

Abstract
Myocardial matrix turnover involves a dynamic balance between collagen synthesis and degradation, which is regulated by matrix metalloproteinases (MMPs). N-acetyl-Ser-Asp-Lys-Pro (Ac-SDKP) is a small peptide that inhibits cardiac inflammation and fibrosis. However, its role in MMP regulation is not known. Thus, we hypothesized that Ac-SDKP promotes MMP activation in cardiac fibroblasts and decreases collagen deposition via this mechanism. To that end, we tested the effects of Ac-SDKP on interleukin-1β (IL-1β; 5 ng/ml)-stimulated adult rat cardiac fibroblasts. We measured total collagenase activity, MMP-2, MMP-9, and MMP-13 expressions, and activity along with their inhibitors, tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2. In order to examine the effects of Ac-SDKP on the signaling pathway that controls MMP transcription, we also measured nuclear factor-κB (NFκB) and p42/44 mitogen-activated protein kinase (MAPK) activation. Ac-SDKP did not alter collagenase or gelatinase activity in cardiac fibroblasts under basal conditions, but blunted the IL-1β-induced increase in total collagenase activity. Similarly, Ac-SDKP normalized the IL-1β-mediated increase in MMP-2 and MMP-9 activities and MMP-13 expression. Inhibition of MMPs by Ac-SDKP was associated with increased TIMP-1 and TIMP-2 expressions. Collagen production was not affected by Ac-SDKP, IL-1β, or a combination of both agents. Ac-SDKP blocked IL-1β-induced p42/44 phosphorylation and NFκB activation in cardiac fibroblasts. We concluded that the Ac-SDKP-inhibited collagenase expression and activation was associated with increased expression of TIMP-1 and TIMP-2. These pharmacological effects of Ac-SDKP may be linked to the inhibition of MAPK and NFκB pathway.
Keywords: Myocardial matrix turnover, Synthesis and degradation, Matrix metalloproteinases, N-acetyl-Ser-Asp-Lys-Pro, Cardiac fibroblasts, NFκB, MAPK
Introduction
Collagen in the myocardial extracellular matrix (ECM) is a major determinant of myocardial structural integrity [42]. Regulation of myocardial ECM turnover involves a dynamic balance between ECM synthesis and degradation. Cardiac fibroblasts are an important site of synthesis of ECM as well as matrix metalloproteinases (MMPs), particularly MMP-2, MMP-9, and MMP-13 [6, 11, 12, 37, 38]. The activity of MMPs is tightly regulated by their activators, such as various pro-inflammatory cytokines [14], and by specific inhibitors, including tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2 [7]. In several cell types, inflammatory cytokines like interleukin-1β (IL-1β) can activate MMPs or regulate the expression of TIMPs, thus inhibiting MMP activity [14].
N-acetyl-Ser-Asp-Lys-Pro (Ac-SDKP) is a naturally occurring peptide with anti-inflammatory and anti-fibrotic properties [34]. We previously reported that Ac-SDKP inhibits myocardial collagen deposition following hypertension and myocardial infarction [28, 45]. We hypothesized that the Ac-SDKP-mediated decrease in organ fibrosis [4, 25, 26, 28, 29, 32, 45] could be a result of reduced collagen synthesis coupled with increased collagen breakdown. Previous in vitro observations made by us and others showed that Ac-SDKP inhibited collagen synthesis in rat cardiac fibroblasts (RCFs) [31, 35], but whether Ac-SDKP inhibits or augments IL-1β-induced collagen degradation by MMPs is not known.
The primary goal of this study is to determine the effect of Ac-SDKP on IL-1β-stimulated MMP activity in cardiac fibroblasts. Contrary to our hypothesis, Ac-SDKP did not increase MMP activity in cardiac fibroblasts, but rather inhibited IL-1β-mediated increases in collagenase and gelatinase. It also blocked MMP-13 expression and attenuated reduction of TIMP-1 and TIMP-2 levels induced by IL-1β. It is well-documented that IL-1β stimulates the production and activation of MMPs via cellular signaling pathways linked to p42/44 mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NFκB) [5, 18, 24, 43]. Here, we found that IL-1β-induced p42/44 and NFκB signaling was also inhibited by Ac-SDKP.
Methods
Isolation of adult rat cardiac fibroblasts
Cardiac fibroblasts were isolated from the hearts of adult male Sprague Dawley rats (200 to 220 g) following the protocol described previously [35]. After the third passage, cells were plated at a non-confluent density on plastic culture dishes (Costar) and grown for 18 h in Dulbecco’s modified Eagle’s medium containing 10 % fetal calf serum and 1 % penicillin/streptomycin. Before drug treatment, cells were synchronized by serum starvation for 48 h.
Drug treatments
Fibroblasts were treated with vehicle (control), Ac-SDKP (0.1–100 nM), IL-1β (5 ng/ml; RDI), or Ac-SDKP plus IL-1β for 30 min to assess p-p42/44 and for 72 h to assess the activity and expression level of MMPs and TIMPs. Cells were preincubated with Ac-SDKP for 20 min before adding IL-1β. An angiotensin-converting enzyme (ACE) inhibitor captopril (10−6 M) was also added to all groups to prevent breakdown of Ac-SDKP via ACE expressed by cardiac fibroblasts. We have chosen the dose of 1 nM Ac-SDKP based on our previous studies of cultured cardiac fibroblasts [31, 35].
