

Abstract
We previously reported that N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) reduces fibrosis and inflammation (macrophages and mast cells). However, it is not known whether Ac-SDKP decreases collagen cross-linking and lymphocyte infiltration; lymphocytes modulate both collagen cross-linking and extracellular matrix formation in hypertension. Thus, we hypothesized that 1) in angiotensin (Ang) II-induced hypertension, Ac-SDKP prevents increases in cross-linked and total collagen by down-regulating lysyl oxidase (LOX), the enzyme responsible for cross-linking, and 2) these effects are associated with decreased a) pro-fibrotic cytokine TGF-β and b) the proinflammatory transcription factor nuclear factor κB (NFκB), and c) CD4+/CD8+ lymphocyte infiltration. We induced hypertension in rats by infusing Ang II either alone or combined with Ac-SDKP for 3 weeks. While Ac-SDKP failed to lower blood pressure or left ventricular hypertrophy, it did prevent Ang II-induced increases in 1) cross-linked and total collagen, 2) LOX mRNA expression and LOXL1 protein, 3) TGF-β expression, 4) nuclear translocation of NFκB, 5) CD4+/CD8+ lymphocyte infiltration and 6) CD68+ macrophages infiltration. In addition, we found a positive correlation between CD4+ infiltration and LOXL1 expression. In conclusion, the effect of Ac-SDKP on collagen cross-linking and total collagen may be due to reduced TGF-β1, LOXL1 and lymphocyte and macrophages infiltration, and its effect on inflammation could be due to lower NFκB.
Keywords: Ac-SDKP, Angiotensin II, Inflammation, Collagen, NFκB, Lysyl oxidase, Fibrosis, Lymphocytes
INTRODUCTION
Hypertension is a major risk factor for cardiovascular disease. During the evolution of hypertension, the myocardium undergoes several morphological changes, including hypertrophy, inflammation, and remodeling of the extracellular matrix (ECM), leading to cardiac dysfunction and heart failure. Chronic recruitment of inflammatory mononuclear leukocytes plays an important role in stimulating cardiac collagen synthesis and remodeling of the ECM [21, 47]. Increased synthesis, degradation and/or maturation of collagen fibers as well as changes in collagen subtypes and collagen cross-linking have been proposed to participate in the development of pathological remodeling and each has specific implications for altered myocardial function [6, 12, 43]. However, increments in myocardial collagen content may occur without chamber remodeling [24], indicating that tissue fibrosis may not be a prerequisite for remodeling; whereas collagen maturation (mainly changes in collagen cross-linking) may contribute directly to chamber remodeling and systolic dysfunction. Indeed, Norton et al [28] showed that increments in cross-linking contribute to enhanced myocardial stiffness, whereas Woodiwiss et al [45] reported that less cross-linking contributes to chamber remodeling. We found that N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP), a naturally occurring endogenous bioactive tetrapeptide released from thymosin-β4 by prolyl-oligopeptidase [8], reduced fibrosis and slightly impaired systolic function without affecting diastolic function in spontaneously hypertensive rats (SHR) [11], suggesting that lowering collagen content does not necessarily improve diastolic capacity.
Ac-SDKP is hydrolyzed mainly by angiotensin-converting enzyme (ACE) and its circulating levels were found to increase 5- to 6-fold in patients treated with ACE inhibitors for hypertension or heart failure [9]. Thus, it has been proposed that the antiinflammatory, antifibrotic and antiproliferative effects of ACE inhibitors may be mediated partly by increasing Ac-SDKP [8]. We have shown that Ac-SDKP prevents and even reverses fibrosis, inflammation (macrophage and mast cells) and cytokine expression in animal models of hypertension [20, 32, 35, 37, 38]; moreover all of these effects occurred without altering blood pressure or left ventricular hypertrophy. However, whether Ac-SDKP reduces collagen cross-linking, LOX expression, translocation of nuclear factor κB (NFκB) or cardiac T-lymphocyte infiltration is unknown. It is widely accepted that during ECM remodeling cardiac myofibroblasts are primarily responsible for total collagen synthesis, while the enzyme lysyl oxidase (LOX) and T lymphocytes play an important role in modulating cardiac ECM and collagen cross-linking in hypertension [47] [42].
Therefore, we tested the hypothesis that in Angiotensin (Ang) II-induced hypertension, Ac-SDKP prevents increases in cross-linked and total collagen by decreasing LOX and CD4+/CD8+ lymphocyte infiltration. These effects are associated with decreases in the profibrotic cytokine TGF-β and translocation of the proinflammatory transcription factor NFκB. To test these hypotheses, hypertension was induced in rats by infusing Ang II either alone or combined with Ac-SDKP for 3 weeks.
METHODS
Adult male Lewis rats weighing 240–320 g (Charles River Laboratories, Wilmington, MA) were housed under controlled conditions. This investigation conformed to the recommendation in the Guide for the Care and Use of Laboratory animals published by US National Institute of Health (NIH Publication 8th Edition, revised 2011) and the protocol was approved by the Henry Ford Hospital Institutional Animal Care and Use Committee (IACUC, A3148-01).
Experimental groups
Male Lewis rats weighing 240–320 g (Charles River Laboratories, Wilmington, MA) were housed under controlled conditions and randomly divided into four groups: 1) control (vehicle); 2) control + Ac-SDKP (800 μg.kg−1.day−1); 3) Ang II (750 μg.kg−1.day−1); and 4) Ang II + Ac-SDKP. Vehicle, Ac-SDKP and Ang II were infused for 3 weeks using a subcutaneous osmotic minipump (Alzet Model 2ML4). Ac-SDKP and Ang II were dissolved in 0.01 N acetic acid in saline. Anesthesia for surgical pump implantation was achieved with pentobarbital (Nembutal, Lundbeck; 50 mg.kg−1, i.p.). Dosages of Ang II and Ac-SDKP were based on previous studies [21, 37]. All treatments were carried out for 3 weeks. This protocol was approved by the Henry Ford Hospital Institutional Animal Care and Use Committee (IACUC).
