

Introduction
Heart failure is a highly prevalent and increasing health problem for the developed and developing worlds. At 40 years of age, the lifetime risk of heart failure is 1 in 5 for both men and women, and despite advances in heart failure therapies, 50% individuals diagnosed with heart failure will die within 5 years [1,2]. Dilated cardiomyopathy (DCM) is the most common form of cardiomyopathy and is a major cause of heart failure [2]. DCM is a myocardial disorder defined by ventricular chamber enlargement and systolic dysfunction, that can occur as a primary cardiomyopathy or in association with other factors, such as coronary artery disease, infection, autoimmune disorders, alcohol excess, chemotherapeutic drugs or nutritional deficiencies. Like many common cardiovascular disorders, DCM is generally regarded as a complex trait with genetic and acquired (environmental) components [3]. Despite the enormous clinical importance of DCM, surprisingly little is known about its genetic basis. Studies of families in which DCM segregates as a Mendelian trait have been instrumental in deciphering fundamental molecular defects that cause impairment of cardiac contractile function. This group of patients with familial DCM is the subject of this review. Current perspectives on the insights gained from genetics studies of familial DCM, implications for clinical practice, and challenges for clinicians and for researchers will be discussed.
Prevalence of familial DCM
Patients with a new diagnosis of DCM can be generally classified into one of two groups. In approximately 50% cases, an acquired cause of DCM can be identified, while in the remaining 50% cases, DCM is termed idiopathic. With careful history-taking and clinical evaluation of first-degree relatives, it has been found that approximately 1 in 4 people with “idiopathic” DCM will have a family history of DCM [4]. Familial clustering of DCM has also been observed in community-based population studies. For example, prospective evaluation of participants in the Framingham Heart Study demonstrated that individuals who had at least one parent with heart failure were twice as likely to develop left ventricular systolic dysfunction when compared to those without a parental history [5]. While familial aggregation could be explained by shared environment, these observations collectively provide strong support for a role for inherited genetic factors. The discovery of gene mutations in families has now established the importance of gene defects in the pathogenesis of DCM.
Clinical presentation
In families with DCM, affected individuals may present with symptoms attributable to heart failure or arrhythmias, such as dyspnoea, fatigue, and palpitations, or may be asymptomatic. The diagnosis of DCM is based on conventional echocardiographic evidence of left ventricular dilatation and reduced systolic function. There may be associated ECG changes or structural heart defects, such as, conduction-system abnormalities, atrial or ventricular arrhythmias, valvular abnormalities, left ventricular non-compaction, or extra-cardiac manifestations (e.g. skeletal myopathy, partial lipodystrophy, sensorineural deafness). When a new diagnosis of DCM is made, affected individuals should be fully investigated to exclude coronary artery disease and causes of DCM other than familial cardiomyopathy.
0.1. Family screening
In individuals who have a family history of DCM and in those with idiopathic DCM, clinical screening of all first-degree family members with physical examination, 12-lead ECG and transthoracic echocardiography is recommended to identify familial disease and to determine the number of affected individuals within families [6]. Familial DCM is suspected when DCM is a predominant disease manifestation in two or more family members. A familial pattern of disease may not be recognized if there is variability in the phenotypic features between members of the same family or if gene defects are non-penetrant in some individuals, and a high level of clinical suspicion may be required. Families with DCM most commonly show an autosomal dominant mode of inheritance although autosomal recessive and X-linked inheritance can also be observed. Apart from family history, there are no specific clinical features that reliably differentiate familial DCM from acquired or non-familial causes of DCM.
0.2. Natural history
The natural history of familial DCM is variable. While the majority of individuals with heart failure are stable on medical therapy, some experience progressive heart failure and ultimately require cardiac transplantation. Individuals with conduction-system abnormalities may develop high-grade atrio-ventricular conduction block and require implantation of pacemakers. Insertion of implantable cardioverter-defibrillator devices (ICD) may be indicated in those family members who have depressed left ventricular ejection fraction (<35%) due to their increased propensity for malignant ventricular arrhythmias. No gene-based indications for ICDs have been developed and standard guidelines should be used [6]. It has been suggested, however, that some mutations, such as those in the LMNA gene, may confer an increased risk of sudden death, and patient genotype may need to be incorporated into clinical decision-making about ICD timing [7,8]. Criteria for risk stratification in familial DCM populations have not been defined, and the extent to which genotype per se is a determinant of outcome has yet to be determined. Clinical practice points for familial DCM are shown in Fig. 1.
