

Abstract
Background
Alcohol and tobacco are often used together, and alcoholism is much more common among smokers compared to non smokers. Studies in humans suggest that nicotine (an active ingredient in cigarette smoke) can increase the consumption of alcohol. Research on rats and mice demonstrated mixed results; some studies report that nicotine increases alcohol consumption, while others show a decrease in drinking. Since cigarette smoke includes many other chemicals, these also may play a significant role on alcohol consumption. For example, two of these other constituents, monoamine oxidase (MAO) inhibitors and acetaldehyde, increase alcohol tolerance and/or alcohol consumption in rodents. The present study was designed to investigate how tobacco smoke may modify self-administration of alcoholin C57BL/6 mice.
Methods
C57BL/6 male mice (4-5 wk old) were acclimated for 3 weeks to consume a 10% (w/v) alcohol solution during a 2-hr daily access during the dark phase. Subsequently, half the animals were exposed to cigarette smoke for 6 hours per day for 16 days. The remaining animals (control) were placed in a smoke-free adjacent chamber. Immediately following the 6‐hr period in the chambers, the control and smoke exposed mice were given access to the 10% alcohol solution for 2 hours.
Results
Animals exposed to cigarette smoke for 6-hr per day consumed approximately 3-5 fold more alcohol than the mice in the control group throughout the 16-day study. The mice in the smoke group had a blood alcohol concentration (BAC) that was nearly 4-fold that of the control mice.
Conclusions
Tobacco smoke increases alcohol consumption several fold higher than reported studies using nicotine treatment alone in rodents. Thus, this model should help to determine the roles of other bioactive components in cigarette smoke that may be important in the high co-abuse of smoking and alcohol consumption.
Keywords: Alcohol Consumption, Tobacco Smoke Exposure, Drug Co-abuse, Adolescent, Nicotine
Introduction
Two of the most abused drugs in the United States, alcohol and tobacco, are often co-abused; about 7% of the US population are co-dependent (Istvan and Matarazzo, 1984; Anthony and Echeagaray-Wagner, 2000). Additionally, alcoholism is estimated to be ten times more common among smokers than among non smokers (Di Franza and Guerrera, 1990), and concurrent use of these drugs poses significant public health concerns and a large financial burden on society. A survey of persons treated for alcoholism and other drug addictions revealed that 26% had died over a 12‐year period; one-third of these deaths were attributed to alcohol-related causes, and one-half were related to smoking (Hurt et al., 1996). These studies help to show the magnitude of the health risks that are associated with the use of these drugs. Therefore, a better understanding of alcohol and tobacco smoke co-abuse is important on many levels.
Results from animal and human studies have shown multiple mechanisms for alcohol and nicotine co-use. For example, a study by Gulick and Gould (2007) found that in rodents, co-treatment may be influenced by each drug ameliorating the aversive effects of the other. Supporting this concept, neuro imaging studies on persons in alcohol withdrawal with continued tobacco smoking had decreased severity of the withdrawal-related changes in GABA neurotransmitter activity (Cosgrove et al., 2011). Also, co-abuse may depend on cross-tolerance between these two drugs. Our laboratory has found a functional cross-tolerance between chronic nicotine and alcohol in neurotransmitter activity in mouse hippo campal brain slices (Proctor et al., 2011). In humans, smokers reported that they felt less intoxicated compared to non-smokers after administration of an identical dose of alcohol (Madden et al., 1995), suggesting that smokers are more tolerant to alcohol's effects. Also, nicotine and alcohol together cause unique changes in neurotransmitter expression, subunit composition, metabolism, and receptor distribution which may affect sensitivity and preference for these two substances (Lajtha and Sershen, 2010).Other possibilities for drug interactions are genetic factors that are involved in the concurrent abuse of tobacco and alcohol (Madden and Heath, 2002), providing an inherent predisposition for co-abuse of these drugs. Thus, many factors are likely involved that underlie alcohol and tobacco co-addiction, including the reduction of side effects, tolerance to alcohol, modulation of synaptic neurotransmission, genetic predisposition and common reward pathways.
In attempts to understand the high co-abuse of these drugs in humans, numerous studies have utilized various animal models to determine the underlying mechanisms. In experiments on rodents, the correlation of alcohol and nicotine use has been some what inconsistent, which may be dependent in part by the various nicotine delivery modes. Some of these studies showed nicotine increased alcohol intake (Lê et al., 2000; Clark et al., 2001; Lê et al., 2003; Lê et al., 2010), and some showed a negative correlation (Sharpe and Samson, 2002; Hendrickson et al., 2009). This is likely due to the variety of experimental methods, including whether the drugs were administered acutely or chronically, the route of nicotine and alcohol administration, drug concentrations, and other factors.
The vast majority of prior studies have used nicotine to mimic tobacco smoke exposure, which may not fully trigger the molecular and physiological changes by the smoke. A study on the behavioral effects of tobacco smoke versus nicotine found that after four hours of whole body tobacco smoke exposure, there was a decrease in brain concentration of nicotine, and lower intracranial self-stimulation thresholds compared to subcutaneous nicotine administration in rats (Harris et al., 2010). Further more, equating nicotine applications to inhaled tobacco smoke involves different pathway kinetics (Rose et al., 1999). Additionally, tobacco smoke contains sensory stimuli that are not triggered by the use of pure nicotine delivery systems (Rose, 2006).
