. 8). As in the case of insertions this can lead to minor (e.g. single amino acid changes) or major protein defects (e.g. reading frame modifications with implications for the entire downstream amino acid sequence of the mutant protein). When larger chromosomal regions are deleted, multiple genes can be lost and/or previously distant DNA sequences can now be juxtaposed (such juxtapositions can lead, for example, to the production of abnormal proteins containing sequences from different genes that have now been “merged” or abnormal expression of otherwise normal proteins by deletions affecting their upstream regulatory regions).
D) amplifications leading to multiple copies of chromosomal regions and consequently to an increased number of copies of the genes located within them and increased levels of the corresponding proteins.
E) inversions involving the reversal of the orientation of a DNA segment, with variable implications for the protein product, similar to the ones described above (Fig. 8).
F) translocations where regions from non-homologous chromosomes are interchanged.
Mutations can affect the expression of a transcript and its corresponding protein, or modify the structure of the resulting protein therefore impairing its function [33]. Depending on their functional effect, mutations can be classified as dominant negative (the mutant gene product acts antagonistically to the wild-type allele), gain-of-function (the mutant gene product gains a new and abnormal function), and loss-of-function (the mutant gene product has less or no function). Loss-of-function mutations can be associated with haploinsufficiency, a common occurrence in the molecular cardiomyopathy setting.
Haploinsufficiency occurs when the gene product of one of the two alleles in an individual is lost due to a DNA deletion or to instability/degradation of the mutant protein. Other terms used to describe the effect of a mutation on the fitness of the carrier are: harmful or deleterious mutations (decreases the fitness of the carrier), beneficial or advantageous mutations (increases the fitness of the carrier), and lethal mutations (leading to the death of the individual carrying them).
Distinguishing polymorphisms from mutations in the research setting
In the field of cardiovascular genetics, when a new genetic variant is identified – a common occurrence given the large number of genes and different variants thereof being screened – it is crucial to first determine whether it represents a benign polymorphism or a pathogenic mutation. Identifying pathogenic mutations enables the characterization of the molecular mechanisms of pathogenesis, and more importantly for the clinical setting, it allows the development of genetic tests for mutation detection in other family members (including pre-symptomatically) as well as unrelated patients with similar phenotypes (see section on ‘Cardiovascular genetics in clinical practice’).
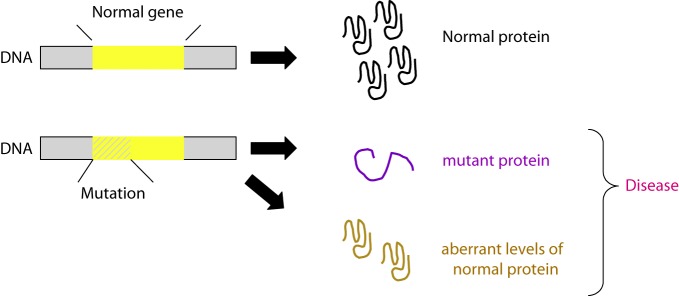
The Clinical Molecular Genetics Society1 and the American College of Medical Genetics2 have issued guidelines to facilitate the determination of the potential pathogenic role of a novel/unclassified variant (Fig. 9). The Human Gene Mutation Database,3 along with locus-specific or disease-specific mutation databases, are valuable resources for first deciphering whether a detected genetic variant represents a known mutation. The databases Online Mendelian Inheritance in Man,4 dbSNP5 and Ensembl,6 along with thorough searches of the literature via PubMED, Google Scholar, Scopus or the Web of Science, can also provide valuable information. From thereon carefully matched controls need to be included in the study populations, co-occurrence with known (in trans) deleterious mutations in the same gene needs to be ruled out, co-segregation with the disease in the family represents useful information, and occurrence of the novel variant concurrent with the incidence of a sporadic disease can be a strong indicator. Bioinformatically, it is important to determine if the unclassified variant leads to an animo acid change and how different the biophysical properties of the new amino acid are: the greater the difference, the higher the likelihood to possess a pathogenic role. Similarly, the more conserved a DNA region is across species, the greater an impact any variations therein are likely to have. A range of in silico analysis tools can also be used for the predication of a pathogenic effect (e.g. Align GVGD, Sorting Intolerant From Tolerant [SIFT], Polyphen, and Alamut) or the prediction of splice sites. One of the best means of determining pathogenicity, however, is the use of suitable functional assays and transgenic animal models [34,35].
Figure 9. From mutation to disease. A DNA mutation can cause qualitative or quantitative changes at the protein level, leading to either a dysfunctional/non-functional protein product and/or aberrant protein expression levels. Both mechanisms can in turn lead to CVD.