Figure 1. Clinical practice points in familial DCM.

Genetics
Numerous studies to identify gene variants responsible for familial DCM have been performed over the past decade. Two main approaches have been used. In large families, genome-wide linkage analysis has been undertaken to define a chromosomal locus, followed by screening of candidate genes in the interval. In smaller families that lack sufficient power for linkage analysis, the most common strategy has been to directly screen candidate genes in cohorts of family probands. Candidate genes are selected on the basis of their known expression patterns and functions. To be considered a potential candidate, genes need to be expressed in the heart and contribute to a biological process that could forseeably affect cardiac contractile function. For mutation detection, gene-coding sequences are analyzed for variants using direct re-sequencing or techniques to identify sequence mismatches, such as high performance liquid chromatography. Any sequence variants identified are then evaluated further to determine whether they are novel, segregate with disease status in family members (i.e. present in all affected individuals and absent in unaffected individuals), are absent from a healthy control population, and have predicted or experimentally demonstrated functional effects. More than 40 chromosomal loci and disease genes have now been associated with adult-onset DCM (Table 1) [9,10]. While mutations in most of these disease genes have been identified in families, some mutations have been found only in sporadic cases.
Table 1. Disease genes associated with adult-onset DCM. 1.
| Prevalence | Genes |
| Most common | LMNA, TTN, DMD |
| Less common | ANKRD1, BAG3, Cypher/ZASP, MYBPC3, MYH6, MYH7, MYPN, RBM20, SCN5A, TNNT2 |
| Uncommon | ACTC, ACTN2, CHRM2, CRYAB, CSRP3, DES, DSP, EYA4, FHL2, GATAD1, ILK, LAMA4, LAMP2, MURC, NEBL, NEXN, PKP2, PLB, PSEN1, PSEN2, SGCD, SUR2, TCAP, TMPO, TNNC1, TNNI3, TPMI, VCL |
Comprehensive prevalence data have yet to be determined. Prevalence has been estimated based on numbers of reported mutations and cohort screening results for selected genes.
0.3. Prevalence of mutations in known disease genes
Few studies have systematically evaluated multiple genes in the same cohort of patients and accurate estimates for the relative prevalence of variants in each of the known disease genes are lacking. There are a number of factors that have led to some bias in available prevalence data. For example, LMNA mutations are the most frequently-reported genetic cause of familial DCM [9,10]. However, LMNA mutations are associated with a very distinctive phenotype of DCM and conduction-system disease [11] and thus the high numbers of gene mutations may be explained in part by the fact that this gene has been more frequently screened than most other disease genes. DCM and conduction-system disease can also result from mutations in the SCN5A gene that encodes the cardiac sodium channel [12,13]. Other phenotypes that give clues to underlying genotypes include mutations in the DMD gene that encodes the cytoskeletal protein dystrophin in which DCM has an X-linked inheritance pattern and can be accompanied by mild or subclinical skeletal myopathy [14,15], and mutations in the transcription factor, EYA4, that cause DCM and sensorineural deafness [16]. While genes with characteristic phenotypes may be screened more often, very large genes have been screened much less frequently, due to the costs and time involved using conventional sequencing methods. A good example of this is the TTN gene that encodes titin, the largest known human protein that has 363 exons. TTN mutations were first reported in families with DCM a decade ago [17,18], but have been very little investigated further until recently. In the largest cohort study performed to date, Herman and colleagues sequenced the TTN gene using next-generation sequencing (NGS) or Sanger sequencing in 312 subjects with DCM, 231 subjects with hypertrophic cardiomyopathy and 249 control subjects. Nonsense, frame-shift or copy number TTN mutations were in found in 67 subjects with DCM (21%) [19]. When only those who had undergone NGS were considered, there was a higher prevalence of these TTN variants, i.e. 54 of 203 DCM subjects (27%). While these findings need to be replicated in other patient cohorts, they do indicate that TTN mutations are far more common than previously suspected and are an important cause of DCM. Prior to this TTN report, it had been estimated that pathogenic variants in the known disease genes accounted for DCM in only a minority (30%) of familial cases [9,10]. These estimates clearly need to be re-evaluated and further studies are required to comprehensively evaluate the prevalence of mutations in all known disease genes.