Detection of MMPs
Total collagenase activity in the cultured supernatant was determined with a commercial kit (Chondrex) according to manufacturer’s instructions. This kit uses collagen I as a substrate, enabling us to measure collagenases including MMP-13 in the cultured supernatant of cardiac fibroblasts. MMP-2 and MMP-9 activities were determined by in-gel zymography as described previously [15]. Conditioned medium was collected and protein content was determined by the Bradford assay (Bio-Rad protein dye reagent) against a bovine serum albumin standard. Measurement of MMP activity per 10 μg protein was obtained by in-gel zymography with 10 % gelatin (Bio-Rad) as the substrate. Unstained digested regions representing MMP activity were quantified using an imaging densitometer (Scion image analyzer, NIH). MMP identity was determined using estimated molecular weights against pre-stained molecular weight markers and loading a positive control of active MMP-2 and MMP-9. MMP-2, MMP-9, MMP-13, TIMP-1, and TIMP-2 protein levels were assessed by Western blotting of the cultured supernatant using anti-MMP-2 (1:1,000; Anaspec), anti-MMP-9 (1:800; Anaspec), anti-MMP-13 (1:3,000; Biogene), anti-TIMP-1 (1:2,000; RDI), and anti-TIMP-2 (1:3,000; RDI) antibodies, respectively. The membranes were re-probed with an anti-GAPDH antibody (1:5,000; Abcam), and the density of the resulting bands was used to normalize the density of MMP and TIMP proteins.
Collagen synthesis
Quiescent cells were pretreated with Ac-SDKP (1 nM) or vehicle for 30 min before exposure to IL-1β (5 ng/ml) for an additional 72 h at 37 °C; cell media was harvested for collagen measurement using hydroxyproline assay [35, 39], and cells were harvested by trypsinizing the wells to determine the number of cells using trypan blue exclusion assay [36]. Collagen content was expressed as micrograms per 105 cells.
Phosphorylation of extracellular signal-regulated kinase (ERK)
Cells were collected in lysis buffer (1 % Triton X-100, 0.5 % Nonidet P-40, 10 mmol/L Tris, 1 mmol/L EDTA, 1 mmol/L EGTA, 150 mmol/L NaCl, 0.4 mmol/L PMSF, 0.2 mmol/L sodium orthovanadate, and 1 g/L leupeptin). Protein concentration was determined using the Bradford assay (Bio-Rad). Proteins from each group were separated by SDS-PAGE on 4–20 % gel, transferred to a PVDF transfer membrane (Amersham), and probed with anti-phospho-ERK1/2 (ERK1/2; 1:1,000; Cell Signaling) and anti-total ERK1/2 (1:1,000; Cell Signaling) antibodies.
Electrophoretic mobility shift assay (EMSA) for NFκB
NFκB expression was assessed in RCFs seeded in a 100-mm culture dish (5×06 cells per dish), serum-deprived for 48 h, and preincubated with or without Ac-SDKP (1 nM) for 30 min. They were then challenged with IL-1β (5 ng/ml) for 2.5 h at 37 °C. Cells were harvested following the protocol in a kit from Active Motif (Carlsbad, CA, USA) to extract nuclear proteins. EMSA was conducted to measure nuclear translocation of NFκB using nuclear extracts from fibroblasts and EMSA “Gel Shift” Protocol for NFκB using a nonradioactive kit from Affymetrix (Santa Clara, CA, USA) according to the manufacturer’s instructions. Optical density of the bands was compared as described previously [13].
Statistical analysis
Data are presented as the mean ± SEM. Differences among treatment groups were determined by ANOVA with Tukey’s post hoc comparisons or Hochberg’s method to correct for multiple testing. p<0.05 was considered significant.
Results
Effects of Ac-SDKP on gelatinase activity in cardiac fibroblasts
In-gel zymography of the supernatant from cultured fibroblasts showed a specific band corresponding to the molecular weight of MMP-2 and MMP-9 (66 and 92 kDa, respectively; Fig. 1a). At the basal level, there was no change in MMP-2 activity following the addition of Ac-SDKP. IL-1β increased MMP-2 activity 2.81±0.25-fold (p=0.001), and this effect was significantly blocked by concurrent treatment with Ac-SDKP (Fig. 1a, b). On the other hand, the band corresponding to MMP-9 activity was absent under basal conditions or in cells treated with Ac-SDKP alone; however, MMP-9 activity was very apparent when cells were stimulated with IL-1β, and it was dose-dependently inhibited by concurrent Ac-SDKP treatment (Fig. 1a, c).
Fig. 1.

Effects of IL-1β and Ac-SDKP on gelatinase activity in cardiac fibroblasts. a Representative zymogram gel showing the effects of IL-1β and Ac-SDKP on MMP-2 and MMP-9. Conditioned medium was electrophoresed in 10 % gelatin gel and stained with Coomassie blue. Unstained digested regions (black lytic bands) corresponding to the molecular weight of MMP-2 (66 kDa) and MMP-9 (92 kDa) were visible after multiple washes. Quantification of b MMP-2 and c MMP-9 in cells treated with vehicle, Ac-SDKP, IL-1β, or Ac-SDKP+IL-1β. Bands corresponding to the molecular weight of MMP-2 and MMP-9 were quantified. Bar graph represents pooled data from five different experiments, each done in triplicate
Effects of Ac-SDKP on MMP-2 and MMP-9 protein levels in cardiac fibroblasts
Ac-SDKP did not alter MMP-2 (Fig. 2a) and MMP-9 (Fig. 2b) protein levels in the basal condition. IL-1β increased the levels of MMP-2 (controls, 1.3±0.5 densitometric units; IL-1β, 3.4± 0.2 densitometric units; p<0.05), which was inhibited by Ac-SDKP (IL-1β+Ac-SDKP, 1.6±0.14; p<0.05, IL-1β vs. IL-1β+Ac-SDKP). Similarly, IL-1β increased the levels of MMP-9 (controls, 0.5±0.06 densitometric units; IL-1β, 1.2± 0.1 densitometric units; p<0.05), which was inhibited by Ac-SDKP (IL-1β+Ac-SDKP, 0.83±0.06 densitometric units; p< 0.05, IL-1β vs. IL-1β+Ac-SDKP).