Mode of inheritance—clinical and genetic heterogeneity
Once a mutation has been directly associated with a pathological phenotype a number of additional parameters need to be evaluated in order to maximize its value in the clinical setting. These parameters relate to the mode of inheritance of a mutation, which impacts directly the chances of detecting it in other family members of the patient, or his/her offspring. The categorization gonosomal or autosomal depends on whether the mutations are located on either of the sex chromosomes or not. For example a mutation on the Y chromosome will only affect males. The dominant or recessive nature relates to the need of one or both alleles, respectively, to carry the mutation for the pathogenic phenotype to develop. In hypertrophic cardiomyopathy (HCM) a number of cases have been reported with homozygosity for the pathogenic mutation. Nishi et al. first reported homozygosity for a MYH7 mutation in two brothers with HCM [36]. Homozygous mutations were also detected in MyBPC in HCM patients [37]. The patients who harbour homozygous mutations present with a more severe clinical phenotype than their heterozygous family members. These observations support the notion of a mutation dosage effect, in which a larger amount of the defective protein leads to a greater disruption of the sarcomere function and results in a more severe clinical outcome. For example, in our Egyptian HCM cohort, none of the mutation-positive patients were homozygous for the mutation detected (data not published) which might be explained either by the rarity of its occurrence in the specific cohort or due to technical limitations in the mutation screening method (Fig. 10).
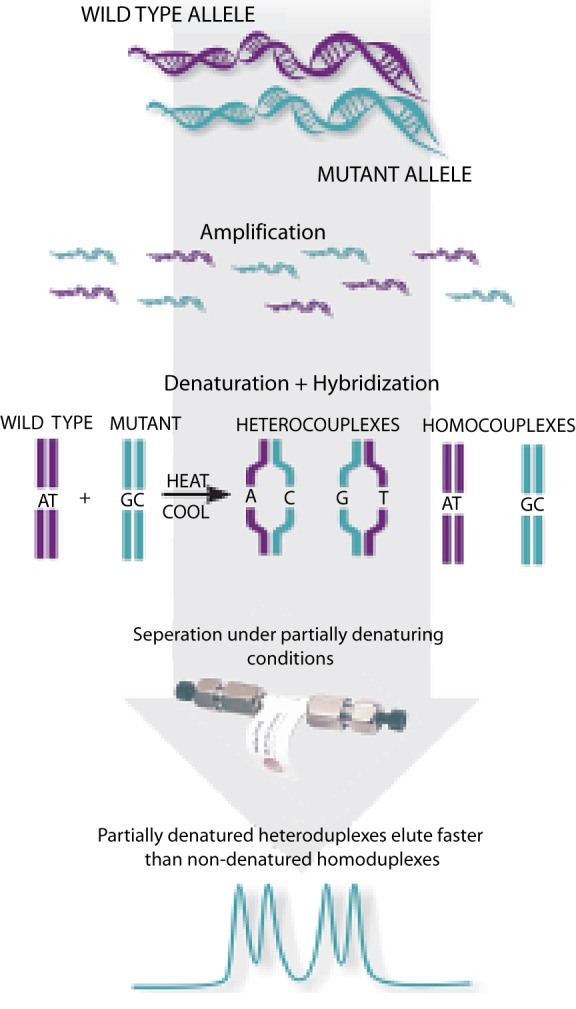
Figure 10. Mutation screening by denaturing high performance liquid chromatography (dHPLC) using WAVE™, Transgenomics. dHPLC can be used as an initial mutation screening method, being dependent on heteroduplex (wild type-mutant) formation, and variant profiles from the wild pattern are subsequently sequenced. Note however, that dHPLC is not capable of detecting homozygosity.
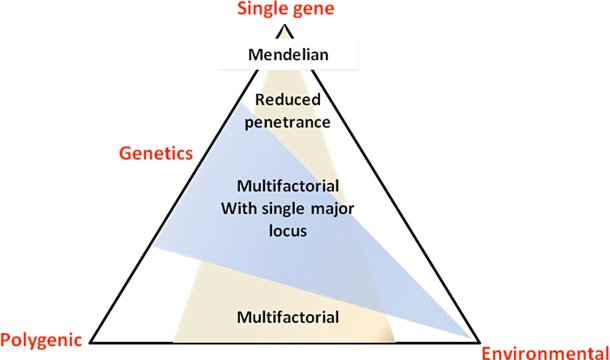
Importantly, a number of exceptions apply to the aforementioned inheritance mode rules, such as in the case of incomplete penetrance (a percentage of the individuals carrying the mutation fail to present the corresponding trait) where mutation carriers may not present with any symptoms even in the presence of a dominant mutation. Furthermore, the phenomena of variable expressivity (variations in a phenotype among individuals carrying a particular genotype) and epistasis (one gene is modified by one or several other genes, e.g. modifier genes) can lead to a range of pathological characteristics despite the presence of the same mutation. These parameters, potentially in combination with environmental factors, can often lead to significant clinical heterogeneity in most inherited CVDs, between unrelated individuals as well as family members carrying the same mutation (Fig. 11) [38].
Figure 11. Role of genetic and environmental factors in determining the spectrum of the disease phenotype. (Strachan T, Read AP. Genes in pedigrees and populations in Human molecular genetics 3. 3rd ed. London; New York: Garland Press; 2004).