0.4. Genetic testing
Due to the high costs and low yield, comprehensive genetic testing for variants in known DCM disease genes has not been incorporated to date into routine patient management. Selected genes of interest have been screened on a research basis by groups in various countries. More recently, commercial genetic testing has become available. This testing is performed using high-throughput re-sequencing or oligonucleotide hybridization arrays to evaluate different subsets of up to 20 of the most commonly-mutated disease genes. While the development of these techniques is a definite step forward, there are still important issues of cost (approximately US$3,000 to $5,500) and yield (usually 20–30%), which have limited their widespread use.
Genetic testing should ideally be performed in the setting of a cardiovascular genetics clinic where experienced molecular cardiologists, clinical geneticists and genetic counsellors can contribute to data interpretation and family management. Knowing which individuals in a family carry a pathogenic variant has important medical and psychosocial implications, particularly for those families with early-onset and aggressive forms of disease. In clinically-affected individuals, genotype results can guide selection and timing of drug therapies and interventions such as cardiac transplantation. This can also give an explanation as to why the disease arose. For asymptomatic family members, especially in younger generations, genotype results can pinpoint those at risk of developing disease, in whom close medical follow-up and lifestyle advice is indicated. Genotype information can be used as a basis for counselling family members contemplating pregnancies, and effects on both the mother and the fetus considered. Genetic results can have implications for participation in sports, employment, and life insurance that should be discussed with family members.
0.5. Genotyping challenges
A major challenge is finding the gene defects responsible for DCM in the substantial proportion of families who remain un-genotyped. This will require a comprehensive list of disease genes and better and more affordable methods for testing. One possibility is that there is a very long list of DCM disease genes, and that perturbation of diverse cardiomyocyte components may result in impaired contraction. Alternatively, some variants in the known disease genes may have been missed. By concentrating on gene coding regions, variants that are located in regulatory elements in promoters, introns or intergenic regions, may not be found. Most of the screening techniques currently utilized are also unable to detect copy number variations, such as deletions and duplications, which are increasingly being recognized to contribute to the pathogenesis of many cardiovascular disorders. The current method for selection of candidate genes for screening is based on known gene functions and preconceived notions of disease pathogenesis. A non-biased approach to variant discovery will be required to gain new perspectives on the types of molecular defects that can cause DCM.
Interpretation of the clinical significance of DNA sequence variants is not always straightforward and clinicians should be aware that a novel non-synonymous variant (i.e. one that results in an amino acid change) discovered in a single individual (usually the proband) is not necessarily the cause of DCM in the family. A number of criteria have been devised to provide genetic evidence of association with disease, such as segregation with affection status in families and absence from control subjects. Genotype-phenotype concordance can be difficult to assess in small families or in those in which DNA samples from affected and unaffected family members are unable to be evaluated for any reason (e.g. deceased individuals, unwillingness to be tested, etc.). To exclude the presence of rare variants in healthy control populations, power calculations indicate that very large numbers of individuals would be required. The most difficult aspect of determining pathogenicity of novel variants usually lies in assessment of potential functional effects. While experimental confirmation of altered gene or protein function is ideal, it can take months to years to evaluate a single variant and this becomes impractical on a wider scale. Several software programs, of varying efficacy, have been developed to predict the consequences of sequence variants based on factors such as the differences in the biophysical properties of the normal and substituted amino acids and sequence conservation across multiple species at the mutation site. These programs are unable to account for the location of variants with respect to functional domains specific to each gene, which will be an important determinant of the likelihood of adverse effects. Prediction programs are also limited by the extent of comparative sequence information, e.g. numbers of different protein isoforms characterized in different tissues, numbers of species in which homologous protein sequences are available, etc.. This problem of interpreting the likely pathogenicity of variants will escalate as new technologies dramatically increase the number of sequence variants found in family DNA samples.