Telemetry for blood pressure assessment
Mean blood pressure (MBP) and heart rate (HR) were continuously monitored by telemetry (Data Sciences International). Rats were anesthetized with ketamine (90 mg.kg−1) and xylazine (10 mg.kg−1) and a telemeter catheter implanted in left carotid artery according with the manufacturer’s specifications and institutional procedures. After allowing the rats to recover, MBP and HR were measured for 1 week using a Data Science acquisition program, which enables us to collect BP readings every 10 seconds for 24 hours. Then, osmotic minipumps were implanted as described aboved and BP was recorded for 3 weeks.
Echocardiography
After 3 weeks, the rats were anesthetized with pentobarbital (Nembutal, Lundbeck; 50 mg.kg−1; i.p.) and subjected to echocardiography and doppler sonography (Acuson Sequoia C 256 with a 15-MHz transducer) and electrocardiography (ECG) for studying cardiac remodeling and function. Rats were euthanized thereafter, and the heart removed.
Hydroxyproline assay and collagen cross-linking
Myocardial total collagen content and insoluble collagen were determined by hydroxyproline assay in undigested and cyanogen bromide (CNBr)-digested samples as described previously [26, 34, 45].
Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) for LOX
Quantitative realtime PCR was performed using a real-time PCR system (Applied Biosystems 7500; Foster, CA) with SYBR green dye and specific primers for LOX and β-actin.
Immunoblotting for LOXL1 and TGF-β1
cardiac LOXL1 and TGF-β1 were determined by immunoblotting. Briefly, proteins from the LV extracts were loaded in SDS-PAGE gels under non-reducing conditions and electrotransferred to a nitrocellulose membrane. Membranes were incubated with an anti-LOXL1 antibody (Abcam, Cambridge, MA) or with a mouse anti-TGF-β1 antibody (R&D Systems, Minneapolis, MN), then incubated with secondary antibodies conjugated with horseradish peroxidase (Cell Signaling Technology, Danvers, MA). Films were examined with a Perfection 3200 scanner (Epson America, Long Beach, CA) and band density and intensity quantified by densitometry. Increases in LOXL1 and TGF-β1 were normalized to GAPDH and expressed as fold increase vs control.
Electrophoretic mobility shift assay for NFκB
Nuclear translocation of NFκB was measured by electrophoretic mobility shift assay (EMSA) using nuclear extracts from 0.5 g LV tissue and a kit from Panomics (Fremont, CA) according to the manufacturer’s instructions. Optical density of the bands was compared as described previously [14].
Immunohistochemistry for lymphocytes and macrophages
LV was cut into three slices and snapfrozen in liquid nitrogen-precooled isopentane and stored at −80°C. Frozen sections (6 μm) were immunostained with anti-rat CD4+ (a marker for T lymphocytes; OX-38, BD Pharmingen) or anti-rat CD8+ antibodies (a marker for cytoxic lymphocytes; OX-8, AbD Serotec) or anti-rat CD68 (a marker for macrophages; OX-38, BD Pharmingen). A Vectastain Elite ABC-peroxidase kit (Vector Laboratories) and the AEC substrate 3-amino-9-ethylcarbazole (Invitrogen) were used to detect positive immunoreactivity. Negative controls were processed in a similar fashion except that they were not incubated with the primary antibody. Positive cells (stained reddish brown) were counted in 40× high-power fields in each section and expressed as number of cells per mm2.
Statistics
All values are expressed as mean ± SEM. We employed analysis of variance (ANOVA) for all four groups, comparing differences between Ang II vs. control and Ang II vs. Ang II + Ac-SDKP. Hochberg’s adjustment for multiple testing was used to determine significance. Blood pressure was analyzed by repeated-measures ANOVA, while linear regression analysis and Pearson’s correlation were used to assess the relationship between CD4+ and CD8+ lymphocyte infiltration and LOXL1 expression, taking p < 0.05 as significant.
For detailed Methods, see supplemental expanded methods section.
RESULTS
Mean blood pressure and myocardial hypertrophy
Ang II significantly increased mean blood pressure (MBP) and caused myocardial hypertrophy (Figure 1). Telemetry data analysis showed that Ac-SDKP did not affect BP in normotensive animals. In Ang II-treated animals Ac-SDKP tended to slightly increase MBP at some periods (p = NS) and it neither influence the fall of BP observed during sleep (dipping pattern) nor heart rate (Figure 1 supplemental material). Ac-SDKP did not affect myocardial hypertrophy in either normotensive or hypertensive animals.
Figure 1. Effect of Ac-SDKP on Ang II-induced hypertension and LV hypertrophy.