Another exception is this of compound heterozygotes (carriers of two different mutations on the two alleles of the same gene) or double heterozygotes (carriers of mutations in two different genes), which carry one copy of each mutation, yet they can develop the disease. Notably, the concomitant presence of multiple genetic defects contributing to the same disease is usually associated with a more severe clinical phenotype. For example, in HCM the presence of multiple pathogenic mutations could be included amongst the risk stratification criteria [39]. Multiple mutations have been observed in about 5% of HCM patients and they are usually associated with higher septal thickness and worse clinical outcomes, such as heart failure and sudden death [40–43]. Double heterozygosity is commonly detected in the Myosin heavy chain (MYH7) and Myosin binding protein C (MyBPC) genes, probably because they represent the most commonly involved genes in the pathogenesis of HCM. Compound heterozygosity in MyBPC however, leading to the absence of a normal protein, has been reported to results in neonatal death in two independent cases, where the parents were each heterozygous for one of the mutations [44]. Similarly to HCM, double heterozygosity has been reported in other CVDs such as long QT, with a similar frequency of 5% [45].
Translating genetic research findings to the cardiovascular clinic
Hereditary CVDs include a variety of different aspects and structures of the cardiovascular system such as inherited cardiomyopathies, arrhythmias, metabolic disorders affecting the heart, congenital heart diseases, as well as vascular disorders such as Marfan syndrome [46–48]. Over the past two decades significant progress has been made towards the identification of the genetic basis of CVD, with tens of genes now known to be implicated in almost all of the different disorders. The magnitude of the role of genetics however, remains elusive. Although in some cases the pathogenesis appears to involve complex mechanisms and multifactorial (genetic and environmental) aetiology, multigenic inheritance (e.g. familial hypercholesterolemia: LDLR, APOB, ABCG5, ABCG8, ARH, PCSK9; hypertrophic cardiomyopathy: MYH7, TNNT2, TPM1, TNNI3, MYL2, MYBPC3, ACTC, MYL3) or even monogenic, also known as Mendelian, inheritance (e.g. Marfan syndrome: FNB1) has been described. Pinpointing the gene(s) and their specific mutations that lead to each pathological phenotype can give rise to valuable, complementary genetic diagnostic/prognostic tools for significantly improved clinical management of CVD patients and their families.
For example, the recently published consensus statement on the state of genetic testing for cardiomyopathies and channelopathies has elegantly presented the list of different genes which contribute by >5% to these inherited disorders. [49,50]. There are more than 50 distinct channelopathy/cardiomyopathy-associated genes with hundreds of mutations discovered to date. Each of these mutations/genes usually accounts for a small percentage of the reported cases, while in many cases the causative mutation/gene is never identified. For example, in channelopathies a mutation is found in <20% of short QT syndrome cases and up to 75% in long QT syndrome cases. An exceptional scenario is this of mutations in the cardiac ryanodine receptor (RYR2) gene in catecholaminergic polymorphic ventricular tachychardia, which account for up to 65% of affected patients [51]. In cardiomyopathies, positive genetic testing results range in frequency from <20% in restrictive cardiomyopathy to 60% in familial HCM. Despite the fact that two decades ago, HCM was termed “a disease of the sarcomere” involving at least 8 causative genes, the rate of mutation detection ranged in frequency from 25–30% in MyBPC and MYH7 to ˜5% in TNNT2 and TNNTI3, and ˜1% in other sarcomeric genes [42,52]. Additionally, there are HCM phenocopies (same phenotype) associated with non-sarcomeric gene mutations and different modes of inheritance, which may on occasion be difficult to exclude from sarcomeric HCM based on clinical evaluation alone. Therefore, multiple genes need to be screened for a multigenic disease such as HCM.
Overall, our understanding of the genetic basis of CVD has been rapidly expanding over the years with important lessons learned both on monogenic as well as complex disease forms [53]. However, the true value of these findings lies in their translation to the clinical setting and their utilisation towards improved CVD diagnosis, prognosis and treatment. Along these lines, genetic testing is currently available for a number of CVDs in the form of clinical service in most Western countries, and increasingly in the developing world.
Significance of genetic testing in cardiovascular medicine
Genetic testing can serve three main goals in the clinical practice: first to determine the mode of inheritance of the specific disease in the specific family and identify if there is risk for other family members; second to organize the clinical assessment of unaffected family members through predictive genetic testing so as to distinguish those who are at risk for the disease and should have regular cardiac follow-up (mutation carriers) and those who are not (mutation non-carriers); third, following the establishment of distinct genotype–phenotype correlations, the application of genetic testing in disease diagnosis, prognosis and personalized treatment (i.e. identification of the drugs to which each patient will respond best) [54].