0.6. Molecular mechanisms of familial DCM
Unlike hypertrophic cardiomyopathy, which has been termed a “disease of the sarcomere”, no single pathogenic mechanism has been found to be responsible for familial DCM [20,21]. The disease genes for familial DCM encode a wide range of proteins located in the sarcomere, cytoskeleton, sarcolemma, nucleus and sarcoplasmic reticulum (Fig. 2) [20], with diverse consequences for cardiomyocyte structure and function (Fig. 3). No gene-specific therapies have yet been identified for familial DCM. It remains to be determined whether defects induced by mutant proteins may converge to a smaller number of downstream pathways that might be more amenable to therapy.
Figure 2. Subcellular organization of the cardiomyocyte. Disease genes for familial DCM encode protein components of the sarcomere, cytoskeleton, sarcolemma, nucleus, and sarcoplasmic reticulum. Fatkin, Physiological Reviews 2002, Am Physiol Soc, used with permission.

Figure 3. Molecular defects associated with familial DCM. Schematic showing that pathogenic gene variants in families can promote DCM by perturbing diverse aspects of cardiac myocyte structure and function.
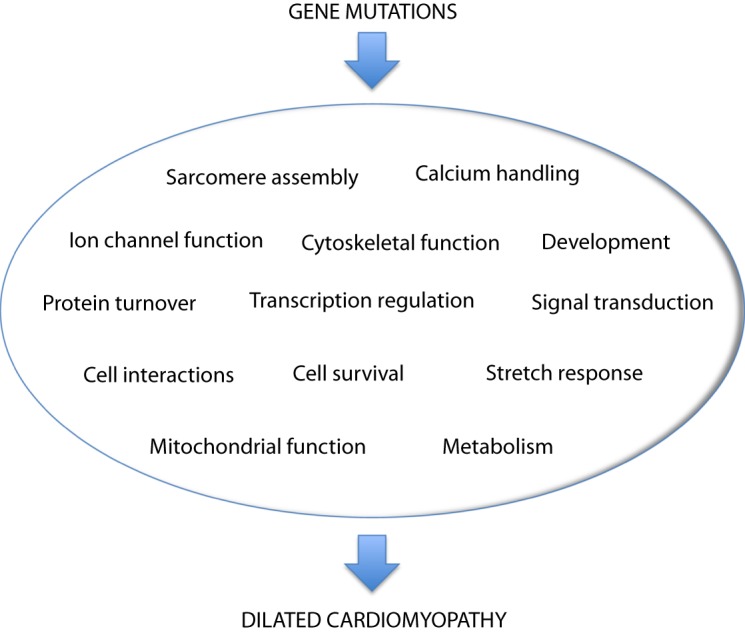
Clinical evaluation of families has shown that there may be marked variability in phenotypes of family members who carry the same genotype. These observations suggest that factors in addition to the major family mutation may contribute to the clinical manifestations of individual family members, including concurrent genetic variants of lesser effect size, co-morbid conditions and lifestyle factors, such as diet, alcohol intake and exercise.
Early disease
One of the main advantages of recognizing familial disease is that there is an opportunity for asymptomatic individuals at risk to undergo monitoring and receive early intervention that may ameliorate the course of disease. Ideally, a program of regular monitoring would be restricted to those asymptomatic family members who are known to carry the family gene mutation, and genotype-negative individuals could be dismissed from screening. However, the practical reality for most families is that genotype information is not available, and in this setting, doctors need to rely on clinical markers of early disease and all asymptomatic offspring of affected individuals should be included in screening programs.