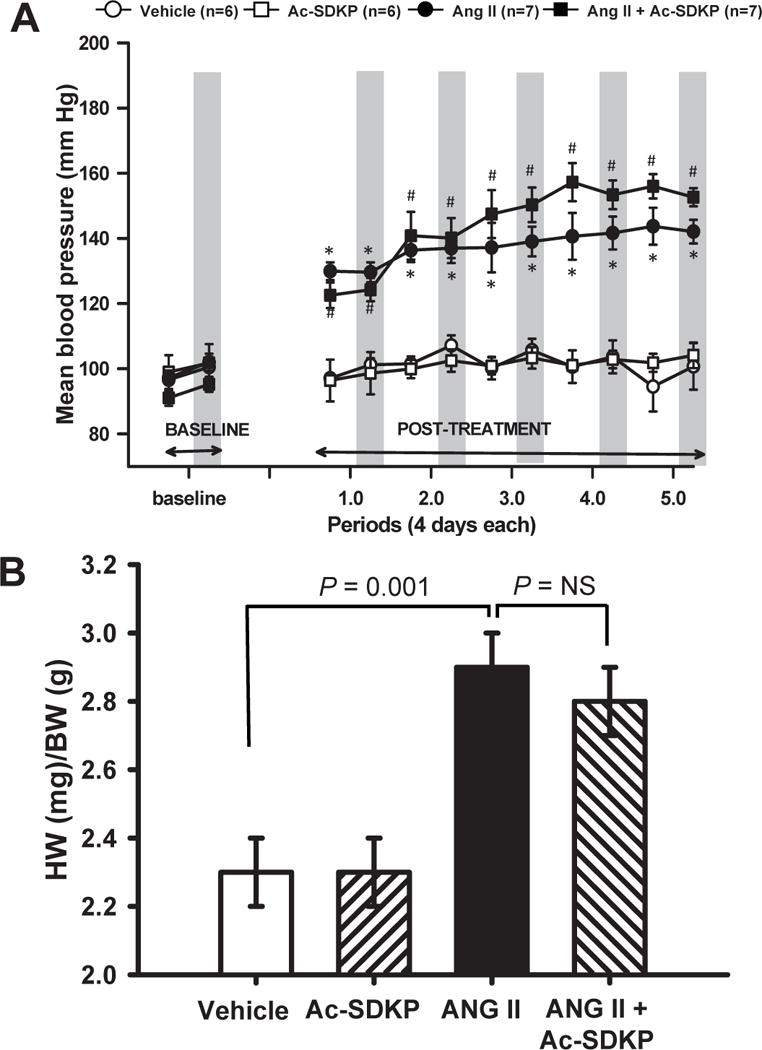
A) Mean blood pressure (MBP; recorded by telemetry) and B) left ventricular weight/body weight (LVW/BW) in rats given vehicle, Ac-SDKP (800 μg/kg/day), angiotensin II (Ang II, 750 μg/kg/day) or Ang II + Ac-SDKP. For MBP, periods of 4 days are separated for 12-hour intervals; and shading indicates the 12-hour nocturnal interval. Ac-SDKP tended to increase MBP, but did not alter left ventricular hypertrophy. * p < 0.05, vehicle vs Ang II; # p < 0.05, vehicle vs Ang II + Ac-SDKP.
Urine levels of Ac-SDKP
Ac-SDKP excretion in the urine increased 12- to 16-fold (from 1.1 ± 0.2 μg/24 h in vehicle to 12.2 ± 1.0 and 17.7 ± 1.2 μg/24 h (P<0.05 vs Vehicle) in Ac-SDKP and Ang II+Ac-SDKP respectively. The infusion of Ang II did not affect the urine excretion of Ac-SDKP (0.9 ± 0.2 μg/24 h, P= NS vs Vehicle).
Effect of Ac-SDKP on body weight, lung weight and Left Ventricle (LV) function and remodeling
None of these parameters were altered by Ac-SDKP in the controls (Table 1). Ang II reduced body weight but had no effect on lung weight. It also caused concentric hypertrophy as indicated by increased thickness of the interventricular septum (IVSTs/d) and posterior wall. Ac-SDKP failed to prevent the Ang II-induced reduction in body weight, but only partially prevented the increase in IVSTs. In addition, Ang II tended to increase the shortening fraction (SF), ejection fraction and cardiac output; and Ac-SDKP partially prevented all three increases, but only SF was statistically significant. Deceleration time of the early mitral filling wave (EDT) and E/A ratio as an indication of LV relaxation were not affected by Ang II with or without Ac-SDKP.
Table 1.
Body weight, lung weight and echocardiography
| Control | Ac-SDKP | Ang II | Ang II + Ac-SDKP | |
|---|---|---|---|---|
| Number of rats | 14 | 13 | 16 | 14 |
| BW(g) | 319 ± 7 | 312 ± 8 | 263 ± 5** | 272 ± 5 |
| LWW/TL (mg/cm) | 54.0 ± 3.5 | 61.6 ± 3.8 | 55.5 ± 2.7 | 56.0 ± 1.8 |
| HR | 383 ± 10 | 358 ± 16 | 353 ± 11 | 331 ± 11 |
| IVSTs (mm) | 2.81 ± 0.10 | 2.79 ± 0.06 | 3.29 ± 0.06*** | 2.97 ± 0.07†† |
| IVSTd (mm) | 1.59 ± 0.04 | 1.65 ± 0.04 | 1.97 ± 0.03*** | 1.89 ± 0.05 |
| PWTs (mm) | 2.79 ± 0.05 | 2.76 ± 0.08 | 3.09 ± 0.06*** | 3.07 ± 0.08 |
| PWTd (mm) | 1.63 ±0.03 | 1.66 ± 0.04 | 2.08 ± 0.05*** | 2.09 ± 0.08 |
| LVDs (mm) | 2.62 ± 0.15 | 2.96 ± 0.16 | 1.97 ± 0.14** | 2.42 ± 0.12† |
| LVDd (mm) | 5.60 ± 0.16 | 5.70 ± 0.19 | 4.81 ± 0.15** | 4.98 ± 0.19 |
| LVAs (mm2) | 6.75 ± 0.61 | 7.29 ± 0.81 | 4.83 ± 0.54* | 5.78 ± 0.72 |
| LVAd (mm2) | 24.65 ± 1.06 | 24.22 ± 1.72 | 20.40 ± 1.25* | 21.19 ± 1.16 |
| SF (%) | 53.4 ± 2.2 | 48.3 ± 2.0 | 59.3 ± 2.2 | 51.5 ± 1.2†† |
| EF (%) | 72.6 ± 2.0 | 70.6 ± 1.7 | 76.9 ± 1.5 | 73.4 ± 1.9 |
| CO (ml/min/100 g) | 29.9 ± 2.8 | 30.1 ± 3.5 | 33.8 ± 2.8 | 30.3 ± 2.1 |
| E/A ratio | 1.46 ± 0.08 | 1.58 ± 0.03 | 1.23 ± 0.12 | 1.46 ± 0.08 |
| EDT(ms) | 29.0 ± 1.7 | 28.8 ± 1.3 | 27.4 ± 1.8 | 29.3 ± 0.9 |
BW = body weight (g); HR = heart rate (beats/min); LWW = lung wet weight, TL = tibia length. IVSTs/d = interventricular septum thickness in systole and diastole; PWTs/d = posterior wall thickness in systole and diastole; LVDs/d = left ventricular dimension in systole and diastole; LVAs/d = left ventricular area in systole and diastole; SF (%) = shortening fraction; EF (%) = ejection fraction; CO = cardiac output/100 g; E/A ratio = ratio between E/A waves. EDT: deceleration time of the early mitral filling wave (ms = milliseconds).
p < 0.05,
p<0.01, and
p < 0.001, Ang II vs. control;
p < 0.05 and
p < 0.01, Ang II + Ac-SDKP vs. Ang II.