The clinical value of genetic screening of a cardiovascular disease patient is therefore valuable initially at the diagnostic/prognostic/therapeutic level, provided the genotype–phenotype associations have been established first. These associations vary considerably among different cardiovascular diseases, different genes and different mutations thereof. The relevance of genetic testing towards these three levels of clinical management is possibly best shown in the setting of the long QT syndrome [49,50]. It is critical to note however, that genetic testing in the cardiovascular disease setting cannot be the basis for clinical management of patients, but can serve a complementary role to the comprehensive clinical evaluation to better address the patient's and his/her family's needs.
Identifying the causative mutation of a proband further allows the genetic screening of its family members, a process of marked predictive power and therefore high importance in the cardiovascular clinic [55]. The significance of such pre-symptomatic genetic testing for the probands family members ranges from ensuring that unaffected mutation carriers receive regular clinical follow-up and prophylactic treatment (where available) to reassurance that clinically ‘suspicious’ findings are unlikely to be indicative of the specific form of the disease in the absence of the specific family mutation (e.g. Fig. 12) [56,57]. Importantly however, a negative genetic test result in the proband's family members cannot by itself exclude the presence of disease in general, since a large number of different genes and a variety of mutations thereof can contribute to the same or a different pathological cardiovascular phenotype – and by chance, a family member could be a carrier of a different gene mutation.
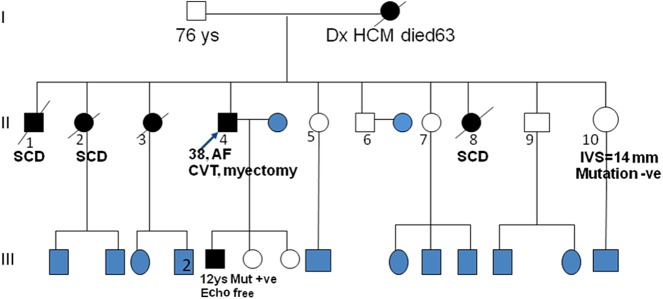
Figure 12. Pedigree of an HCM positive family from the BA HCM Study. A pathogenic mutation in MYH7 exon 23 (Glu927Lys) was detected in the proband II-4. Echo screening of all siblings was undertaken, and sister II-10 was found to have an interventricular septal measurement of 14 mm. Genetic screening of all family members excluded HCM diagnosis for the sister (II-10). However, the symptom free and echo clear son of the proband, was positive for the mutation and therefore given a pre-symptomatic diagnosis of HCM at the age of 12 years. Symbols in white represent unaffected individuals, in black are individuals with HCM based on clinical or genetic findings, and in blue are individuals who have not been screened by echo or genetic testing (unpublished data).
Genetic testing of children in the family has always posed an ethical concern, particularly for adult-onset diseases. Therefore pre-symptomatic testing of children should be extensively discussed with the family after a mutation has been identified in the proband, and in the context of the specific cardiovascular disease [58]. In cases where pre-symptomatic genetic screening and mutation identification has direct implications on the child's clinical follow-up, lifestyle adaptations and preventive treatments, it would be valuable to proceed with genetic testing, upon the parents’ approval. For example, for long QT syndrome and catecholaminergic polymorphic ventricular tachychardia, and occasionally in high risk HCM families, in which preventive measures or prophylactic therapy is advisable for asymptomatic mutation positive family members, genetic testing should be undertaken in early childhood, i.e. regardless of age. On the other hand, for late-onset and/or reduced penetrance diseases, it is reasonable to proceed with clinical monitoring as needed during childhood, leaving the genetic testing option open for when the individual reaches adulthood [49,50]. When a child has already presented with a CVD, the use of genetic testing is complementary to all other clinical tests, and especially valuable for identifying other family members at risk, since childhood-onset cases, even when presumed as sporadic, can often have a genetic aetiology. For example, approximately half of the presumed sporadic cases of childhood-onset hypertrophy have genetic causes [59].
Bridging the cardiovascular and the genetics clinics
Although the translation of molecular genetics to routine clinical practice is slow, a series of certified genetic testing centers (www.genetests.org) have been established, and guidelines have already been issued for a number of cardiovascular diseases such as HCM, dilated cardiomyopathy (DCM) and arrhythmogenic right ventricular cardiomyopathy (ARVC) [60,61]. The consensus is that a minimum of three to four generation family history needs to be obtained, the relatives at risk need to be identified and directed for clinical screening, the potential genetic nature of the disease needs to be explained, and the possibility of genetic testing should be discussed where appropriate.