0.7. Echocardiographic indices of early disease
Echocardiographic screening of large cohorts of asymptomatic relatives in families with DCM has shown that a substantial proportion of individuals (˜25%) have abnormalities such as left ventricular dilation or reduced contraction [22–24]. Isolated left ventricular dilation is the most common of these abnormalities. Unless the context of a positive family history is taken into account, this finding is all too frequently reported as “upper limit of normal”, particularly in young fit individuals, and ignored. Studies by our group and others have shown that approximately 1 in 10 asymptomatic family members with left ventricular dilation will progress to DCM within a 5-year period [25,26]. These observations clearly show that at least in some family members, left ventricular dilation is a marker of early cardiomyopathy, and raise important questions as to how the subgroup of family members at greatest risk of progression can be identified. Various echocardiographic parameters and biomarkers have been proposed to detect pre-clinical defects of myocardial contraction, including tissue Doppler imaging [27], exercise echo [28], brain natriuretic peptide levels [29] and cardiac autoantibodies [30]. We have recently evaluated several of these parameters in a cohort of asymptomatic relatives with left ventricular dilation and age- and gender-matched control subjects [26]. We found that for most parameters, there was a substantial scatter of data and overlap between relatives and healthy control subjects. The best discrimination between these groups was obtained with simple M-mode–based measurements of left ventricular dimensions (adjusted for age, height and weight) and fractional shortening. Promising data have been obtained using cine magnetic resonance imaging and this technique warrants further evaluation for detection of preclinical left ventricular dysfunction [31].
0.8. Challenges in diagnosis and management of early disease
Additional prospective studies are required to evaluate markers of early myocardial dysfunction so that a high-risk subgroup can be more accurately defined. Even when clinical criteria for risk stratification are established however, there is currently no data to guide physicians on who to treat, when to treat, and how to treat. Two recent studies in mouse models of DCM have suggested that prophylactic intervention with the -blocker, carvedilol [32], or MAPK inhibitors [33] may attenuate the development of contractile dysfunction. There is a pressing need for clinical-trial based data in large cohorts of asymptomatic family members to further evaluate these, and other therapies. In designing clinical trials for early disease, two important factors need to be addressed. First, in order to rationalize the numbers of study subjects and trial duration, and to maximize the likelihood of a positive outcome, such studies need to be performed in high-risk cohorts (as discussed above). Second, clinical trials should ideally be restricted to asymptomatic family members who are known to be genotype-positive, to avoid potentially confounding effects of inclusion of some individuals who have left ventricular dilation due to other causes. Unless substantial progress can be made in genotyping families, recruiting adequate numbers of subjects for early disease trials will not be possible. Family genotyping is currently a rate-limiting step towards progress in this field. Priorities for research in familial DCM are summarized in Fig. 4.
Figure 4. Current research priorities for familial DCM.

Future directions
The development of NGS technologies is a major breakthrough that is revolutionizing studies of the genetic basis of human disease. NGS enables large tracts of genomic DNA to be sequenced in parallel, and thus the human genome can be sequenced more quickly, more accurately and more cost-effectively than previously possible. NGS is revitalizing the genetics of DCM and will dramatically increase the yield of mutation identification in families. A number of NGS options and platforms are currently available. Whole-genome sequencing of genes and intergenic regions provides comprehensive coverage but generates enormous amounts of data that need to be stored and analyzed. An alternative strategy that is gaining widespread popularity is exome sequencing that focusses on the 1% of the human genome that is protein-coding. Since the vast majority of disease-causing variants identified to date have been located in protein-coding sequences [34], and the majority of rare coding sequence variants are predicted to have deleterious functional effects [35], exome sequencing is a powerful tool for mutation discovery [36]. Exome sequencing studies are revealing that even healthy people have in excess of 20,000 coding-sequence variants, 50% of which are missense, nonsense or splice site variants that could have functional effects [36,37]. The rate-limiting step in NGS is now finding ways to prioritise these variants for further analysis. Other genomic techniques that are complementary to exome sequencing include arrays of single nucleotide polymorphisms and non-polymorphic markers that enable large copy number variations such as deletions and duplications to be detected and mapped. Identification of pathogenic variants will rely on robust family data, development of better function prediction software programs, and high-throughput in vivo models, such as genetically engineered zebrafish.