Tobacco smoke contains numerous bioactive compounds such as acetaldehyde, levulinic acid, mono amine oxidase (MAO) inhibitors and carbon monoxide that may add and/or modify the effects of nicotine alone. Acetaldehyde is formed in high concentrations when cigarettes are burned. Animal research demonstrates that rats prefer a combination of acetaldehyde and nicotine compared to either substance alone (Rabinoff et al., 2007). Also, rats trained to self-administer acetaldehyde have been shown to consume more alcohol than control rats, indicating that acetaldehyde formed by smoking cigarettes and alcohol consumption may have a synergistic interaction (Amit and Smith, 1985). In rats, levulinic acid 1) enhances the binding of nicotine to brain nicotinic receptors, 2) increases peak plasma nicotine levels, and 3) desensitizes the upper respiratory tract allowing smoke to be inhaled deeper into the lungs (Keithly et al., 2005; Rabinoff et al., 2007). MAO inhibitors may also play a role in drug co-abuse because MAO is an enzyme involved in the breakdown of neurotransmitters, such as dopamine. Dopamine and its reward pathways are thought to play a major role in drug addiction, including tobacco and alcohol. For example, inhibition of the MAO enzyme increases the availability of dopamine in the brain (van Amsterdam J. et al., 2006).It has been shown that there are several chemicals in tobacco smoke that reduced the activity of MAO, whereas nicotine alone did not (Fowler et al., 2003; Castagnoli and Murugesan, 2004; Lewis et al., 2007). Also, MAO inhibitors actually increase tolerance to alcohol in mice (Popova et al., 2000).Combustion of cigarettes from carbon monoxide may alter neuronal activity depending on the severity of the exposure (Raub and Benignus, 2002). Therefore, besides nicotine, the complex array of bioactive chemicals in cigarette smoke may significantly contribute to addiction and play a role in co-abuse with alcohol consumption.
In support of this concept, a study on tobacco cessation treatments found that denicotinized cigarettes decreased usage of nicotine‐ containing cigarettes significantly more than nicotine gum (Johnson et al., 2004).This suggests that humans prefer nicotine-free tobacco smoke over nicotine replacement when trying to quit smoking. Further more, most nicotine replacement treatments have a low success rate for smoking cessation (Hajek et al., 2009). In fact, smokers do not voluntarily self-administer pure nicotine even when they are deprived of cigarettes (Dar and Frenk, 2004). Perhaps the other components in cigarette smoke have additional properties that cannot be accounted for by nicotine alone in tobacco dependence, especially as it relates to increased alcohol consumption. Given the differences of nicotine effects on alcohol consumption in various animal preparations, the present study was designed to test for changes on the self-administration of alcohol consumption by tobacco smoke exposure as a more physiological model to better mimic the human smoking conditions.
Methods
Animals
Male C57BL/6 mice 4-5 weeks old were housed four animals per cage with bedding, free access to mouse chow and water except as noted below. The animals' 12‐hrlight/dark periods were shifted with lights on at 2:00 AM and lights off at 2:00 PM. The mice were weighed every seven days and observed daily for health status. Prior to the smoke exposure, the mice were randomly selected so that half the mice were assigned to the smoke group and the other half to the control (no smoke) group for the remainder of the study. The mice were housed and tested at the Denver Veterans Affairs Medical Center. All experiments were conducted with an approved animal protocol from the VA Institutional Animal Care and Use Committee, and were in accordance with all animal guidelines enforced by the VA Medical Center.
Tobacco smoking machine
The smoke delivery system, manufactured by Teague Enterprises (Lexington, KY), is a microprocessor-controlled smoking machine that produced a mixture of main stream and side stream smoke from filtered research cigarettes. Cigarettes were loaded into a gravity-fed magazine where they were pneumatically pushed into a vertical wheel and lighted. Each cigarette was “puffed” 9 times by suction and the main and side stream smoke was captured in a small, Plexiglas mixing chamber. Each puff had a volume of 37 cc, was two seconds in duration, and had a flow rate of 1.1 liters per minute. The smoke was then transported to one of the two smoke chambers that can house up to 8 mouse cages each. The second chamber without smoke was used as the control environment. The 3R4F research cigarette was used in this smoke delivery system, purchased from the University of Kentucky, College of Agriculture (Lexington, KY). These cigarettes have a tar content of 9.5 mg and 0.73 mg of nicotine per cigarette, comparable to commercially available cigarettes.
Smoke exposure
The mice were placed in the “smoke chambers” with no smoke exposure for 6 hours per day (10 AM to 4 PM) for 3 weeks to acclimate to this environment. At 10:00 am each day, the animals were transferred to secondary (test) cages with the same 4 cage-mates as in their home cage. These cages were placed into one of the two smoke chambers, one of which would later be per fused with cigarette smoke. After this 3‐week acclimation period, main stream and side stream tobacco smoke was introduced into one of the smoke chambers during the 6-hr chamber environment (10AM to 4PM) daily for the next 16 days. This resulted in whole body exposure for the smoke-exposed group at a rate of 1 to 3 cigarettes per 10-min cycle; the control chamber (holding the control group) remained smoke-free during the entire 16-day period (see Time-line in Fig. 1). This exposure time was selected based on a prior study that administered tobacco smoke to mice for 6 hours using the same smoke delivery system to induce cancer in rodents (Witschi et al., 2000). Total smoke particulate (TSP) in the smoke chamber was measured to ensure a constant smoke concentration and to relate to the animal nicotine blood levels.
Figure 1.
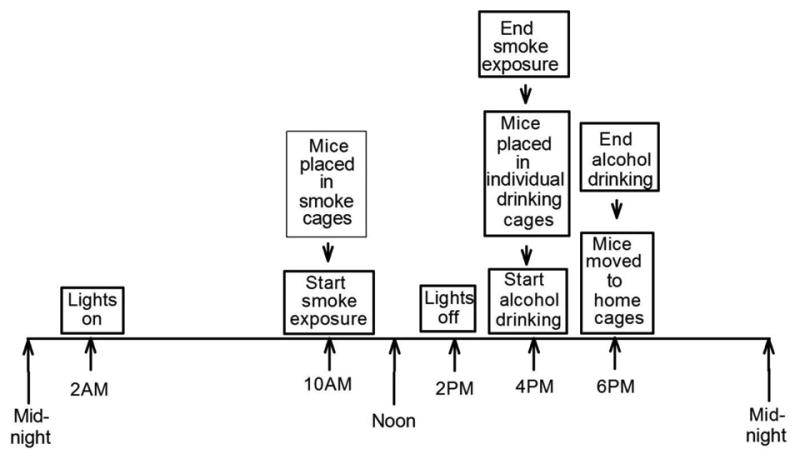
The 24-hour timeline showing that the dark cycle began at 2:00 PM and ended at 2:00 AM. From 10:00 AM to 4:00 PM, the smoke-exposed animals were exposed to smoke (the * indicates that the control animals were not subjected to smoke during this time). Immediately after the smoke exposure period, both groups had free access to alcohol starting 2 hours into the dark cycle beginning at 4:00 PM and ending at 6:00 PM.