Effect of Ac-SDKP on myocardial fibrosis and collagen cross-linking
Ang II significantly increased myocardial fibrosis and this increase was prevented by Ac-SDKP; however, it had no effect on total collagen deposition in the normotensive controls (Figure 2 A–E). Ang II increased total and insoluble (cross-linked) but not soluble collagen and this was prevented by Ac-SDKP (Figure 2 F–G).
Figure 2. Effect of Ac-SDKP on Ang II-induced myocardial fibrosis and collagen crosslinking.
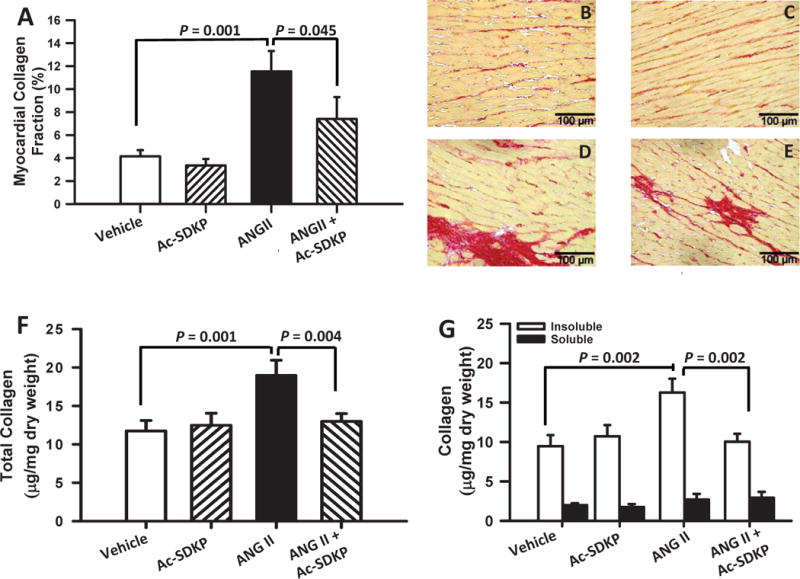
The upper panel (A) shows myocardial collagen fraction (%) quantification measured in picrosirius red staining section for all groups. Ang II significantly increased fibrosis and Ac-SDKP prevented this increase. Upper right panels (B–E) are representative images for vehicle (B), Ac-SDKP (C), Ang II (D) and Ang II + Ac-SDKP (E). Lower panels show cardiac collagen and cross-linking in rats given vehicle, Ac-SDKP, Ang II or Ang II + Ac-SDKP. F) Total collagen; G) Insoluble and soluble collagen. Ac-SDKP prevented the increase in total and insoluble (cross-linked) collagen with no apparent changes in soluble (non-cross-linked) collagen.
Effect of Ac-SDKP on cardiac lysyl oxidase, TGF-β1 expression and nuclear translocation of NFκB
Ang II significantly increased LOX mRNA and LOXL1 protein expression and this increase was prevented by Ac-SDKP (Figure 3). TGF-β1 protein expression also increased significantly and this was prevented by Ac-SDKP (Figure 4). Chronic Ang II infusion significantly increased nuclear translocation of NFκB and this was also prevented by Ac-SDKP (Figure 5). None of these parameters were influenced by Ac-SDKP in the controls.
Figure 3. Effect of Ac-SDKP on Ang II-induced LOX expression.
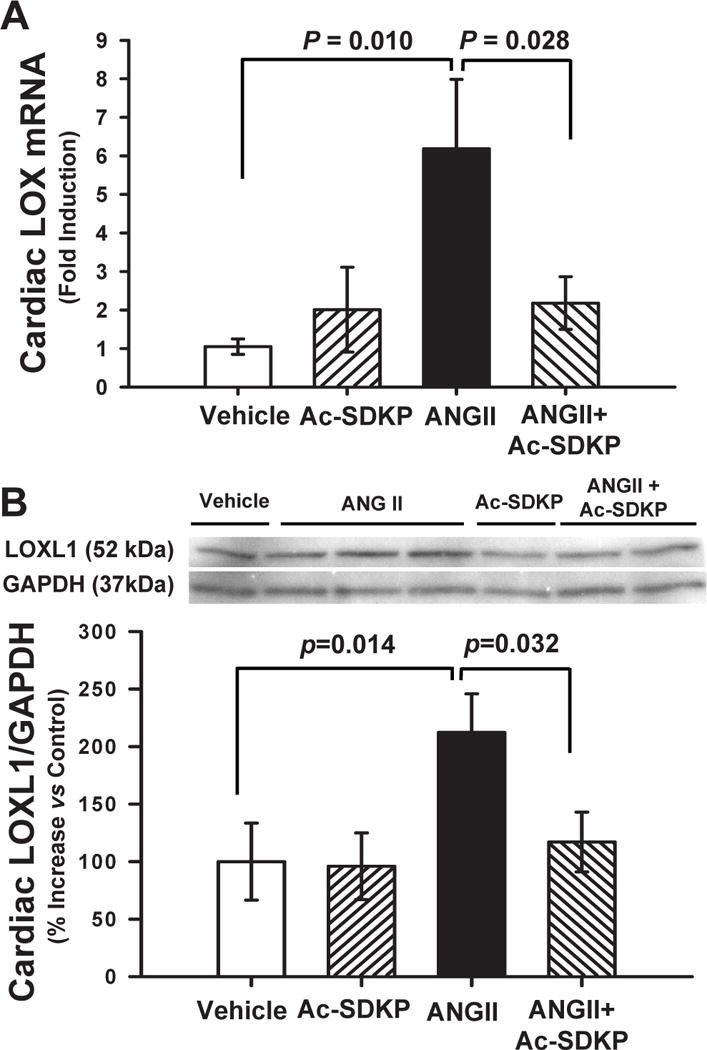
A) Cardiac LOX mRNA and B) LOXL1 protein in rats given vehicle, Ac-SDKP, Ang II or Ang II + Ac-SDKP. Ac-SDKP reduced cardiac LOX expression. In Panel B top and bottom show representative blots and quantitative analysis, respectively, for LOXL1 expression versus GAPDH.