Fig. 2.

Effects of IL-1β and Ac-SDKP on MMP-2 protein levels in cardiac fibroblasts. Cardiac fibroblasts were synchronized and treated with either vehicle, Ac-SDKP, IL-1β, or Ac-SDKP+IL-1β. MMP-2 was detected by immunoblotting. a, c Representative blots and b, d bar graphs demonstrating the effects of IL-1β and Ac-SDKP on MMP-2 level; n=3–4
Effects of Ac-SDKP on collagenase activity in cardiac fibroblasts
We treated cardiac fibroblasts with Ac-SDKP, IL-1β, or both. At the basal level, Ac-SDKP did not alter collagenase activity (control, 6.0±0.6 U/mg protein; Ac-SDKP, 4.5±0.5 U/mg protein; p=NS, control vs. Ac-SDKP); however, IL-1β significantly increased collagenase activity (IL-1β, 9.4±0.6 U/ml; p<0.01, IL-1β vs. controls). This effect of IL-1β was significantly decreased by Ac-SDKP (5.3±0.7 U/mg protein; p<0.01, IL-1β vs. IL-1β+Ac-SDKP) (Fig. 3).
Fig. 3.

Effects of IL-1β and Ac-SDKP on collagenase activity in cardiac fibroblasts. Cardiac fibroblasts were synchronized and treated with either vehicle, Ac-SDKP, IL-1β, or Ac-SDKP+IL-1β for 72 h. Collagenase activity was measured in the conditioned medium using a commercial kit. The bar graph represents the pooled data from four separate experiments, each done in quadruplicate
Effects of Ac-SDKP on MMP-13 levels in cardiac fibroblasts
MMP-13 was produced by cardiac fibroblasts at the basal level. IL-1β increased MMP-13 further (controls, 1.5±0.32 densitometric units; IL-1β, 3.6±0.7 densitometric units; p<0.05), and its expression was significantly inhibited by Ac-SDKP (2.0±0.41 densitometric units; p<0.05, IL-1β vs. IL-1β+Ac-SDKP). Ac-SDKP did not alter MMP-13 levels under basal conditions (Fig. 4).
Fig. 4.

Effects of IL-1β and Ac-SDKP on MMP-13 protein levels in cardiac fibroblasts. Cardiac fibroblasts were synchronized and treated with vehicle, Ac-SDKP, IL-1β, or Ac-SDKP+IL-1β. MMP-13 in the cultured supernatant was detected by immunoblotting using an anti-MMP-13 antibody. The bar graph represents the pooled data from three separate experiments, each done in triplicate
Effects of Ac-SDKP on TIMP-1 and TIMP-2 levels in cardiac fibroblasts
Western blotting of the cultured supernatant showed that cardiac fibroblasts treated with IL-1β had significantly reduced TIMP-1 (controls, 3.1±0.87 densitometric units; IL-1β, 1.1±0.1 densitometric units; p<0.01). Ac-SDKP treatment led to a small but significant increase in TIMP-1 to 1.7± 0.7 densitometric units (p<0.05, IL-1β vs. IL-1β+Ac-SDKP) (Fig. 5b). In addition, IL-1β decreased TIMP-2 (controls, 2.1±0.5 densitometric units; IL-1β, 0.7±0.3 densitometric units; p<0.05). Ac-SDKP partially reversed the inhibitory effect of IL-1β (1.4±0.4 densitometric units; p<0.05, IL-1β vs. IL-1β+Ac-SDKP) (Fig. 5d).
Fig. 5.

Effects of IL-1β and Ac-SDKP on TIMP-1 and TIMP-2 levels in cardiac fibroblasts. Cardiac fibroblasts were synchronized and treated with either vehicle, Ac-SDKP, IL-1β, or Ac-SDKP+IL-1β. TIMP-1 and TIMP-2 in the conditioned medium were detected by immunoblotting. a, c Bar diagrams representing the effects of IL-1β and Ac-SDKP on TIMP-1 level. b, d Bar graphs representing the effects of IL-1β and Ac-SDKP on TIMP-2 level. Each bar represents pooled data from three separate experiments
Effects of Ac-SDKP on collagen synthesis
We tested whether IL-1β alone or in combination affects collagen production in fibroblast culture. We found that none of the treatments significantly affected total collagen production compared to the control (Fig. 6).
Fig. 6.