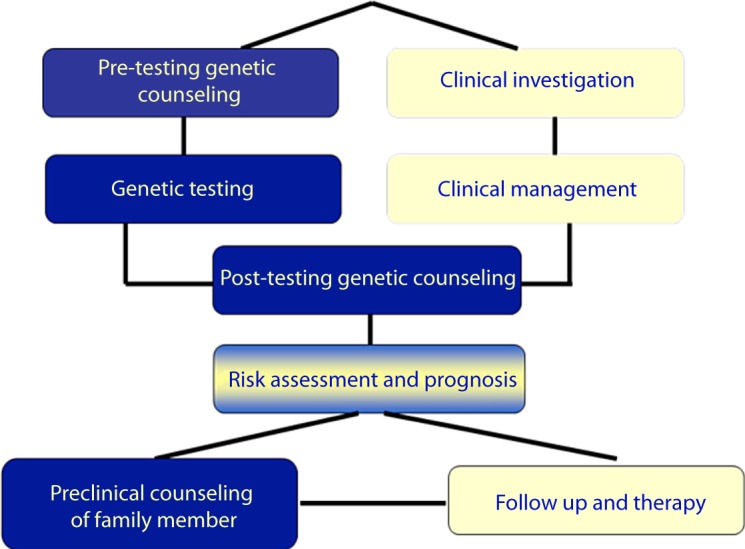
In order to follow these guidelines, cardiology clinics around the world need to ensure that cardiologists are provided with appropriate training in key genetic concepts, along with information on the latest developments in cardiovascular genetics and the best means to apply them in the clinic. Importantly, the close interaction between cardiologists, geneticists and genetic counsellors, especially in complex cases, will significantly expedite the ‘benchside-to-bedside’ translation of the latest genetic discoveries and optimize the clinical care provided to the patient [62,63]. For example, when routine cardiovascular genetic screening fails to detect the causative mutations, screening can be extended to include broad gene panels and/or application of high throughput technologies. Similarly, in cases where new mutations are identified, targeted genetic tests can be designed, if needed, for screening family members at risk. Currently, different modes of cardiologist-geneticist interactions are being adopted in clinical settings around the world, a process that requires time, continuing education and to some extent, reorganization of health systems [64]. An example of such an evolving system of interdisciplinary interactions is that of the Egyptian National Genetic study of HCM (Fig. 13).
Figure 13. Combined clinical and genetic evaluation of CVD patients will allow for improved disease management and patient care.
In conclusion, cardiovascular genetic testing is valuable for improving the standards of care for CVD patients and their families at the diagnostic, prognostic and therapeutic level. Importantly, for healthcare systems worldwide, it further represents a cost-effective approach by enabling the timely identification of individuals at risk, ensuring regular follow-up only for the individuals at risk and early disease detection, as well as enabling, where possible, the use of disease preventative measures in order to minimize the environmental contributing factors [65]. To this end, clinical cardiovascular genetics is increasingly emphasized in undergraduate and postgraduate medical education and incorporated in cardiological clinics worldwide [54].
Future directions
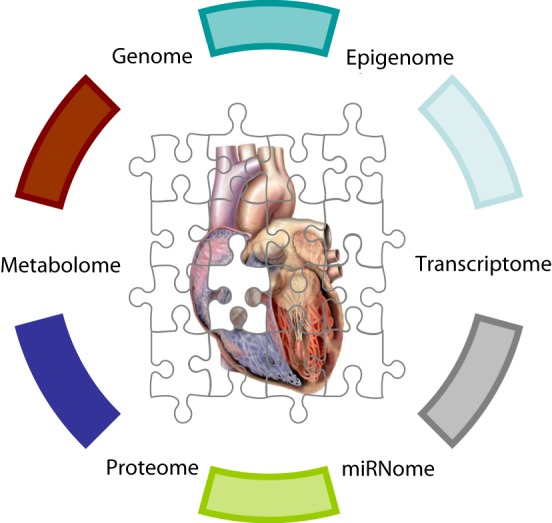
The tremendous technological advancements over the past decade have empowered the discovery of new biological concepts and the emergence of entirely new scientific fields. Among them, cardiac systems genetics – a systems-based analysis of genetic variants considering all different levels spanning from their effect on the cardiac transcriptome, proteome, metabolome to organ physiology/pathophysiology (phenome) (Fig. 14). The global analysis of the downstream functional – molecular and cellular – implications of different genetic variants, will allow the meaningful integration of molecular and clinical data in a powerful way.
Figure 14. To fully unravel the intricate pathways regulating cardiac physiology and pathophysiology the global studies of the human genome will need to be extended to similar studies at the epigenome (chemical changes to the DNA and histone affecting the chromatin structure and function of the genome), transcriptome (the full set of transcripts produced from the human DNA), miRNome (the full set of microRNAs produced from the human DNA), proteome (the full set of proteins) and metabolome (full set of metabolites) levels.
Systems genetics will in turn, serve as an integral part of network medicine, an advanced form of molecular medicine, where perturbations, rather than individual molecules, are investigated as the underlying causes of complex diseases [66]. In cardiology, examples of important first steps in this direction are the identification of cardiac gene expression signatures related with response to left ventricular assist device implantation [67,68] and peripheral leukocyte expression signatures indicative of post-cardiac transplantation tissue rejection [69]. Parameters such as epigenetics and microRNAs are increasingly integrated in network medicine, adding new dimensions to the intricate mechanisms of cardiovascular disease (e.g. [70,71]). A likely next addition to network medicine, based on emerging new data [72,73], could be this of metagenomics – the genomic investigation of micro-organisms inside the human body, and their effect on the global networks orchestrating human cardiac physiology/pathophysiology.