Figure 4. Effect of Ac-SDKP on Ang II-induced cardiac TGF-β1 expression.
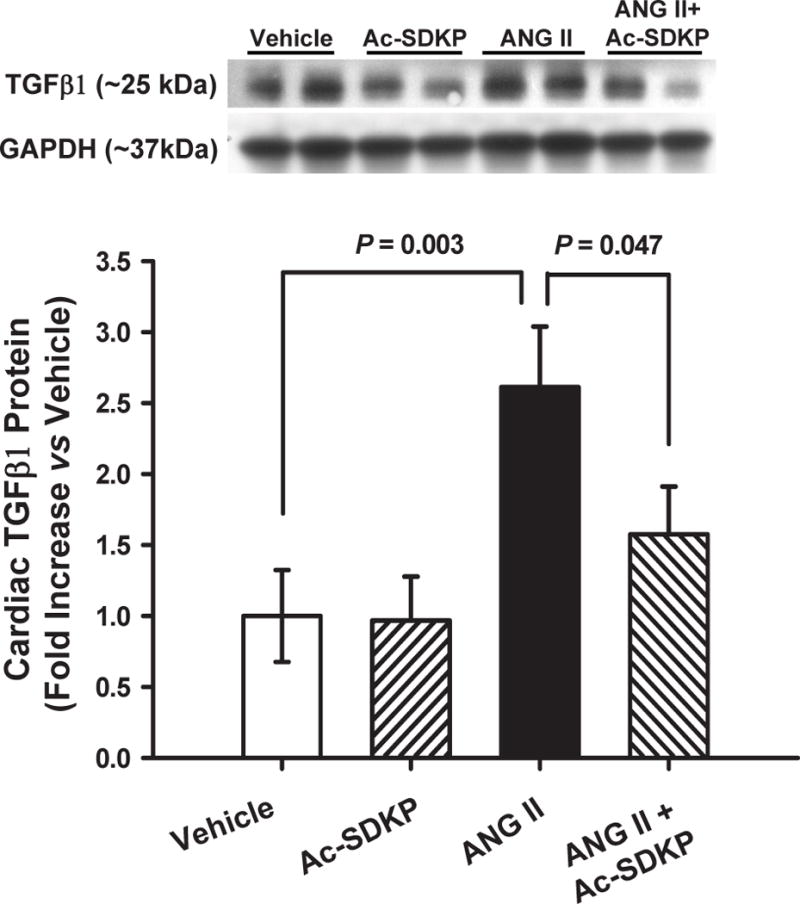
Expression of TGF-β1 in rats given vehicle, Ac-SDKP, Ang II or Ang II + Ac-SDKP. Ac-SDKP prevented cardiac TGF-β1 expression from increasing.
Figure 5. Effect of Ac-SDKP on Ang II-induced cardiac NFκB expression.
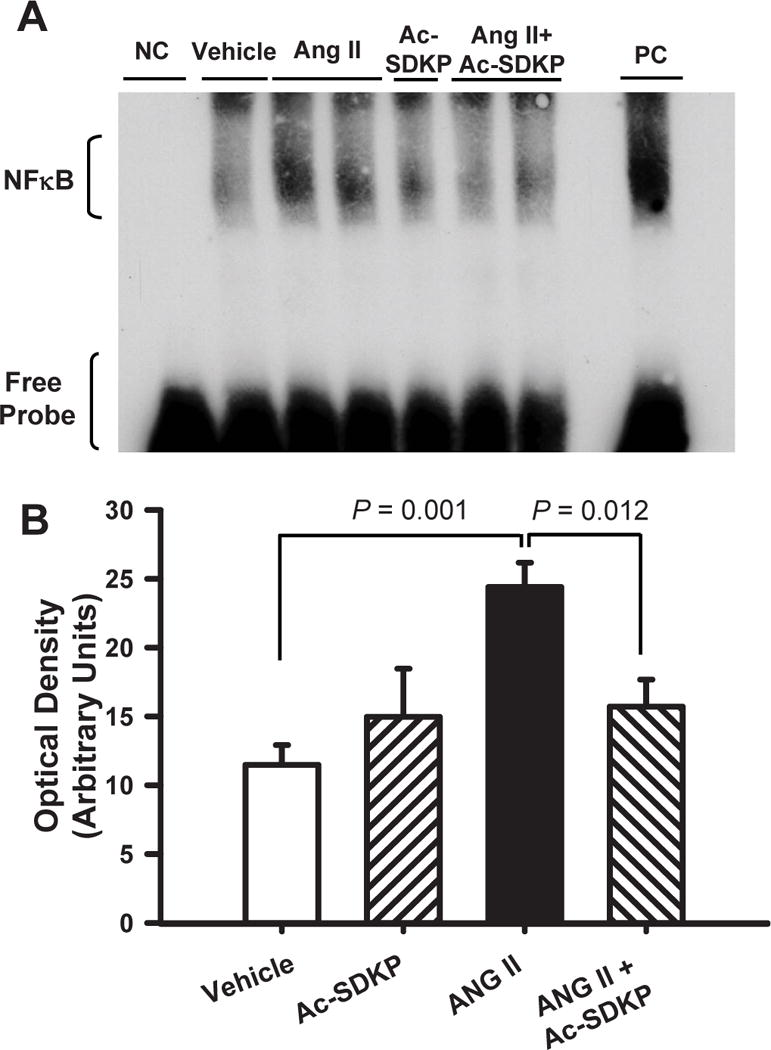
Cardiac NFκB nuclear translocation as quantified by electromobility shift assay (EMSA) in rats given vehicle, Ac-SDKP, Ang II or Ang II + Ac-SDKP. A) Representative EMSA (NC: negative control; PC: positive control) and B) NFκB quantitative analysis data. NFκB was increased at 3 weeks post-Ang II. Ac-SDKP significantly reduced cardiac nuclear translocation of NFκB.