Effects of IL-1β and Ac-SDKP on collagen production in cardiac fibroblasts. Cardiac fibroblasts were synchronized and treated with vehicle, Ac-SDKP, IL-1β, or Ac-SDKP+IL-1β for 72 min. Collagen production in the cell media was assessed by hydroxyproline assay. Total collagen production was not affected by any treatment. The bar graph represents the pooled data from six separate experiments
Effect of Ac-SDKP on IL-1β-induced ERK1/2 signaling in cardiac fibroblasts
IL-1β reportedly increases MMP activity by inducing phosphorylation of ERK1/2 (p42/44 MAPK) [5]. We tested whether the Ac-SDKP-mediated decrease in MMP activity is related to the inhibition of p42/44 phosphorylation in IL-1β-stimulated cells. We observed that IL-1β significantly stimulated phosphorylation of p42/44 MAPK (controls, 0. 50±0.19 densitometric units; IL-1β, 2.2±0.8 densitometric units; p<0.05). In basal conditions, Ac-SDKP had no effect, whereas in IL-1β-treated fibroblasts, Ac-SDKP significantly reduced p42/44 MAPK phosphorylation (0.98±0.34 densitometric units; p<0.05, IL-1β vs. IL-1β+Ac-SDKP) (Fig. 7).
Fig. 7.

Effects of IL-1β and Ac-SDKP on ERK1/2 phosphorylation in cardiac fibroblasts. Cardiac fibroblasts were synchronized and treated with vehicle, Ac-SDKP, IL-1β, or Ac-SDKP+IL-1β for 30 min. ERK1/2 in the cell lysate was detected by immunoblotting using an anti-phospho-ERK1/2 antibody. The same membrane was probed with an anti-ERK1/2 antibody and protein levels of phospho-ERK1/2 normalized to total ERK1/2. The bar graph represents the pooled data from three separate experiments
Effects of Ac-SDKP on IL-1β-activated NFκB in cardiac fibroblasts
IL-1β is a potent activator of NFκB in cardiac fibroblasts [43]. We tested whether the Ac-SDKP-mediated decrease in MMP activity is related to the inhibition of NFκB activation in IL-1β-stimulated fibroblasts. IL-1β significantly stimulated NFκB binding activity (controls, 3.01±0.5-fold increase vs. control; p<0.005). Conversely, pretreatment with Ac-SDKP significantly reduced IL-β-induced NFκB binding activity (2.02±0.32-fold vs. control; p<0.01) (Fig. 8). Ac-SDKP itself had no effect on NFκB activity. This result suggests that Ac-SDKP blunted NFκB activation in IL-1β signaling.
Fig. 8.
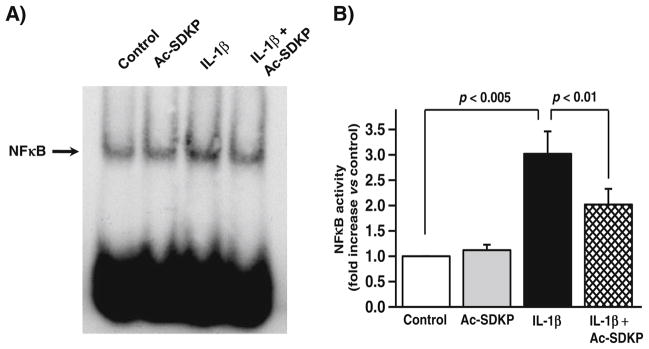
Effects of Ac-SDKP on IL-1β-activated NFκB in cardiac fibroblasts. Cardiac fibroblasts were synchronized and treated with vehicle, Ac-SDKP, IL-1β, or Ac-SDKP+ IL-1β for 2.5 h. NFκB activity was assessed by electrophoretic mobility gel shift assay. a Representative gel shift; b quantitative data for NFκB activity; n=10 for each treatment
Discussion
We have previously shown that Ac-SDKP inhibits collagen synthesis [35] and decreases cardiac fibrosis [28, 32]. Our aim was to investigate whether Ac-SDKP also activates collagenolytic enzymes in cardiac fibroblasts. The major finding of this study is that Ac-SDKP had no effect on total collagenase and MMP-2 and MMP-9 activities or protein levels and MMP-13 at the basal level. Rather, it counteracted the effects of IL-1β on gelatinase and total collagenase activity, MMP-2, MMP-9, MMP-13, TIMP-1, and TIMP-2 protein levels, p42/p44 phosphorylation, and NFκB activation. Thus, these data imply that decreased collagen deposition caused by Ac-SDKP may not be necessarily due to increased collagen degradation, but rather to decreased collagen synthesis [35].
MMP activity is regulated by both transcriptional and posttranslational mechanisms. Posttranslational regulation occurs through the activation of latent proenzymes (proMMPs) by factors such as serum, heparin, prostaglandin E2, and pro-inflammatory cytokines [2, 22, 40]. IL-1β is a crucial MMP-regulatory cytokine and has been shown to increase MMP expression and activity in various cell types, including chondrocytes [3], vascular smooth muscle cells [33], and fibroblasts [24, 30, 38]. We know that Ac-SDKP blocks tissue infiltration by monocytes and macrophages [34, 45], which can produce and/or activate various cytokines including IL-1β. Here, we found that Ac-SDKP opposes the stimulatory effects of IL-1β on MMP activity.
MMPs are important enzymes involved in the breakdown of ECM components, including various collagen types. MMPs can be divided into different subgroups, among them collagenase (MMP-1, MMP-8, and MMP-13), gelatinase (MMP-2 and MMP-9), stromelysin (MMP-3 and MMP-7), membrane-type MMPs (MMP-14), and others [20, 23]. MMP-1, MMP-8, and MMP-13 (collagenases) specifically split the triple helix of fibrillar collagens [23]. The resultant denatured collagen molecule is then susceptible to degradation by other proteases, particularly the gelatinases MMP-2 and MMP-9. Thus, the role of collagenases might be crucial for the initiation of ECM remodeling. Of all the collagenases, we chose to measure MMP-13 because it is the only MMP of the collagenase family shown thus far to be expressed in RCFs [38]. MMP-13 is a collagenase found in the cardiac interstitium [46]; it degrades fibrillar collagens of various types, including types I and III, the major constituents of the cardiac ECM [1, 19].