Cardiology is rapidly transformed with powerful new technologies expediting the acquisition of new knowledge and exciting new discoveries enriching our understanding of the intricate genotype–phenotype correlations. The close interaction of cardiologists and geneticists is facilitating the transition of novel findings to clinical practice and vice versa. It is also enabling the rapid establishment of appropriate research strategies to address emerging clinical questions. Ultimately the convergence of the two disciplines promises to transform the way we perceive, manage and treat CVD.
Disclosure of Conflict of Interest
None.
Acknowledgements
Molecular genetics study of HCM in Egypt is supported by the Magdi Yacoub Foundation Serving Egypt and Bibliotheca Alexandrina. We would like to thank the physicians Maha Saber, Gehan Magdy, Sarah Moharam, Hala Mahfouz and Ahmed Elguindy for their clinical and scientific collaboration in the BA HCM study (of which a separate manuscript is under preparation). DS is supported by the European Community's Seventh Framework Programme FP7/2007–2013 under grant agreement #HEALTH-F2-2009-241526 “EUTrigTreat”, the Hellenic Cardiological Society and the John S. Latsis Public Benefit Foundation.
Abbreviations
HCM: hypertrophic cardiomyopathy, CV: cardiovascular, LQT: long QT, UV: unclassified variant.
Footnotes
Notes
References
- [1].Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- [2].Watson JD, Crick FH. The structure of DNA. Cold Spring Harb Symp Quant Biol. 1953;18:123–131. doi: 10.1101/sqb.1953.018.01.020. [DOI] [PubMed] [Google Scholar]
- [3].Thoma F, Koller T. Influence of histone H1 on chromatin structure. Cell. 1977;12(1):101–107. doi: 10.1016/0092-8674(77)90188-x. [DOI] [PubMed] [Google Scholar]
- [4].Franklin RE, Gosling RG. Evidence for 2-chain helix in crystalline structure of sodium deoxyribonucleate. Nature. 1953;172(4369):156–157. doi: 10.1038/172156a0. [DOI] [PubMed] [Google Scholar]
- [5].Franklin RE, Gosling RG. Molecular configuration in sodium thymonucleate. Nature. 1953;171(4356):740–741. doi: 10.1038/171740a0. [DOI] [PubMed] [Google Scholar]
- [6].Wilkins MH, et al. Helical structure of crystalline deoxypentose nucleic acid. Nature. 1953;172(4382):759–762. doi: 10.1038/172759b0. [DOI] [PubMed] [Google Scholar]
- [7].Wilkins MH, Stokes AR, Wilson HR. Molecular structure of deoxypentose nucleic acids. Nature. 1953;171(4356):738–740. doi: 10.1038/171738a0. [DOI] [PubMed] [Google Scholar]
- [8].Crick FH. The Complementary Structure of DNA. Proc Natl Acad Sci USA. 1954;40(8):756–758. doi: 10.1073/pnas.40.8.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Spector DL. The dynamics of chromosome organization and gene regulation. Annu Rev Biochem. 2003;72:573–608. doi: 10.1146/annurev.biochem.72.121801.161724. [DOI] [PubMed] [Google Scholar]
- [10].Lamond AI, Earnshaw WC. Structure and function in the nucleus. Science. 1998;280(5363):547–53. doi: 10.1126/science.280.5363.547. [DOI] [PubMed] [Google Scholar]
- [11].Antequera F, Bird A. Predicting the total number of human genes. Nat Genet. 1994;8(2):114. doi: 10.1038/ng1094-114a. [DOI] [PubMed] [Google Scholar]
- [12].Nowak R. Mining treasures from ’junk DNA’. Science. 1994;263(5147):608–610. doi: 10.1126/science.7508142. [DOI] [PubMed] [Google Scholar]
- [13].Small D, Nelkin B, Vogelstein B. Nonrandom distribution of repeated DNA sequences with respect to supercoiled loops and the nuclear matrix. Proc Natl Acad Sci U S A. 1982;79(19):5911–5915. doi: 10.1073/pnas.79.19.5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tsongalis GJ, et al. Partial characterization of nuclear matrix attachment regions from human fibroblast DNA using Alu-polymerase chain reaction. Cancer Res. 1992;52(13):3807–3810. [PubMed] [Google Scholar]
- [15].Finishing the euchromatic sequence of the human genome, Nature. 2004;431:7011,931–945. [DOI] [PubMed]
- [16].Pennisi E. Genomics. DNA study forces rethink of what it means to be a gene. Science. 2007;316(5831):1556–1557. doi: 10.1126/science.316.5831.1556. [DOI] [PubMed] [Google Scholar]
- [17].Crick FH. On protein synthesis. Symp Soc Exp Biol. 1958;12:138–163. [PubMed] [Google Scholar]
- [18].Crick F. Central dogma of molecular biology. Nature. 1970;227(5258):561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- [19].Kollmar R, Farnham PJ. Site-specific initiation of transcription by RNA polymerase II. Proc Soc Exp Biol Med. 1993;203(2):127–139. doi: 10.3181/00379727-203-43583. [DOI] [PubMed] [Google Scholar]
- [20].Balvay L, Libri D, Fiszman MY. Pre-mRNA secondary structure and the regulation of splicing. Bioessays. 1993;15(3):165–169. doi: 10.1002/bies.950150304. [DOI] [PubMed] [Google Scholar]
- [21].Gorlach M, Burd CG, Dreyfuss G. The mRNA poly(A)-binding protein: localization, abundance, and RNA-binding specificity. Exp Cell Res. 1994;211(2):400–407. doi: 10.1006/excr.1994.1104. [DOI] [PubMed] [Google Scholar]