Effect of Ac-SDKP on Ang II-induced CD4+/CD8+ lymphocyte and CD68+ macrophages infiltration
Chronic Ang II infusion increased CD4+ (Figure 6 A–E) and CD8+ (Figure 6 F–J) lymphocyte infiltration in the heart. Ac-SDKP tended to prevent the increase in CD4+ and completely prevented the increase in CD8+. Similarly, Ang II infusion increased myocardial CD68+ macrophages infiltration (Figure 6 K) and this increase was prevented with Ac-SDKP.
Figure 6. Effect of Ac-SDKP on Ang II-induced cardiac T cell infiltration.
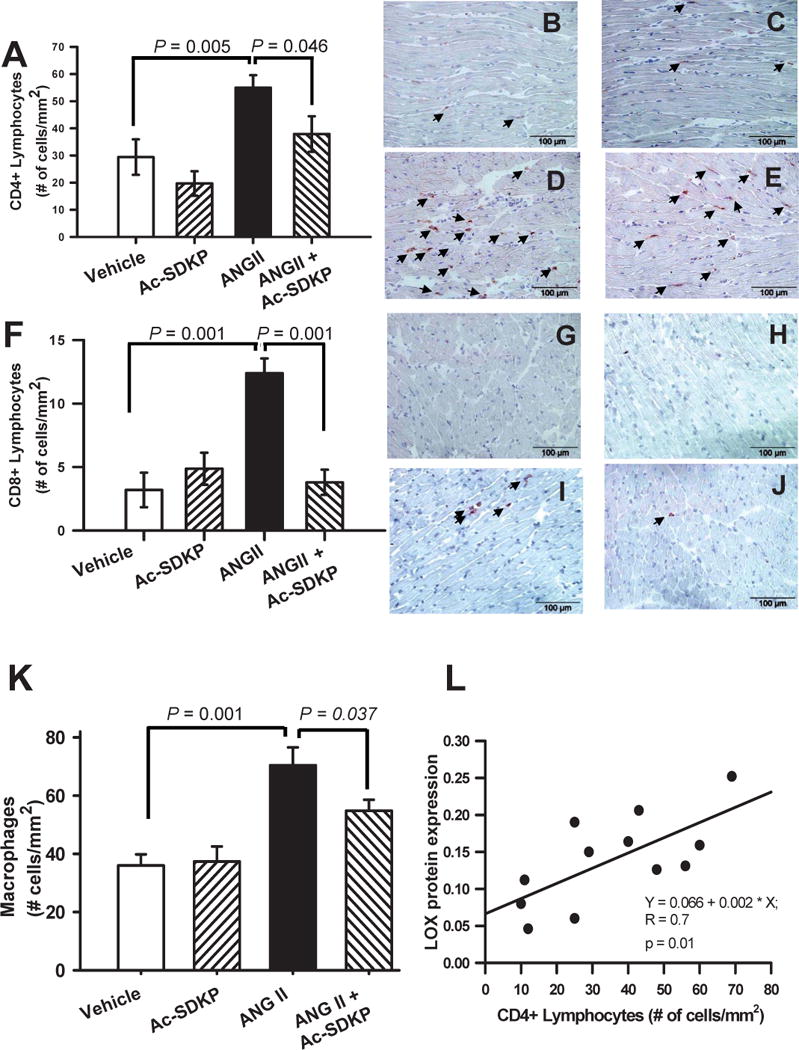
Cardiac CD4+ and CD-8 T-lymphocyte and CD68+ macrophages infiltration in rats given vehicle, Ac-SDKP, Ang II or Ang II + Ac-SDKP. Upper panels (A–E) correspond to CD4+ (T-helper) quantitative analysis (A) of the number of cardiac CD4+ lymphocytes and representative pictures for vehicle (B), Ac-SDKP (C), Ang II (D) and Ang II + Ac-SDKP (E). Middle panels (F–J) represent CD8+ (T-cytotoxic) for quantitative analysis (F) of the number of cardiac CD4+ lymphocytes and representative pictures for vehicle (G), Ac-SDKP (H), Ang II (I) and Ang II + Ac-SDKP (J). Arrows indicate stained cells (dark brown staining). Ac-SDKP prevented cardiac CD4+ and CD8+ lymphocyte infiltration. Lower panels (K) represent quantitative analysis of cardiac CD68+ cells (macrophages). Ac-SDKP prevented cardiac macrophages infiltration. Changes in the number of CD4+ lymphocytes infiltrating the myocardium correlated positively with cardiac LOXL1 expression (p = 0.01). (Lower panel (L), AU. = arbitrary units).
Correlation between CD4+ lymphocytes and LOXL1 protein expression
CD4+ lymphocyte infiltration correlated positively with LOXL1 protein expression (Figure 6 L). We found no significant correlation between CD8+ lymphocytes and LOXL1 protein expression.