Although IL-1β has been shown to activate gelatinases in cardiac fibroblasts [37, 38], its role in collagenase activation is not known. We found that IL-1β also increased collagenase activity in adult RCFs. As MMP-13 is the major collagenase secreted by cardiac fibroblasts, the decrease in collagenase activity by Ac-SDKP was at least partly due to the inhibition of MMP-13 expression.
Cytokine upsurge accompanied by MMP-2 and MMP-9 activation has been implicated in various myocardial pathologies including hypertension, myocardial infarction, and dilated cardiomyopathy [8, 37]. In vitro, cytokine stimulation of cultured cardiac fibroblasts regulates MMP expression and activity [38]. As reported earlier, IL-1β increased MMP-2 protein expression and activity as measured by in-gel zymography [38]. Ac-SDKP inhibited the IL-1β-mediated increase in MMP-2 activity and protein expression, suggesting that the decreases in MMP-2 zymographic activity were at least partly due to decreased protein expression. This finding would fit well with an original preliminary observation in which Ac-SDKP prevented cardiac rupture in mice subjected to myocardial infarction [44]; in this model, Ac-SDKP would prevent not only the invasion of the myocardium by inflammatory cells, but also MMP-2 and MMP-9 expressions and activities.
MMP activity is inhibited by endogenous TIMPs [41]. TIMP-1 works better than any other TIMPs against MMP-1, MMP-3, MMP-9, and MMP-13, while TIMP-2 has a greater affinity for MMP-2. Since we observed that IL-1β-induced MMP-2 activity is inhibited by Ac-SDKP, we focused on measuring TIMP-1 and TIMP-2. As reported previously, IL-1β reduced TIMP-1 and TIMP-2 levels in cardiac fibroblasts. This effect was partially blocked when the cells were co-treated with Ac-SDKP, indicating that the inhibitory effect of Ac-SDKP on MMP-2 activity is mediated in part by an increase in TIMP-1 and TIMP-2.
Various signaling cascades are involved in MMP regulation. Ac-SDKP inhibits Smad2/3 and ERK1/2 MAPK signaling in cardiac fibroblasts without affecting p38 and JNK activity [35]. IL-1β was found to stimulate MMP-9 synthesis and release by activating PI3K-Akt and MEK1-ERK signaling cascades, while p38 signaling was not involved [5]. Moreover, ERK1/2 signaling was shown to be directly involved in MMP regulation, as inhibition of its phosphorylation by U0126, an inhibitor of mitogen-activated protein kinase kinase, led to decreased MMP-2 and MMP-9 activities [9]. The current study shows that Ac-SDKP inhibits the effects of IL-1β on ERK1/2 phosphorylation, suggesting that Ac-SDKP-mediated regulation of MMP-2 activity could be related to the inhibition of the ERK1/2 signaling cascade. The inhibitory effect of Ac-SDKP on ERK1/2 could likely involve the activation of Src homology 2-containing protein tyrosine phosphatase-2 [27], which has been shown to regulate IL-1β signaling in fibroblasts [17]. NFκB is another important downstream regulator of MMP expression and activity in cardiac fibroblasts [43] and other fibroblast types [18, 24]. This transcription factor is composed of two subunits of 50 and 65 kDa, which are sequestered in the cytoplasm in an inactive form by IκB. Upon activation, IκB undergoes phosphorylation and degradation, and the NFκB heterodimer translocates into the nucleus, where it binds to DNA and activates transcription [10, 16, 21]. Indeed, specific inhibition of NFκB in RCFs has been show to blunt IL-1β-stimulated increases in MMP-2 and MMP-9 expressions and activities [43]. Our present study also shows that inhibition by Ac-SDKP of MMPs was paralleled by a significant blockade of NFκB activity. As depicted in Fig. 9, Ac-SDKP interfered with IL-1β-induced MMPs via inhibition of MAPK and NFκB activation; however, it remains unclear how phosphorylated p42/p44 is associated with NFκB activation in MMP expression and activation [10, 16, 21].
Fig. 9.

Schematic representations of signaling pathways that are affected by Ac-SDKP. IL-1β-stimulated increase in MMP-2/MMP-9 and other collagenase protein levels and activity is blocked by Ac-SDKP, interfering with the activation of ERK1/2 and NFκB signaling pathways
Adverse myocardial remodeling in dilated cardiomyopathy and post-myocardial infarction [8, 37] is preceded by inflammatory cell infiltration, cytokine release, and activation of collagenase and gelatinase [8, 37]. In our previous study, we observed that Ac-SDKP inhibited infarct rupture in a mouse model of myocardial infarction [44]; this may have resulted from Ac-SDKP inhibiting cytokine-mediated MMP expression and activation in cardiac fibroblasts. Therefore, Ac-SDKP or its analogs that are resistant to ACE or other enzymatic degradation could potentially be used as a new therapeutic agent that counteracts the adverse effects associated with MMP activation in pro-inflammatory conditions including infarction.
Acknowledgments
This work was supported by National Institutes of Health grants HL-28982 (OAC) and HL-071806 (NER).