In the next decade and beyond, we can expect that many more families will be able to be genotyped. Large families with autosomal dominant DCM and any families with a recessive inheritance pattern will continue to be a valuable resource and should be encouraged to participate in gene discovery research programs. While mutation screening strategies to date have focussed on single genes, NGS data will enable a more global analysis of genetic variation in each individual, and may challenge current paradigms of disease causation. It is likely that unique individual genetic profiles consisting of combinations of common and rare genetic variants of varying effect size may collectively contribute to cardiac phenotypes, even in families with a single major variant. Elucidation of the functional consequences of variants that segregate in families should provide fresh perspectives on molecular mechanisms of DCM and may open new avenues of research into novel drug targets and specific gene-targeted treatment. The ultimate goal of genetics studies is “bench to bedside” translation with more effective approaches to family management. Given the substantial economic and social costs of heart failure, greater emphasis needs to be placed on early diagnosis and preventative intervention.
Acknowledgments
Dr. Fatkin is supported by the National Health and Medical Research Council of Australia.
0.9. Disclosures
None.
References
- [1].Lloyd-Jones DM, Larson MG, Leip EP, et al. Lifetime risk for developing congestive heart failure. Circulation. 2002;106:3068–3072. doi: 10.1161/01.cir.0000039105.49749.6f. [DOI] [PubMed] [Google Scholar]
- [2].Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics 2011 update: A report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies. An American Heart Association scientific statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- [4].Petretta M, Pirozzi F, Sasso L, Paglia A, Bonaduce D. Review and metaanalysis of the frequency of familial dilated cardiomyoapthy. Am J Cardiol. 2011;108:1171–1176. doi: 10.1016/j.amjcard.2011.06.022. [DOI] [PubMed] [Google Scholar]
- [5].Lee DS, Pencina MJ, Benjamin EJ, et al. Association of parental heart failure with risk of heart failure in offspring. N Engl J Med. 2006;355:138–147. doi: 10.1056/NEJMoa052948. [DOI] [PubMed] [Google Scholar]
- [6].Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy – a Heart Failure Society of America Practice Guideline. J Cardiac Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- [7].Van Berlo JH, de Voogt WG, van der Kooi AJ, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med. 2005;83:79–83. doi: 10.1007/s00109-004-0589-1. [DOI] [PubMed] [Google Scholar]
- [8].Pasotti M, Klersy C, Pilotto A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52:1250–1260. doi: 10.1016/j.jacc.2008.06.044. [DOI] [PubMed] [Google Scholar]
- [9].Fatkin D, Otway R, Richmond Z. Genetics of dilated cardiomyopathy. Heart Fail Clin. 2010;6:129–140. doi: 10.1016/j.hfc.2009.11.003. [DOI] [PubMed] [Google Scholar]
- [10].Hershberger RE, Siegfried JD. Update 2011: Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2011;57:1641–1649. doi: 10.1016/j.jacc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- [12].McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E, Mestroni L. Familial Cardiomyopathy Registry Research Group. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110:2163–2167. doi: 10.1161/01.CIR.0000144458.58660.BB. [DOI] [PubMed] [Google Scholar]
- [13].Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Arbustini E, Diegoli M, Morbini P, et al. Prevalence and characteristics of dystrophin defects in adult male patients with dilated cardiomyopathy. J Am Coll Cardiol. 2000;35(7):1760–1768. doi: 10.1016/s0735-1097(00)00650-1. [DOI] [PubMed] [Google Scholar]
- [15].Feng J, Yan J, Buzin CH, et al. Comprehensive mutation screening of the dystrophin gene in patients with nonsyndromic X-linked dilated cardiomyopathy. J Am Coll Cardiol. 2002;40:1120–1124. doi: 10.1016/s0735-1097(02)02126-5. [DOI] [PubMed] [Google Scholar]