Alcohol access
Immediately following the 6-hr exposure to smoke (or no-smoke), the mice were placedinto individual cages with food and bedding, but the water bottle was replaced with a 10% (w/v) alcohol solution in a 13 mL mouse volumetric drinking unit with a valved-sipper tube (Med-Associates, St. Albans, VT) for 2 hours. The 2-hr drinking window (4PM – 6PM; Fig. 1) corresponded to the2-4 hour period after the beginning of the dark cycle (drinking in the dark, DID) when these animals do most of their drinking (Rhodes et al., 2005). After this 2-hr alcohol access period, the mice were returned to their home cages with their same cage-mates. The sipper tube units were weighed on an analytical scale before and after the drinking session to measure the amount of the alcohol solution consumed. These male C57BL/6 mice have a genetic predisposition for alcohol preference (Mc Clearn and Rogers DA, 1959; Rhodes et al., 2005) that contributed to their selection for this study. The 2-hr alcohol access time was utilized throughout the study, starting when the mice began the 3-week acclimation period.
Blood alcohol concentration and nicotine blood levels
Blood alcohol concentration (BAC) and nicotine levels were determined on Day 12 and Day 16 via intra cardiac blood samples taken from groups of mice from both the smoke-exposed and control groups following gas CO2 euthanasia. Samples were centrifuged at 5000 × g for 15 min at 0° C in heparinized tubes. The supernatants were used to determine both BAC (enzyme method, Analox Instruments Ltd., London, UK) and blood nicotine levels (HPLC/mass spectrometer, iC42 Laboratory, Aurora, CO).
Data Analysis
Daily alcohol consumption, weekly body weight, BAC and blood nicotine level data from the smoke-exposed and control groups were recorded. Statistical analyses were carried out using Student's t-test or ANOVA with post hoc pair wise comparisons (Holm-Sidak).
Results
Acclimation to the alcohol drinking paradigm
During the final 6 days of the 3-week acclimation time to the light/dark shift and self-administration of alcohol (without prior smoke exposure), the animals drank a stable amount of alcohol during the 2-hr access window, with an average of 0.48 ± 0.05g/kg (mean ± SEM; n=39). Average weight of the animals increased from 22.6 to 23.1 g (n=39) during those 6 days.
Exposure to cigarette smoke reduced growth in body weight
During the following 16 days of cigarette smoke exposure in the smoke-exposed group, there was an average of a 3% decrease in body weight (22.5 to 21.9 g) from their initial baseline weights. During this same time period, the control mice had gained 9% in body weight (23.7 to 25.8 g). Thus, the animals receiving the smoke had a 12% overall reduced body weight relative to the control group by the end of the study (Fig. 2). The initial baseline weights of both groups were not significantly different: 22.3 g for the smoke group (n=20) and 22.9 g (n=19; p>0.3) for the control group.
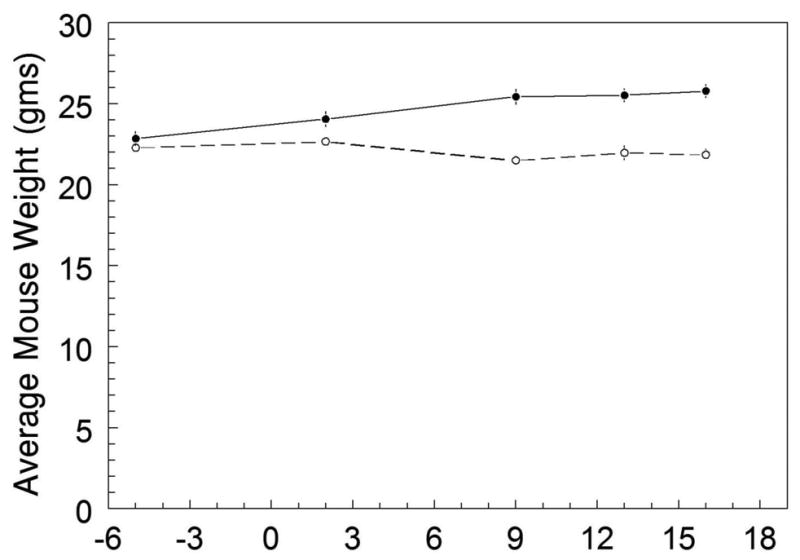
Figure 2.
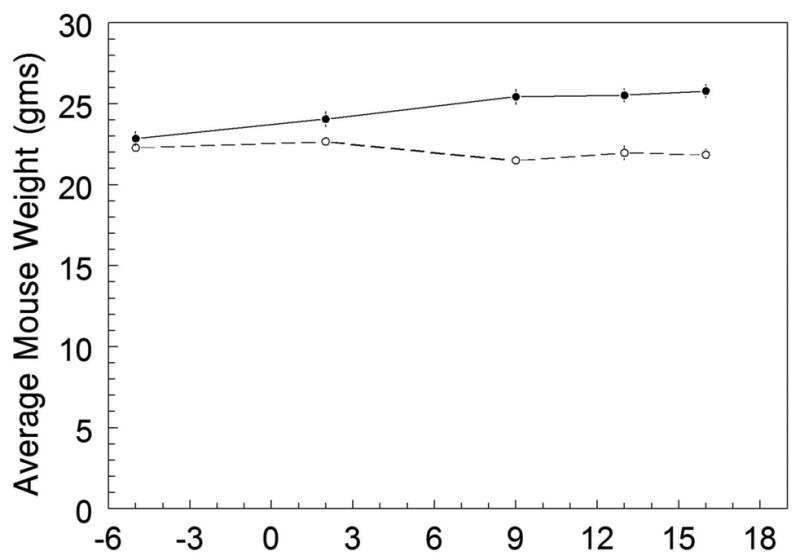
The average mouse body weights for the smoke-exposed (n=20) and control groups (n=19) plotted throughout the experiment. The arrow represents the beginning of smoke exposure on Day 1, which continued until Day 16. Days -6 to 0 represent the average body weight baseline prior to the smoke exposure. Weights were measured on Days -6, 2, 9, 13, and 16. The mouse weights for the smoke-exposed and control groups were not significantly different prior to the smoke exposure. After smoke exposure, the smoke group gradually lost 3% of their initial weight, whereas the control group gained 9%.