- [22].Munroe D, Jacobson A. Tales of poly(A): a review. Gene. 1990;91(2):151–158. doi: 10.1016/0378-1119(90)90082-3. [DOI] [PubMed] [Google Scholar]
- [23].Varani G. A cap for all occasions. Structure. 1997;5(7):855–858. doi: 10.1016/s0969-2126(97)00239-6. [DOI] [PubMed] [Google Scholar]
- [24].Staley JP, Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998;92(3):315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- [25].Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5(10):827–835. doi: 10.1038/nrm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].McCarthy MI, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9(5):356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- [27].Arvanitis DA, et al. The Ser96Ala variant in histidine-rich calcium-binding protein is associated with life-threatening ventricular arrhythmias in idiopathic dilated cardiomyopathy. Eur Heart J. 2008;29(20):2514–2525. doi: 10.1093/eurheartj/ehn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sachidanandam R, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 6822;(409):928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- [29].International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007 Oct 18;449(7164):851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Weber JL, et al. Human diallelic insertion/deletion polymorphisms. Am J Hum Genet. 2002 Oct;71(4):854–862. doi: 10.1086/342727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mills RE, et al. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006 Sep;16(9):1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hastings PJ, et al. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10(8):551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Olivotto I, et al. Developmental origins of hypertrophic cardiomyopathy phenotypes: a unifying hypothesis. Nat Rev Cardiol. 2009;6(4):317–321. doi: 10.1038/nrcardio.2009.9. [DOI] [PubMed] [Google Scholar]
- [34].Shephard R, Semsarian C. Role of animal models in HCM research. J Cardiovasc Transl Res. 2009;2(4):471–482. doi: 10.1007/s12265-009-9120-y. [DOI] [PubMed] [Google Scholar]
- [35].Vignier N, et al. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ Res. 2009;105(3):239–248. doi: 10.1161/CIRCRESAHA.109.201251. [DOI] [PubMed] [Google Scholar]
- [36].Nishi H, et al. Possible gene dose effect of a mutant cardiac beta-myosin heavy chain gene on the clinical expression of familial hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 1994;200(1):549–556. doi: 10.1006/bbrc.1994.1483. [DOI] [PubMed] [Google Scholar]
- [37].Nanni L, et al. Hypertrophic cardiomyopathy: two homozygous cases with “typical” hypertrophic cardiomyopathy and three new mutations in cases with progression to dilated cardiomyopathy. Biochem Biophys Res Commun. 2003;309(2):391–398. doi: 10.1016/j.bbrc.2003.08.014. [DOI] [PubMed] [Google Scholar]
- [38].Frey N, Luedde M, Katus HA. Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol. 9(2):91–100. doi: 10.1038/nrcardio.2011.159. [DOI] [PubMed] [Google Scholar]
- [39].Kelly M, Semsarian C. Multiple mutations in genetic cardiovascular disease: a marker of disease severity? Circ Cardiovasc Genet. 2009;2(2):182–190. doi: 10.1161/CIRCGENETICS.108.836478. [DOI] [PubMed] [Google Scholar]
- [40].Girolami F, et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010;55(14):1444–1453. doi: 10.1016/j.jacc.2009.11.062. [DOI] [PubMed] [Google Scholar]
- [41].Ingles J, et al. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005;42(10):e59. doi: 10.1136/jmg.2005.033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Richard P, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107(17):2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- [43].Van Driest SL, et al. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44(9):1903–1910. doi: 10.1016/j.jacc.2004.07.045. [DOI] [PubMed] [Google Scholar]
- [44].Lekanne Deprez RH, et al. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J Med Genet. 2006;43(10):829–832. doi: 10.1136/jmg.2005.040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tester DJ, et al. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2(5):507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- [46].Roger VL, et al. Executive summary: heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2012;125(1):188–197. doi: 10.1161/CIR.0b013e3182456d46. [DOI] [PubMed] [Google Scholar]
- [47].Roger VL, et al. Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Maron BJ, et al. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur Heart J. 2003;24(21):1965–1991. doi: 10.1016/s0195-668x(03)00479-2. [DOI] [PubMed] [Google Scholar]
- [49].Ackerman MJ, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Europace. 13(8):1077–1109. doi: 10.1093/europace/eur245. [DOI] [PubMed] [Google Scholar]