- [16].Schonberger J, Wang L, Shin JT, et al. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat Genet. 2005;37:418–422. doi: 10.1038/ng1527. [DOI] [PubMed] [Google Scholar]
- [17].Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30:201–204. doi: 10.1038/ng815. [DOI] [PubMed] [Google Scholar]
- [18].Itoh-Satoh M, Hayashi T, Nishi T, et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;291:385–393. doi: 10.1006/bbrc.2002.6448. [DOI] [PubMed] [Google Scholar]
- [19].Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev. 2002;82:945–980. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- [21].Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011;364:1643–1656. doi: 10.1056/NEJMra0902923. [DOI] [PubMed] [Google Scholar]
- [22].Baig MK, Goldman JH, Caforio AL, Coonar AS, Keeling PJ, McKenna WJ. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol. 1998;31:195–201. doi: 10.1016/s0735-1097(97)00433-6. [DOI] [PubMed] [Google Scholar]
- [23].Crispell KA, Wray A, Ni H, Nauman DJ, Hershberger RE. Clinical profiles of four large pedigrees with familial dilated cardiomyopathy. Preliminary recommendations for clinical practice. J Am Coll Cardiol. 1999;34:837–847. doi: 10.1016/s0735-1097(99)00276-4. [DOI] [PubMed] [Google Scholar]
- [24].Michels VV, Olson TM, Miller FA, Ballman KV, Rosales AG, Driscoll DJ. Frequency of development of idiopathic dilated cardiomyopathy among relatives of patients with idiopathic dilated cardiomyopathy. Am J Cardiol. 2003;91:1389–1392. doi: 10.1016/s0002-9149(03)00341-2. [DOI] [PubMed] [Google Scholar]
- [25].Mahon NG, Murphy RT, MacRae CA, Caforio AL, Elliott PM, McKenna WJ. Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann Intern Med. 2005;143:108–115. doi: 10.7326/0003-4819-143-2-200507190-00009. [DOI] [PubMed] [Google Scholar]
- [26].Fatkin D, Yeoh T, Hayward CS, et al. Evaluation of left ventricular enlargement as a marker of early disease in familial dilated cardiomyopathy. Circ Cardiovasc Genet. 2011;4:342–348. doi: 10.1161/CIRCGENETICS.110.958918. [DOI] [PubMed] [Google Scholar]
- [27].Matsumura Y, Elliott PM, Mahon NG, Virdee MS, Doi Y, McKenna WJ. Familial dilated cardiomyopathy: assessment of left ventricular systolic and diastolic function using Doppler tissue imaging in asymptomatic relatives with left ventricular enlargement. Heart. 2006;92:405–406. doi: 10.1136/hrt.2005.065474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sakata K, Ino H, Fujino N, et al. Exercise-induced systolic dysfunction in patients with non-obstructive hypertrophic cardiomyopathy and mutations in the cardiac troponin genes. Heart. 2008;94:1282–1287. doi: 10.1136/hrt.2007.116970. [DOI] [PubMed] [Google Scholar]
- [29].McDonagh TA, Robb SD, Murdoch DR, et al. Biochemical detection of left ventricular systolic dysfunction. Lancet. 1998;351:9–13. doi: 10.1016/s0140-6736(97)03034-1. [DOI] [PubMed] [Google Scholar]
- [30].Caforio AL, Mahon NG, Baig MK, et al. Prospective familial assessment in dilated cardiomyopathy. Cardiac autoantibodies predict disease development in asymptomatic relatives. Circulation. 2007;115:76–83. doi: 10.1161/CIRCULATIONAHA.106.641472. [DOI] [PubMed] [Google Scholar]
- [31].Koikkalainen JR, Antial M, Lotjonen JMP, et al. Early familial dilated cardiomyopathy: identification with determination of disease state parameter from cine MR image data. Radiology. 2008;249:88–96. doi: 10.1148/radiol.2491071584. [DOI] [PubMed] [Google Scholar]
- [32].Chandar S, Yeo LS, Leimena C, et al. Effects of mechanical stress and carvedilol in lamin A/C-deficient dilated cardiomyopathy. Circ Res. 2010;106:573–582. doi: 10.1161/CIRCRESAHA.109.204388. [DOI] [PubMed] [Google Scholar]
- [33].Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulate kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009;18:241–247. doi: 10.1093/hmg/ddn343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stenson PD, Ball EV, Howells K, et al. The Human Gene Mutation database: providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Hum Genomics. 2009;4:69–72. doi: 10.1186/1479-7364-4-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- [37].The 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]