‘Binge’ smoke exposure
To simulate a possible teenager “binge” exposure to cigarette smoke, the smoke concentration was quickly ramped up to approximately 2.5-fold the test level during the first three days of smoke exposure. On Day 1, 1 cigarette per cycle was used (giving a 3.5 μg/m3 concentration of nicotine in the whole body smoke exposure); on Day 2, 2 cigarettes per cycle were given (4.7 μg/m3 nicotine concentration in the smoke), followed on Day 3 by 3 cigarettes per cycle (8.6 μg/m3 nicotine concentration in the smoke).
Over these first three days, there were no significant differences in alcohol consumption between control animals (0.6 ± 0.01 g/kg) and the smoke-exposed group (0.8 ± 0.19 g/kg; p>0.35).
Exposure to the test cigarette smoke greatly enhanced the self-administration of alcohol
On Day 4 through Day 12, the smoke-exposed mice received the test level of smoke exposure, 1 cigarette per 10 min cycle (3.5 μg/m3 nicotine concentration in the smoke) for 6 hours. During Day 4 through Day 7, the animals exposed to this smoke concentration greatly increased their alcohol consumption to a peak value about 5-fold (2.71 ± 0.22 g/kg) more than the control group (0.59 ± 0.04 g/kg; p< 0.001; Fig 3). Interestingly, on Days 8 and 9, there was a drop in alcohol intake to a level approximately 3-fold (1.45 ± 0.05 g/kg) above the control mice (0.48 ± 0.04 g/kg; p<0.001). For the remainder of the experiment, the alcohol intake remained about 3-fold higher than the alcohol consumption in the control animals (Fig. 3).
Figure 3.
Daily average alcohol intake during the 2-hour alcohol access period from 6 days prior to smoke exposure (Day -6 to Day 0) until 16 days after the tobacco smoke exposure. The arrow represents the first day of smoke exposure at 1 cigarette per 10-min cycle. Days 1-3 were designed as the adolescent “binge” of smoking (2 cigarettes per cycle on Day 2, and 3 cigarettes per cycle on Day 3), which did not cause a significant change in alcohol intake for the smoke-exposed group (open circles; 0.8 ± 0.19 g/kg, n=20) relative to the control group (closed circles; 0.6 ± 0.005, n=19; p>0.3). On Days 4 through 12, one cigarette per cycle was used, with an initial 5-foldpeak increase in drinking that decreased to a 3-fold level above the control group. From Day 13 to Day 16, 2 cigarettes per 10-min cycle were used (initiated on the day indicated by the triangle), yet the alcohol intake remained stable at about 3-fold above the control values.*p < 0.01.
Increasing the amount of test smoke
Following the stable drinking values during Days 9-12, we wanted to determine if increased smoke concentration could affect this 3-fold increase in alcohol consumption. Smoke concentration was increased to 2 cigarettes per 10‐min cycle on Days 13-16 in the smoke-exposed group. During this phase of increased smoke concentration, the alcohol consumption continued to be approximately 3-fold higher than the alcohol consumption of the control animals (Fig. 3), with no significant difference in the average intake between Days 9-12 (1.4 ± 0.05 g/kg) and Days 13-16 (1.3 ± 0.15 g/kg; p>0.05).
Individual alcohol consumption was variable from day to day
Daily alcohol consumption for each individual mouse in the smoke-exposed (n=20) and control groups (n=19) was quite variable. However, the average alcohol intake per mouse for the control group remained stable for the entire 16-day period of testing (Fig. 4a).The individual consumption in the smoke-exposed group (Fig.4b) demonstrates variability, but an overall stable trend of alcohol intake for 6 days prior to smoke administration and for the next 3 days of smoke exposure. From Days 4 to 7, the majority of animals had a marked increase in consumption. On Days 8-9, there was a decrease in intake which then stabilized for the remainder of the experiment. Thus, despite the daily variation in individual mouse consumption, the trend shows a stable intake in the control group (Fig. 4a), and a consistent increase in alcohol consumption in the smoke-exposed group during the cigarette smoke exposure (Fig. 4b).
Figure 4.
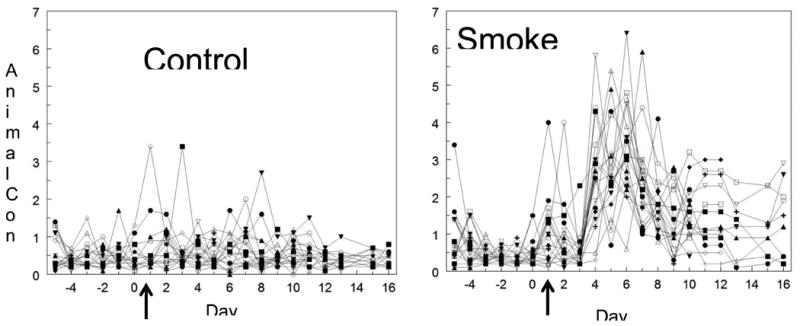
Daily alcohol intake during the 2 hour alcohol access period for each individual mouse. A. A plot of the daily alcohol consumption (g/kg) in each mouse for the control group. The control mice showed fairly stable drinking throughout the study. The arrow represents the first day when smoke was introduced to the smoke-exposed group. B. A plot of the daily alcohol consumption for the mice in the smoke group. The data show the alcohol drinking variability of the mice during the smoke phase. The arrow represents the first day of smoke exposure. Days 1-3 were the adolescent “binge” period of smoking, which did not cause a significant increase in alcohol intake during those initial days. However, on Days 4 through 7 there was an increase in drinking 4-5 times over the controls, followed by a large drop in intake on Days 8-9, that then stabilized to a level about 3-fold above the controls. From Day 13 to Day 16, 2 cigarettes per ten minutes were used without a significant change in alcohol consumption.