- [50].Ackerman MJ, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Heart Rhythm. 8(8):1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- [51].Priori SG, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106(1):69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- [52].Thierfelder L, et al. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77(5):701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- [53].Kathiresan S, Srivastava D. Genetics of human cardiovascular disease. Cell. 148(6):1242–1257. doi: 10.1016/j.cell.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Charron P, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 31(22):2715–2726. doi: 10.1093/eurheartj/ehq271. [DOI] [PubMed] [Google Scholar]
- [55].Skrzynia C, Demo EM, Baxter SM. Genetic counseling and testing for hypertrophic cardiomyopathy: an adult perspective. J Cardiovasc Transl Res. 2009;2(4):493–499. doi: 10.1007/s12265-009-9127-4. [DOI] [PubMed] [Google Scholar]
- [56].Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54(3):201–211. doi: 10.1016/j.jacc.2009.02.075. [DOI] [PubMed] [Google Scholar]
- [57].Girolami F, et al. A molecular screening strategy based on beta-myosin heavy chain, cardiac myosin binding protein C and troponin T genes in Italian patients with hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown) 2006;7(8):601–607. doi: 10.2459/01.JCM.0000237908.26377.d6. [DOI] [PubMed] [Google Scholar]
- [58].Demo EM, Skrzynia C, Baxter S. Genetic counseling and testing for hypertrophic cardiomyopathy: the pediatric perspective. J Cardiovasc Transl Res. 2009;2(4):500–507. doi: 10.1007/s12265-009-9126-5. [DOI] [PubMed] [Google Scholar]
- [59].Morita H, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358(18):1899–1908. doi: 10.1056/NEJMoa075463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hershberger RE, et al. Genetic evaluation of cardiomyopathy–a Heart Failure Society of America practice guideline. J Card Fail. 2009;15(2):83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- [61].Semsarian C. Guidelines for the diagnosis and management of hypertrophic cardiomyopathy. Heart Lung Circ. 2007;16(1):16–18. doi: 10.1016/j.hlc.2006.10.020. [DOI] [PubMed] [Google Scholar]
- [62].Cowan J, et al. Genetic testing and genetic counseling in cardiovascular genetic medicine: overview and preliminary recommendations. Congest Heart Fail. 2008;14(2):97–105. doi: 10.1111/j.1751-7133.2008.08217.x. [DOI] [PubMed] [Google Scholar]
- [63].Hershberger RE. Cardiovascular genetic medicine: evolving concepts, rationale, and implementation. J Cardiovasc Transl Res. 2008;1(2):137–143. doi: 10.1007/s12265-008-9031-3. [DOI] [PubMed] [Google Scholar]
- [64].Olivotto I, Kassem HS, Girolami F. Genetic testing for hypertrophic cardiomyopathy: ongoing voyage from exploration to clinical exploitation. Cardiogenetics. 2011:e5-8. [Google Scholar]
- [65].Wordsworth S, et al. DNA testing for hypertrophic cardiomyopathy: a cost-effectiveness model. Eur Heart J. 2010;31(8):926–935. doi: 10.1093/eurheartj/ehq067. [DOI] [PubMed] [Google Scholar]
- [66].Vidal M, Cusick ME, Barabasi AL. Interactome networks and human disease. Cell. 2011;144(6):986–998. doi: 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hall JL, et al. Molecular signature of recovery following combination left ventricular assist device (LVAD) support and pharmacologic therapy. Eur Heart J. 2007;28(5):613–627. doi: 10.1093/eurheartj/ehl365. [DOI] [PubMed] [Google Scholar]
- [68].Birks EJ, et al. Gene profiling changes in cytoskeletal proteins during clinical recovery after left ventricular-assist device support. Circulation. 2005;112(9 Suppl):I57–I64. doi: 10.1161/CIRCULATIONAHA.104.526137. [DOI] [PubMed] [Google Scholar]
- [69].Pham MX, et al. Gene-expression profiling for rejection surveillance after cardiac transplantation. N Engl J Med. 2010;362(20):1890–1900. doi: 10.1056/NEJMoa0912965. [DOI] [PubMed] [Google Scholar]
- [70].Movassagh M, et al. Distinct epigenomic features in end-stage failing human hearts. Circulation. 2011;124(22):2411–2422. doi: 10.1161/CIRCULATIONAHA.111.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].D'Alessandra Y, et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur Heart J. 2010;31(22):2765–2773. doi: 10.1093/eurheartj/ehq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Willner D, et al. Metagenomic detection of phage-encoded platelet-binding factors in the human oral cavity. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4547–4553. doi: 10.1073/pnas.1000089107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Li S, et al. Signature microRNA expression profile of essential hypertension and its novel link to human cytomegalovirus infection. Circulation. 2011;124(2):175–184. doi: 10.1161/CIRCULATIONAHA.110.012237. [DOI] [PubMed] [Google Scholar]