DISCUSSION
The present study shows that in Ang II-induced hypertension, Ac-SDKP prevents increased total cardiac collagen, myocardial collagen cross-linking and LOXL1 as well as cardiac NFκB activation and infiltration by CD4+ (helper T cells or TH cells) and CD8+ (cytotoxic T cells). These changes occurred without lowering blood pressure (BP), indicating that the effects of Ac-SDKP are independent of BP. Decreased LOX mRNA and LOXL1 expression could be one mechanism by which Ac-SDKP prevents of the augmented collagen cross-linking. Thus, although previous studies have also documented increased LOX and collagen cross-linking in L-NAME-induced hypertension [47, 49], this is the first time that cardiac LOXL1 expression has been shown to be increased in Ang II-induced hypertension and prevented by Ac-SDKP.
Although fibrillar collagen is formed at the interstitial space, further modification ultimately leads to formation of type I and III cross-linked collagen fibers. The final step in cross-linking building, resulting in synthesis of insoluble (cross-linked) collagen [42], is catalyzed by LOX family enzymes (LOXL-1, 2, 3 and 4). LOX is a copper-dependent amine oxidase secreted as a pro-enzyme by fibrogenic cells and proteolytically cleaved in the extracellular space to produce the active enzyme [40]. Like many other enzymes, it can be regulated at three different levels: 1) mRNA; 2) protein synthesis; and 3) activity [23]. Several factors reportedly increase LOX expression, including TGF-β, platelet-derived and fibroblast growth factors, lymphocytes, NFκB, hypoxia-inducible factor 1α and shear stress [16, 23, 40, 48, 49]. Our findings primarily show that Ac-SDKP prevented hypertension-induced cardiac LOX mRNA and protein synthesis. We have previously reported that Ac-SDKP reduced target organ damage by inhibiting: fibroblast proliferation, collagen synthesis, differentiation of fibroblasts into myofibroblasts and cardiac interstitial cell proliferation [33, 36, 37], thus, the decrease in cross-linked and total collagen could be partly due to reduction of total collagen synthesis.
There is also evidence that increased collagen cross-linking exacerbates myocardial stiffness and cardiac dysfunction [3, 45]. We found that the reduction in total and cross-linked collagen induced by Ac-SDKP was associated with decreased shortening fraction in hypertensive animals. These data agree with our previous finding that in SHR treated with Ac-SDKP, reduction of total collagen was accompanied by decreased contractile (systolic) function [11]. This also agrees with Baicu et al [4] who reported that in hypertrophied papillary muscles from cats, acute regression of fibrosis impaired systolic function. Taken together, our data may suggest that in hypertension, increased collagen cross-linking provides scaffolding for myocytes and also plays a crucial role in maintaining geometric structure and coordinating contractile forces in response to increased load. Thus, without changing loading conditions such as hypertension and/or myocyte hypertrophy, but simply reducing collagen and/or collagen cross-linking may not be beneficial to the heart; on the contrary, it may jeopardize optimal cardiomyocyte interaction and function. It is also important to emphasize that the effects of reduced fibrosis on systolic function may differ during other stages of hypertrophy. The 3-week model we studied represents a compensated stage of myocardial hypertrophy in which fibrosis may play a crucial role in coordinating myocytes contractility.
Diastolic dysfunction is a complex alteration characterized by an abnormal LV filling pattern. Although the cause remains unclear [5], diastole can be altered by abnormal relaxation, stiffness, and/or changes in arterial compliance [17]. By increasing myocardial hypertrophy, cross-linked collagen and interstitial fibrosis, Ang II could increase myocardial stiffness and decrease relaxation, impairing diastolic function. We evaluated diastolic function by Doppler mitral flow velocity but observed no significant change in E/A ratio and LV stiffness [indicated by the deceleration time of the early mitral filling wave (EDT)] in response to 3 weeks of Ang II-induced hypertension in the absence and/or presence of Ac-SDKP. We also found absence of any sign for pulmonary congestion as expressed by the ratio LWW/TL ratio. This may be related to the brief treatment period which was not long enough for diastolic dysfunction to develop.
Chronic inflammation, characterized by infiltrating monocytes, macrophages and lymphocytes and increased circulating and/or local cytokines, may play an important role in the development of cardiac fibrosis [21, 44]. We and others have shown that chronic Ac-SDKP infusion reduces macrophage and mast cells infiltration (innate immunity), as well as cytokines and fibrosis [7, 21, 22, 29, 33]. Here, we confirmed that Ac-SDKP prevented macrophages infiltration and observed that Ac-SDKP also reduced cardiac infiltration by CD4+ and CD8+ lymphocytes (adaptive immunity). T lymphocytes are mobilized to sites of injury and regulate the immune response by producing cytokines that activate macrophages and other inflammatory cells. Yu et al [47] reported that cardiac tissue from hypertensive mice deficient in T and B lymphocytes exhibited decreased total and cross-linked collagen and LOX at the same time increased MMP-2, which strongly suggests that lymphocytes play a prominent role in ECM remodeling in hypertension. The role of T-lymphocytes in ECM remodeling is also supported by Shi et al [39] in CCI4-induced hepatic injury. Lymphocytes may induce fibrosis by stimulating release of TGF-β1, one of the most important cytokines in the regulation of fibrosis and LOX expression in the heart [23, 46]. Here we showed that Ac-SDKP prevented Ang II-induced upregulation of TGF-β1 and cardiac increased of CD4+ (T-helper) and CD8+ (T-cytotoxic) lymphocyte infiltration. Simultaneously, Ac-SDKP reduced LOXL1 and total and cross-linked collagen. Therefore, we believe that by lowering both TGF-β1 and T-lymphocyte infiltration, Ac-SDKP prevents the increase in cardiac LOX mRNA and LOXL1 protein expression, thereby reducing cross-linked collagen. This reasoning is further supported by Kvakan et al who showed that adoptive transfer immunosuppressive T regulatory (Treg) cells exerted an anti-fibrotic effect in mice infused with Ang II [19]. While we did not conduct in vitro experiments testing whether Ac-SDKP reduces LOXL1 activity by inhibiting T-lymphocytes, our results are supported by Yu et al [48] who showed both in vitro and in vivo that increasing TH1 lymphocyte activity directly enhances both LOX and cross-linked collagen. Because CD4+ lymphocyte infiltration correlates positively with LOXL1 expression it seems likely that Ac-SDKP can modulate ECM remodeling at least in part by preventing T-lymphocyte infiltration.