Blood levels of alcohol were significantly higher in smoke-exposed mice
As expected, the BAC was significantly higher in mice exposed to the cigarette smoke compared to the levels in the control animals. Following the 2‐hr period of access to the 10% alcohol solution, the average BAC in the smoke exposed animals (5.1 ± 2.10 mM, n=13) was approximately 4-fold higher than the BAC in the control mice (1.3 ± 0.25 mM, n=17; p<0.05). This data supports the consumption of alcohol measured from the amount of daily alcohol intake (Fig.5).
Figure 5.
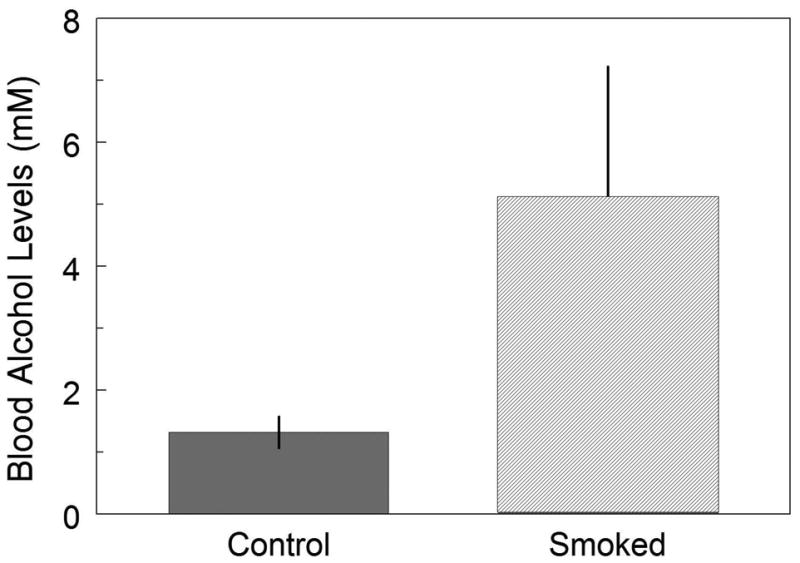
Average BAC after 2 hours of alcohol intake for the control and smoke-exposed groups pooled from Day 12 and Day 16. The bars illustrate the averaged (∼4‐fold) increase in BAC in the smoke-exposed group compared to the control group. *p < 0.05.
Nicotine blood levels
To better relate to other studies, blood nicotine samples were determined. The average nicotine level in the blood of the smoke-exposed group following the 6-hr smoke exposure was 2.92 ± 0.59 ng/mL at a nicotine concentration in the smoke chamber of 4.7 μg/m3. The mice in the control group had blood nicotine levels that were below the quantitative measurement (<0.7 ng/mL).
Discussion
The selection of the drinking parameter
A single-bottle ‘choice’ for the alcohol drinking measurements was available to the animals for two hours following the 6-hr smoke (smoke-exposed) or no-smoke (control) condition. Although a two-bottle choice (the alcohol solution plus a water bottle) could have been used to determine alcohol preference (Macdonall and Marcucella, 1979), these animals readily drink alcohol to pharmacologically significant blood alcohol levels (McClearn and Rogers DA, 1959; Rhodes et al., 2005) without prior training using the DID paradigm. Also, a single bottle choice may be more reflective of human behavior as observations of alcoholics reveal that they prefer alcohol when thirsty, which is likely due to the inhibitory effect of alcohol on the thirst response from osmotic stimulation (Eisenhofer and Johnson, 1983). This is further supported by a study that found humans prefer alcohol over water immediately after smoking a cigarette (Barrett et al., 2006). For animals that do not readily drink an alcohol solution, common training methods to induce alcohol consumption include the sucrose-fade method (Samson, 1986; Tolliver et al., 1988) and water restriction (Finn et al., 2005). These methods can entail longer training periods, making it more difficult to study animals young enough to mimic adolescent drinking behavior. Thus, 1) the single bottle choice, 2) the use of readily drinking young C57BL/6 mice and 3) the optimized DID paradigm were utilized in this study and resulted in a significant increase in self-administered alcohol following exposure to the cigarette smoke.
Weight loss in the animals exposed to the cigarette smoke
The mice from the control and the smoke-exposed groups had similar averaged body weights (smoke-exposed: 22.3 g; control: 22.9 g) that were not significantly different prior to the tobacco smoke exposure. After the daily tobacco exposure (6 hr per day for 16 days), the smoke-exposed group lost weight (∼3%) relative to their initial weight, but remained in a healthy state by observational stress and distress scores. However, compared to the growth in the no-smoke controls (∼9% increase), the smoke-exposed mice had ‘lost’ approximately 12% of their body weight during the 16‐day protocol. In spite of this significant weight reduction, the mice continued to consume high amounts of alcohol each day. Indeed, decreased weight gain from nicotine alone has been reported in rodents (Grunberg et al., 1984), and in humans from smoking cigarettes (Donny et al., 2011).
Binge cigarette smoke exposure did not affect alcohol intake in the first 3 days
At the initiation of the 16-day smoke protocol following a 6-day baseline period, these adolescent mice received a rapid increase in smoke concentration for the first three days, with 1, 2, and 3 cigarettes per ten-min cycle for Days 1, 2, and 3 respectively. This was done to represent a human ‘adolescent binge’. This period did not significantly affect alcohol intake in the smoke-exposed group relative to the control group. After this binge phase, smoke concentration was set at the test condition of 1 cigarette per cycle for the next 9 days.
Tobacco smoke greatly increases the self-administration of alcohol consumption
This animal model paradigm produced a large multi-fold increase in alcohol intake from Day 4 to Day 7, due to the effects of tobacco smoke exposure. After this peak response, there was a significant drop in alcohol intake for the next 2 days, and leveled off to approximately 3-fold above the controls, which lasted for the remainder of the experiment (Fig. 3). This drop in alcohol intake maybe due to a decrease in the reinforcing effects of alcohol consumption, as the amount of alcohol intake is correlated with reward (Spanagel and Holter, 2000). Alternatively, the drop may represent tolerance to the effects of tobacco smoke on the desire to consume alcohol. The precise mechanisms for this biphasic alcohol intake are not known. Despite this reduction in peak alcohol consumption, the data demonstrates that tobacco smoke produced a high multifold increase in alcohol consumption throughout the study.