Footnotes
Conflict of interest Nour-Eddine Rhaleb and Oscar A. Carretero received grant support from F. Hoffman-La Roche Ltd., Basel, Switzerland.
Contributor Information
Nour-Eddine Rhaleb, Email: nrhaleb1@hfhs.org, Hypertension and Vascular Research Division, Department of Internal Medicine, Henry Ford Hospital, E&R 7121, 2799 West Grand Boulevard, Detroit, MI 48202, USA. Department of Physiology, Wayne State University, Detroit, MI, USA.
Saraswati Pokharel, Hypertension and Vascular Research Division, Department of Internal Medicine, Henry Ford Hospital, E&R 7121, 2799 West Grand Boulevard, Detroit, MI 48202, USA.
Umesh C. Sharma, Hypertension and Vascular Research Division, Department of Internal Medicine, Henry Ford Hospital, E&R 7121, 2799 West Grand Boulevard, Detroit, MI 48202, USA
Hongmei Peng, Hypertension and Vascular Research Division, Department of Internal Medicine, Henry Ford Hospital, E&R 7121, 2799 West Grand Boulevard, Detroit, MI 48202, USA.
Edward Peterson, Department of Biostatistics, Henry Ford Hospital, Detroit, MI, USA.
Pamela Harding, Hypertension and Vascular Research Division, Department of Internal Medicine, Henry Ford Hospital, E&R 7121, 2799 West Grand Boulevard, Detroit, MI 48202, USA.
Xiao-Ping Yang, Hypertension and Vascular Research Division, Department of Internal Medicine, Henry Ford Hospital, E&R 7121, 2799 West Grand Boulevard, Detroit, MI 48202, USA.
Oscar A. Carretero, Hypertension and Vascular Research Division, Department of Internal Medicine, Henry Ford Hospital, E&R 7121, 2799 West Grand Boulevard, Detroit, MI 48202, USA
References
- 1.Brilla CG, Maisch B. Regulation of the structural remodelling of the myocardium: from hypertrophy to heart failure. Eur Heart J. 1994;15(Suppl D):45–52. doi: 10.1093/eurheartj/15.suppl_d.45. [DOI] [PubMed] [Google Scholar]
- 2.Brilla CG, Maisch B, Zhou G, Weber KT. Hormonal regulation of cardiac fibroblast function. Eur Heart J. 1995;16(Suppl C):45–50. doi: 10.1093/eurheartj/16.suppl_c.45. [DOI] [PubMed] [Google Scholar]
- 3.Carroll MA, Doumad AB, Li J, Cheng MK, Falck JR, McGiff JC. Adenosine2A receptor vasodilation of rat preglomerular microvessels is mediated by EETs that activate the cAMP/PKA pathway. Am J Physiol Renal Physiol. 2006;291:F155–F161. doi: 10.1152/ajprenal.00231.2005. [DOI] [PubMed] [Google Scholar]
- 4.Cavasin MA, Liao TD, Yang X-P, Yang JJ, Carretero OA. Decreased endogenous levels of Ac-SDKP promote organ fibrosis. Hypertension. 2007;50:130–136. doi: 10.1161/HYPERTENSIONAHA.106.084103. [DOI] [PubMed] [Google Scholar]
- 5.Cheng MK, McGiff JC, Carroll MA. Renal arterial 20-hydroxyeicosatetraenoic acid levels: regulation by cyclooxygenase. Am J Physiol Renal Physiol. 2003;284:F474–F479. doi: 10.1152/ajprenal.00239.2002. [DOI] [PubMed] [Google Scholar]
- 6.Cleutjens JPM, Kandala JC, Guarda E, Guntaka RV, Weber KT. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995;27:1281–1292. doi: 10.1016/s0022-2828(05)82390-9. [DOI] [PubMed] [Google Scholar]
- 7.Coffey CE. Building a system of perfect depression care in behavioral health. Jt Comm J Qual Patient Saf. 2007;33:193–199. doi: 10.1016/s1553-7250(07)33022-5. [DOI] [PubMed] [Google Scholar]
- 8.D’Angelo DD. Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorn GW, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eberhardt W, Huwiler A, Beck KF, Walpen S, Pfeilschifter J. Amplification of IL-1 beta-induced matrix metalloproteinase-9 expression by superoxide in rat glomerular mesangial cells is mediated by increased activities of NF-kappa B and activating protein-1 and involves activation of the mitogen-activated protein kinase pathways. J Immunol. 2000;165:5788–5797. doi: 10.4049/jimmunol.165.10.5788. [DOI] [PubMed] [Google Scholar]
- 11.Eghbali M, Blumenfeld OO, Seifter S, Buttrick PM, Leinwand LA, Robinson TF, Zern MA, Giambrone MA. Localization of types I, III and IV collagen mRNAs in rat heart cells by in situ hybridization. J Mol Cell Cardiol. 1989;21:103–113. doi: 10.1016/0022-2828(89)91498-3. [DOI] [PubMed] [Google Scholar]
- 12.Eghbali M, Czaja MJ, Zeydel M, Weiner FR, Zern MA, Seifter S, Blumenfeld OO. Collagen chain mRNAs in isolated heart cells from young and adult rats. J Mol Cell Cardiol. 1988;20:267–276. doi: 10.1016/s0022-2828(88)80059-2. [DOI] [PubMed] [Google Scholar]
- 13.Fujihara CK, Antunes GR, Mattar AL, Malheiros DM, Vieira JM, Jr, Zatz R. Chronic inhibition of nuclear factor-kappaB attenuates renal injury in the 5/6 renal ablation model. Am J Physiol Renal Physiol. 2007;292:F92–F99. doi: 10.1152/ajprenal.00184.2006. [DOI] [PubMed] [Google Scholar]