Increased smoke concentration did not affect alcohol consumption
Increasing the smoke concentration on Days 13-16 did not cause a significant increase or decrease in alcohol intake relative to Days 9-12 (the steady, 1‐cigarette per cycle level). So, even during a doubling of the smoke exposure (2 cigarettes per cycle), the alcohol intake remained 3-fold above the control group. The reason for this may be due to nicotine receptor desensitization, such that a higher concentration of smoke did not result in a significant behavioral change. Health concerns for the animals (loss of body weight and accompanying metabolic changes), made it impossible to significantly increase the smoke exposure concentration for an extended period. The results from preliminary findings when mice were exposed to the smoke from 3 cigarettes per cycle for 6 hrs caused a high weight ‘loss’(15%) relative to the controls after only 10 days (Burns and Proctor, unpublished data).
Blood alcohol and nicotine levels
At the end of the study, blood alcohol levels were measured to further test for differences in BAC between the smoke-exposed and control groups. This data supports the alcohol consumption values, in that the exposure to tobacco smoke significantly increases alcohol consumption, and hence, blood alcohol levels in these mice. The mice exposed to the cigarette smoke had an average BAC that was approximately 4 fold elevated after the 2-hr alcohol access period compared to the average in the control group of mice.
The average blood nicotine level after the 6-hr smoke exposure was 2.9 ng/mL. This level was somewhat lower than those found in chronic smokers (4-72 ng/mL) (Russell et al., 1980). A limitation of these findings is that it may not precisely represent nicotine levels found in chronic smokers throughout a “typical” day. However, it would be difficult to equate the nicotine levels between this animal paradigm of smoke exposure and those levels found in various populations of smokers due to the highly variable smoking conditions. In animals, using higher smoke concentrations and shorter but more frequent exposure periods to better replicate human smoking conditions, would likely add to the complexity of this study and significantly add to the handling and cage changing stress.
Tobacco smoke versus nicotine exposure
The use of tobacco smoke in this animal model for alcohol self-administration extendsprevious studies by showing differences in behavioral effects when compared to nicotine alone. In the present study, exposure to tobacco smoke caused a 3-5 fold increase in self-administered alcohol consumption that is much higher than what has been demonstrated by other studies using nicotine alone, with a peak increase in drinking from 25 to 200% (Lê et al., 2000; Clark et al., 2001; Lê et al., 2003). Another difference in this study includes the inability of an increased concentration of tobacco smoke (from 1 to 2‐cigarette per cycle level) to cause a change in alcohol consumption compared to other studies showing a dose-dependent relationship between nicotine and drinking (Lê et al., 2000; Clark et al., 2001; Lê et al., 2003). Another difference between tobacco smoke and nicotine exposure was the delivery route as demonstrated by Harris et al. (2010) showing that inhalational delivery of nicotine (from smoke) produced different behavioral effects than by subcutaneous injection of nicotine. In addition, both nicotine and alcohol have dose-dependent effects on learning and memory (Gulick and Gould, 2007). A study on rodents showed an enhanced spatial memory with subcutaneous nicotine, whereas spatial memory was inhibited with tobacco smoke (Nowakowska et al., 2006). Therefore, several studies find that nicotine may not be a good substitute for the effects of tobacco smoke for various behavioral effects.
At least some of these differences between nicotine and tobacco smoke are likely to be from the effects of other bioactive compounds in the smoke and not entirely due to the inhalation of only nicotine delivered in the smoke. Tobacco smoke has compounds such as acetaldehyde and MAO inhibitors, which alone have been shown to increase alcohol intake and/or tolerance in rodents (Amit and Smith, 1985;Popova et al., 2000). A clinical study showed that tobacco smoke alters tolerance to alcohol, which may be due in part to these compounds (Madden et al., 1995). Acetaldehyde, levulinic acid, carbon monoxide and MAO inhibitors represent only a few of the many bioactive compounds and additives found in cigarette smoke (Rabinoff et al., 2007). Thus, tobacco smoke seems to be a more representative behavioral model than nicotine alone when studying smoking and drinking co-abuse.
Implications from this study
Our basic finding that whole-body tobacco smoke exposure greatly increases self-administered alcohol consumption and BAC in C57BL/6 mice indicates the importance of the interactions of these two drugs at several levels. The present animal model should be useful to provide additional information on the mechanisms that underlie these drug effects that modify the drive for increased alcohol consumption when the bioactive compounds in cigarette smoke are included.
There are several other implications of this study. First, we feel that tobacco smoke represents a better model of “nicotine dependence” when the other bioactive compounds are present that likely add to and/or alter the nicotine kinetics and sensory stimuli. Secondly, this animal model supports research in humans that show exposure of adolescents to smoking can trigger enhanced alcohol consumption to levels of alcohol abuse. In fact, adolescents are three times more likely to abuse alcohol later in life if they consume tobacco (Hughes, 1995). Lastly, it would seem evident that alcoholics in a treatment program may have higher success rates when smoking cessation treatments are included in the program. In a human trial, however, attempts at tobacco cessation did not result in a concurrent decrease in alcohol intake after one year (Hurt et al., 1994). In most studies, tobacco cessation programs have a low success rate of long-term abstinence (Hughes, 1995). Another possible contributing failure of tobacco cessation on treating alcohol abuse is the need to replace or modify the bioactive compounds in cigarettes. For example, loss of the MAO inhibitors in smoke may result in depression, and untreated depression is associated with alcohol abuse (Lynskey, 1998).
Therefore, a better understanding of co-abuse using tobacco smoke as a model should help further investigations in understanding the relationships and underlying cellular and biochemical processes that are involved in alcohol and tobacco smoke addiction.