- 14.Galis ZS, Muszynski M, Sukhova GK, Simon-Morrissey E, Unemori EN, Lark MW, Amento E, Libby P. Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ Res. 1994;75:181–189. doi: 10.1161/01.res.75.1.181. [DOI] [PubMed] [Google Scholar]
- 15.Guarda E, Katwa LC, Myers PR, Tyagi SC, Weber KT. Effects of endothelins on collagen turnover in cardiac fibroblasts. Cardiovasc Res. 1993;27:2130–2134. doi: 10.1093/cvr/27.12.2130. [DOI] [PubMed] [Google Scholar]
- 16.Hambleton J, McMahon M, DeFranco AL. Activation of Raf-1 and mitogen-activated protein kinase in murine macrophages partially mimics lipopolysaccharide-induced signaling events. J Exp Med. 1995;182:147–154. doi: 10.1084/jem.182.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrera Abreu MT, Wang Q, Vachon E, Suzuki T, Chow CW, Wang Y, Hong O, Villar J, McCulloch CA, Downey GP. Tyrosine phosphatase SHP-2 regulates IL-1 signaling in fibroblasts through focal adhesions. J Cell Physiol. 2006;207:132–143. doi: 10.1002/jcp.20544. [DOI] [PubMed] [Google Scholar]
- 18.Kessler DJ, Duyao MP, Spicer DB, Sonenshein GE. NF-kB-like factors mediate interleukin 1 induction of c-myc gene transcription in fibroblasts. J Exp Med. 1992;176:787–792. doi: 10.1084/jem.176.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levant A, Levy E, Argaman M, Fleisher-Berkovich S. Kinins and neuroinflammation: dual effect on prostaglandin synthesis. Eur J Pharmacol. 2006;546:197–200. doi: 10.1016/j.ejphar.2006.06.074. [DOI] [PubMed] [Google Scholar]
- 20.Li HJ, Yin H, Yao YY, Shen B, Bader M, Chao L, Chao J. Tissue kallikrein protects against pressure overload-induced cardiac hypertrophy through kinin B2 receptor and glycogen synthase kinase-3beta activation. Cardiovasc Res. 2007;73:130–142. doi: 10.1016/j.cardiores.2006.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang KC, Lee CW, Lin WN, Lin CC, Wu CB, Luo SF, Yang CM. Interleukin-1beta induces MMP-9 expression via p42/p44 MAPK, p38 MAPK, JNK, and nuclear factor-kappaB signaling pathways in human tracheal smooth muscle cells. J Cell Physiol. 2007;211:759–770. doi: 10.1002/jcp.20992. [DOI] [PubMed] [Google Scholar]
- 22.Liclican EL, McGiff JC, Pedraza PL, Ferreri NR, Falck JR, Carroll MA. Exaggerated response to adenosine in kidneys from high salt-fed rats: role of epoxyeicosatrienoic acids. Am J Physiol Renal Physiol. 2005;289:F386–F392. doi: 10.1152/ajprenal.00421.2004. [DOI] [PubMed] [Google Scholar]
- 23.McGiff JC, Ferreri NR. Eicosanoids and the kidney. In: Alpern R, Hebert SC, editors. The kidney: physiology and pathophysiology. Elsevier; New York: 2007. [Google Scholar]
- 24.Noh EM, Kim JS, Hur H, Park BH, Song EK, Han MK, Kwon KB, Yoo WH, Shim IK, Lee SJ, Youn HJ, Lee YR. Cordycepin inhibits IL-1beta-induced MMP-1 and MMP-3 expression in rheumatoid arthritis synovial fibroblasts. Rheumatology (Oxford) 2009;48:45–48. doi: 10.1093/rheumatology/ken417. [DOI] [PubMed] [Google Scholar]
- 25.Peng H, Carretero OA, Brigstock DR, Oja-Tebbe N, Rhaleb N-E. Ac-SDKP reverses cardiac fibrosis in rats with renovascular hypertension. Hypertension. 2003;42:1164–1170. doi: 10.1161/01.HYP.0000100423.24330.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng H, Carretero OA, Liao TD, Peterson EL, Rhaleb N-E. Role of N-acetyl-seryl-aspartyl-lysyl-proline in the antifibrotic and anti-inflammatory effects of the angiotensin-converting enzyme inhibitor captopril in hypertension. Hypertension. 2007;49:695–703. doi: 10.1161/01.HYP.0000258406.66954.4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng H, Carretero OA, Peterson EL, Yang X-P, Santra K, Rhaleb N-E. N-acetyl-seryl-aspartyl-lysyl-proline inhibits ET-1-induced collagen production by preserving Src homology 2-containing protein tyrosine phosphatase-2 activity in cardiac fibroblasts. Pflugers Arch. 2012;464:415–423. doi: 10.1007/s00424-012-1150-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng H, Carretero OA, Raij L, Yang F, Kapke A, Rhaleb NE. Antifibrotic effects of N-acetyl-seryl-aspartyl-lysyl-proline on the heart and kidney in aldosterone-salt hypertensive rats. Hypertension. 2001;37:794–800. doi: 10.1161/01.hyp.37.2.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng H, Carretero OA, Vuljaj N, Liao TD, Motivala A, Peterson EL, Rhaleb N-E. Angiotensin-converting enzyme inhibitors: a new mechanism of action. Circulation. 2005;112:2436–2445. doi: 10.1161/CIRCULATIONAHA.104.528695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pladys P, Sennlaub F, Brault S, Checchin D, Lahaie I, Le NL, Bibeau K, Cambonie G, Abran D, Brochu M, Thibault G, Hardy P, Chemtob S, Nuyt AM. Microvascular rarefaction and decreased angiogenesis in rats with fetal programming of hypertension associated with exposure to a low-protein diet in utero. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1580–R1588. doi: 10.1152/ajpregu.00031.2005. [DOI] [PubMed] [Google Scholar]