Reference List
- Amit Z, Smith BR. A multi-dimensional examination of the positive reinforcing properties of acetaldehyde. Alcohol. 1985;2:367–370. doi: 10.1016/0741-8329(85)90077-1. [DOI] [PubMed] [Google Scholar]
- Anthony JC, Echeagaray-Wagner F. Epidemiologic analysis of alcohol and tobacco use. Alcohol Res Health. 2000;24:201–208. [PMC free article] [PubMed] [Google Scholar]
- Barrett SP, Tichauer M, Leyton M, Pihl RO. Nicotine increases alcohol self-administration in non-dependent male smokers. Drug Alcohol Depend. 2006;81:197–204. doi: 10.1016/j.drugalcdep.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Castagnoli K, Murugesan T. Tobacco leaf, smoke and smoking, MAO inhibitors, Parkinson's disease and neuroprotection; are there links? Neurotoxicology. 2004;25:279–291. doi: 10.1016/S0161-813X(03)00107-4. [DOI] [PubMed] [Google Scholar]
- Clark A, Lindgren S, Brooks SP, Watson WP, Little HJ. Chronic infusion of nicotine can increase operant self-administration of alcohol. Neuropharmacology. 2001;41:108–117. doi: 10.1016/s0028-3908(01)00037-5. [DOI] [PubMed] [Google Scholar]
- Cosgrove KP, Esterlis I, Mason GF, Bois F, O'Malley SS, Krystal JH. Neuroimaging insights into the role of cortical GABA systems and the influence of nicotine on the recovery from alcohol dependence. Neuropharmacology. 2011;60:1318–1325. doi: 10.1016/j.neuropharm.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar R, Frenk H. Do smokers self-administer pure nicotine? A review of the evidence. Psychopharmacology (Berl) 2004;173:18–26. doi: 10.1007/s00213-004-1781-2. [DOI] [PubMed] [Google Scholar]
- DiFranza JR, Guerrera MP. Alcoholism and smoking. J Stud Alcohol. 1990;51:130–135. doi: 10.15288/jsa.1990.51.130. [DOI] [PubMed] [Google Scholar]
- Donny EC, Caggiula AR, Weaver MT, Levin ME, Sved AF. The reinforcement-enhancing effects of nicotine: implications for the relationship between smoking, eating and weight. Physiol Behav. 2011;104:143–148. doi: 10.1016/j.physbeh.2011.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer G, Johnson RH. Effects of ethanol ingestion on thirst and fluid consumption in humans. Am J Physiol. 1983;244:R568–R572. doi: 10.1152/ajpregu.1983.244.4.R568. [DOI] [PubMed] [Google Scholar]
- Finn DA, Belknap JK, Cronise K, Yoneyama N, Murillo A, Crabbe JC. A procedure to produce high alcohol intake in mice. Psychopharmacology (Berl) 2005;178:471–480. doi: 10.1007/s00213-004-2039-8. [DOI] [PubMed] [Google Scholar]
- Fowler JS, Logan J, Wang GJ, Volkow ND. Monoamine oxidase and cigarette smoking. Neurotoxicology. 2003;24:75–82. doi: 10.1016/s0161-813x(02)00109-2. [DOI] [PubMed] [Google Scholar]
- Grunberg NE, Bowen DJ, Morse DE. Effects of nicotine on body weight and food consumption in rats. Psychopharmacology (Berl) 1984;83:93–98. doi: 10.1007/BF00427430. [DOI] [PubMed] [Google Scholar]
- Gulick D, Gould TJ. Acute ethanol has biphasic effects on short- and long-term memory in both foreground and background contextual fear conditioning in C57BL/6 mice. Alcohol Clin Exp Res. 2007;31:1528–1537. doi: 10.1111/j.1530-0277.2007.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajek P, Stead LF, West R, Jarvis M, Lancaster T. Relapse prevention interventions for smoking cessation. Cochrane Database of Systematic Reviews. 2009 doi: 10.1002/14651858.CD003999.pub3. Issue 1Art. No.: CD003999. doi:110.1002/14651858.CD003999.pub3. [DOI] [PubMed] [Google Scholar]
- Harris AC, Mattson C, LeSage MG, Keyler DE, Pentel PR. Comparison of the behavioral effects of cigarette smoke and pure nicotine in rats. Pharmacol Biochem Behav. 2010;96:217–227. doi: 10.1016/j.pbb.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson LM, Zhao-Shea R, Tapper AR. Modulation of ethanol drinking-in-the-dark by mecamylamine and nicotinic acetylcholine receptor agonists in C57BL/6J mice. Psychopharmacology (Berl) 2009;204:563–572. doi: 10.1007/s00213-009-1488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JR. Clinical implications of the association between smoking and alcoholism. In: Fertig JB, Allen JP, editors. Alcohol and Tobacco: From Basic Science to Clinical Practice. Washington, DC: Supt. of Docs., U.S. Govt. Print. Off.: National Institute on Alcohol Abuse and Alcholism; 1995. pp. 171–185. NIAAA Research Monograph No. 30. NIH Pub. No. 95-3931. [Google Scholar]
- Hurt RD, Eberman KM, Croghan IT, Offord KP, Davis LJ, Jr, Morse RM, Palmen MA, Bruce BK. Nicotine dependence treatment during inpatient treatment for other addictions: a prospective intervention trial. Alcohol Clin Exp Res. 1994;18:867–872. doi: 10.1111/j.1530-0277.1994.tb00052.x. [DOI] [PubMed] [Google Scholar]
- Hurt RD, Offord KP, Croghan IT, Gomez-Dahl L, Kottke TE, Morse RM, Melton LJ., III Mortality following inpatient addictions treatment. Role of tobacco use in a community-based cohort. JAMA. 1996;275:1097–1103. doi: 10.1001/jama.275.14.1097. [DOI] [PubMed] [Google Scholar]
- Istvan J, Matarazzo JD. Tobacco, alcohol, and caffeine use: a review of their inter relationships. Psychol Bull. 1984;95:301–326. [PubMed] [Google Scholar]
- Johnson MW, Bickel WK, Kirshenbaum AP. Substitutes for tobacco smoking: a behavioral economic analysis of nicotine gum, denicotinized cigarettes, and nicotine-containing cigarettes. Drug Alcohol Depend. 2004;74:253–264. doi: 10.1016/j.drugalcdep.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Keithly L, Ferris WG, Cullen DM, Connolly GN. Industry research on the use and effects of levulinic acid: a case study in cigarette additives. Nicotine Tob Res. 2005;7:761–771. doi: 10.1080/14622200500259820. [DOI] [PubMed] [Google Scholar]
- Lajtha A, Sershen H. Nicotine: alcohol reward interactions. Neurochem Res. 2010;35:1248–1258. doi: 10.1007/s11064-010-0181-8. [DOI] [PubMed] [Google Scholar]
- Lê AD, Corrigall WA, Harding JW, Juzytsch W, Li TK. Involvement of nicotinic receptors in alcohol self-administration. Alcohol Clin Exp Res. 2000;24:155–163. doi: 10.1111/j.1530-0277.2000.tb04585.x. [DOI] [PubMed] [Google Scholar]
- Lê AD, Lo S, Harding S, Juzytsch W, Marinelli PW, Funk D. Coadministration of intravenous nicotine and oral alcohol in rats. Psychopharmacology (Berl) 2010;208:475–486. doi: 10.1007/s00213-009-1746-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê AD, Wang A, Harding S, Juzytsch W, Shaham Y. Nicotine increases alcohol self-administration and reinstates alcohol seeking in rats. Psychopharmacology (Berl) 2003;168:216–221. doi: 10.1007/s00213-002-1330-9. [DOI] [PubMed] [Google Scholar]
- Lewis A, Miller JH, Lea RA. Monoamine oxidase and tobacco dependence. Neurotoxicology. 2007;28:182–195. doi: 10.1016/j.neuro.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Lynskey MT. The comorbidity of alcohol dependence and affective disorders: treatment implications. Drug Alcohol Depend. 1998;52:201–209. doi: 10.1016/s0376-8716(98)00095-7. [DOI] [PubMed] [Google Scholar]
- Macdonall JS, Marcucella H. Increasing the rate of ethanol consumption in food- and water-satiated rats. Pharmacol Biochem Behav. 1979;10:211–216. doi: 10.1016/0091-3057(79)90089-3. [DOI] [PubMed] [Google Scholar]
- Madden PA, Heath AC. Shared genetic vulnerability in alcohol and cigarette use and dependence. Alcohol Clin Exp Res. 2002;26:1919–1921. doi: 10.1097/01.ALC.0000040960.15151.30. [DOI] [PubMed] [Google Scholar]
- Madden PA, Heath AC, Starmer GA, Whitfield JB, Martin NG. Alcohol sensitivity and smoking history in men and women. Alcohol Clin Exp Res. 1995;19:1111–1120. doi: 10.1111/j.1530-0277.1995.tb01588.x. [DOI] [PubMed] [Google Scholar]
- McClearn G, Rogers DA. Differences in alcohol preference among inbred strains of mice. Q J Stud Alcohol. 1959;20:691–695. [Google Scholar]
- Nowakowska E, Kus K, Florek E, Czubak A, Jodynis-Liebert J. The influence of tobacco smoke and nicotine on antidepressant and memory-improving effects of venlafaxine. Hum Exp Toxicol. 2006;25:199–209. doi: 10.1191/0960327106ht611oa. [DOI] [PubMed] [Google Scholar]
- Popova NK, Vishnivetskaya GB, Ivanova EA, Skrinskaya JA, Seif I. Altered behavior and alcohol tolerance in transgenic mice lacking MAO A: a comparison with effects of MAO A inhibitor clorgyline. Pharmacol Biochem Behav. 2000;67:719–727. doi: 10.1016/s0091-3057(00)00417-2. [DOI] [PubMed] [Google Scholar]
- Proctor WR, Dobelis P, Moritz AT, Wu PH. Chronic nicotine treatment differentially modifies acute nicotine and alcohol actions on GABA(A) and glutamate receptors in hippocampal brain slices. Br J Pharmacol. 2011;162:1351–1363. doi: 10.1111/j.1476-5381.2010.01141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinoff M, Caskey N, Rissling A, Park C. Pharmacological and chemical effects of cigarette additives. Am J Public Health. 2007;97:1981–1991. doi: 10.2105/AJPH.2005.078014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raub JA, Benignus VA. Carbon monoxide and the nervous system. Neurosci Biobehav Rev. 2002;26:925–940. doi: 10.1016/s0149-7634(03)00002-2. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Rose JE. Nicotine and nonnicotine factors in cigarette addiction. Psychopharmacology (Berl) 2006;184:274–285. doi: 10.1007/s00213-005-0250-x. [DOI] [PubMed] [Google Scholar]
- Rose JE, Behm FM, Westman EC, Coleman RE. Arterial nicotine kinetics during cigarette smoking and intravenous nicotine administration: implications for addiction. Drug Alcohol Depend. 1999;56:99–107. doi: 10.1016/s0376-8716(99)00025-3. [DOI] [PubMed] [Google Scholar]
- Russell MA, Jarvis M, Iyer R, Feyerabend C. Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. Br Med J. 1980;280:972–976. doi: 10.1136/bmj.280.6219.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson HH. Initiation of ethanol reinforcement using a sucrose-substitution procedure in food- and water-sated rats. Alcohol Clin Exp Res. 1986;10:436–442. doi: 10.1111/j.1530-0277.1986.tb05120.x. [DOI] [PubMed] [Google Scholar]
- Sharpe AL, Samson HH. Repeated nicotine injections decrease operant ethanol self-administration. Alcohol. 2002;28:1–7. doi: 10.1016/s0741-8329(02)00238-0. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Holter SM. Pharmacological validation of a new animal model of alcoholism. J Neural Transm. 2000;107:669–680. doi: 10.1007/s007020070068. [DOI] [PubMed] [Google Scholar]
- Tolliver GA, Sadeghi KG, Samson HH. Ethanol preference following the sucrose-fading initiation procedure. Alcohol. 1988;5:9–13. doi: 10.1016/0741-8329(88)90036-5. [DOI] [PubMed] [Google Scholar]
- van Amsterdam J, Talhout R, Vleeming W, Opperhuizen A. Contribution of monoamine oxidase (MAO) inhibition to tobacco and alcohol addiction. Life Sci. 2006;79:1969–1973. doi: 10.1016/j.lfs.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Witschi H, Uyeminami D, Moran D, Espiritu I. Chemoprevention of tobacco-smoke lung carcinogenesis in mice after cessation of smoke exposure. Carcinogenesis. 2000;21:977–982. doi: 10.1093/carcin/21.5.977. [DOI] [PubMed] [Google Scholar]