- 31.Pokharel S, Rasoul S, Roks AJ, van Leeuwen RE, van Luyn MJ, Deelman LE, Smits JF, Carretero O, van Gilst WH, Pinto YM. N-acetyl-Ser-Asp-Lys-Pro inhibits phosphorylation of Smad2 in cardiac fibroblasts. Hypertension. 2002;40:155–161. doi: 10.1161/01.hyp.0000025880.56816.fa. [DOI] [PubMed] [Google Scholar]
- 32.Pokharel S, van Geel PP, Sharma UC, Cleutjens JPM, Bohnemeier H, Tian X-L, Schunkert H, Crijns HJGM, Paul M, Pinto YM. Increased myocardial collagen content in transgenic rats overexpressing cardiac angiotensin-converting enzyme is related to enhanced breakdown of N-acetyl-Ser-Asp-Lys-Pro and increased phosphorylation of Smad2/3. Circulation. 2004;110:3129–3135. doi: 10.1161/01.CIR.0000147180.87553.79. [DOI] [PubMed] [Google Scholar]
- 33.Quilley J, McGiff JC. Multiple roles of eicosanoids in blood pressure regulation. In: Lip G, Hall J, editors. Comprehensive hypertension. Mosby Elsevier; Philadelphia: 2007. [Google Scholar]
- 34.Rasoul S, Carretero OA, Peng H, Cavasin MA, Zhuo J, Sanchez-Mendoza A, Brigstock DR, Rhaleb NE. Antifibrotic effect of Ac-SDKP and angiotensin-converting enzyme inhibition in hypertension. J Hypertens. 2004;22:593–603. doi: 10.1097/00004872-200403000-00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rhaleb N-E, Peng H, Harding P, Tayeh M, LaPointe MC, Carretero OA. Effect of N-acetyl-seryl-aspartyl-lysyl-proline on DNA and collagen synthesis in rat cardiac fibroblasts. Hypertension. 2001;37:827–832. doi: 10.1161/01.hyp.37.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robidoux C, Pelé JP, Maclouf J, Pradelles P, Sirois P. Stimulation of release of prostaglandins and thromboxanes from isolated guinea pig lung cells by bradykinin, f-Met-Leu-Phe, phorbol myristate, ionophore A23187, and leukotrienes. Inflammation. 1988;12:285–295. doi: 10.1007/BF00915766. [DOI] [PubMed] [Google Scholar]
- 37.Schillaci G, Pirro M, Pucci G, Ronti T, Vaudo G, Mannarino MR, Porcellati C, Mannarino E. Prognostic value of elevated white blood cell count in hypertension. Am J Hypertens. 2007;20:364–369. doi: 10.1016/j.amjhyper.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 38.Siwik DA, Chang DLF, Colucci WS. Interleukin-1b and tumor necrosis factor-a decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–1265. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 39.Stegemann H, Stalder K. Determination of hydroxyproline. Clin Chim Acta. 1967;18:267–273. doi: 10.1016/0009-8981(67)90167-2. [DOI] [PubMed] [Google Scholar]
- 40.Tyagi SC, Kumar S, Katwa L. Differential regulation of extracellular matrix metalloproteinase and tissue inhibitor by heparin and cholesterol in fibroblast cells. J Mol Cell Cardiol. 1997;29:391–404. doi: 10.1006/jmcc.1996.0283. [DOI] [PubMed] [Google Scholar]
- 41.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases. Structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 42.Weber KT, Sun Y, Tyagi SC, Cleutjens JP. Collagen network of the myocardium: function, structural remodeling and regulatory mechanisms. J Mol Cell Cardiol. 1994;26:279–292. doi: 10.1006/jmcc.1994.1036. [DOI] [PubMed] [Google Scholar]
- 43.Xie Z, Singh M, Singh K. Differential regulation of matrix metalloproteinase-2 and -9 expression and activity in adult rat cardiac fibroblasts in response to interleukin-1. J Biol Chem. 2004;279:39513–39519. doi: 10.1074/jbc.M405844200. [DOI] [PubMed] [Google Scholar]
- 44.Xu J, Carretero OA, Rhaleb N-E, Yang X-P. Effect of Ac-SDKP on cardiac rupture and early remodeling after myocardial infarction in mice (abstract) Circulation. 2004;110:III-27. [Google Scholar]
- 45.Yang F, Yang X-P, Liu YH, Xu J, Cingolani O, Rhaleb N-E, Carretero OA. Ac-SDKP reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension. 2004;43:229–236. doi: 10.1161/01.HYP.0000107777.91185.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao YY, Yin H, Shen B, Chao L, Chao J. Tissue kallikrein infusion prevents cardiomyocyte apoptosis, inflammation and ventricular remodeling after myocardial infarction. Regul Pept. 2007;140:12–20. doi: 10.1016/j.regpep.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]