Here, we also report for the first time that nuclear translocation of NFκB, a transcription factor that plays a pivotal role in inflammation and healing [27, 30], was increased in Ang II-induced hypertension, and prevented by Ac-SDKP. Blockade of NFκB resulted in reduced lymphocyte and macrophage infiltration [25] and cardiac fibrosis after myocardial infarction [18]. The link between reduced NFκB and lymphocyte infiltration is supported by the fact that development, proliferation and infiltration of CD4+ T-lymphocytes depends on NFκB [2, 13]. Thus, our findings may suggest that the anti-inflammatory effects of Ac-SDKP in the heart could be mediated by reducing NFκB activity. According to the present and previous reports in other models [15, 41], it is also possible that NFκB could represent a key downstream signaling for TGF-β1’s effects. This further supports the concept that by blocking NFκB activity and TGF-β expression, Ac-SDKP afforded cardiac protection from inflammation and fibrosis, respectively (Figure 2 supplemental data). However, currently there are not conclusive evidences for supporting the notion that NF-κB regulates Ang II-induced LOX expression. Recent finding from Adams et al [1] showed that Ang II increases LOX via activation of the small G protein Rac1-GTPase and the profibrotic connective tissue growth factor. Furthermore, other studies suggested that LOX gene regulation may be mediated by advanced glycation end products through binding of NFκB and activator protein-1 to the LOX promoter [23, 31]. Thus, it may be that different dependent or independent pathways of NFκB regulate cardiac LOX genes expression. Further investigations are warranty for investigate the pathway by which Ac-SDKP regulates Ang II induced-cardiac LOX expression.
Study limitations
1) Determination of LOX activity. We are aware that findings of increased LOX expression could be strengthened by showing LOX activity. Unfortunately, we were unable to determine LOX activity and we were limited to measurements of LOX mRNA and LOXL1 protein expression instead. Nevertheless, LOX protein expression reportedly correlates very well with LOX activity, and LOXL1 has 85 % homology to LOX [10, 23]; thus, increased LOX expression may serve as an indication of LOX activation.
In conclusion, in Ang II-induced hypertension chronic Ac-SDKP infusion prevented increases in: 1) insoluble and total collagen and cardiac LOX mRNA and LOXL1 expression, which may explain the low levels of cross-linked and total collagen; 2) TGF-β1, which is a known potent fibrogenic growth factor; 3) nuclear translocation of NFκB, which is considered to play a pivotal role in inflammation; and 4) cardiac infiltration of T-lymphocytes, which together with reduction of TGF-β1 may contribute to reduced LOXL1, cross-linked and total collagen. Taken together, our findings support the hypothesis that Ac-SDKP is an endogenous immunomodulator that acts in several ways to prevent target organ damage when cardiac inflammation and fibrosis develop. Central to this effect is reduction of NFκB, which in turn lowers both innate and adaptive immunity.
Clinical Perspectives
In Ang II-induced hypertension treatment with an infusion of Ac-SDKP prevented increases in collagen cross-linking and total collagen, TGF-β1, LOXL1 and lymphocyte and macrophages infiltration, without decreasing blood pressure. Ac-SDKP analogues that are resistant to degradation by ACE or other enzymes could be important not only in the treatment of end organ damage in hypertension but also in other diseases associated with increased collage deposition and activation of the innate and adaptive immunity such as systemic lupus erythematous, autoimmune myocarditis, pulmonary fibrosis and cirrhosis of the liver.
Supplementary Material
Acknowledgments
The technical assistance of Gulser Gurocak, Mingzhu Tian and Derek Smolarek is greatly appreciated.
SOURCES OF FUNDING
This work was supported by NIH grants HL-028982 to OAC and HL-071806 to NER.
Footnotes
CONFLICT(S) OF INTEREST/DISCLOSURE(S) STATEMENT
None
Authors Contribution to the manuscript
Germán E. GONZÁLEZ: performed most of the animal experiments, autopsy, and data analysis and wrote the manuscript.
Nour-Eddine RHALEB: performed data analysis, wrote and corrected the manuscript. Pablo NAKAGAWA: Participated in the experiments and data analysis.
Tang-Dong LIAO: Performed the Immunohistochemistry
Yunhe LIU: Performed the echocardiogram and participated in the experiments.
Pablo LEUNG: Participated in the experiments and data analysis.
Xiangguo DAI: Performed slides preparation, immunohistochemistry and quantification of macrophages.
Xiao-Ping YANG: Performed the echocardiography and participated in the data analysis.
Oscar A. CARRETERO: Directed all the project and wrote the manuscript
References
- 1.Adam O, Theobald K, Lavall D, Grube M, Kroemer HK, Ameling S, Schafers HJ, Bohm M, Laufs U. Increased lysyl oxidase expression and collagen cross-linking during atrial fibrillation. J Mol Cell Cardiol. 2011;50:678–685. doi: 10.1016/j.yjmcc.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 2.Artis D, Speirs K, Joyce K, Goldschmidt M, Caamano J, Hunter CA, Scott P. NF-kappa B1 is required for optimal CD4+ Th1 cell development and resistance to Leishmania major. J Immunol. 2003;170:1995–2003. doi: 10.4049/jimmunol.170.4.1995. [DOI] [PubMed] [Google Scholar]
- 3.Badenhorst D, Maseko M, Tsotetsi OJ, Naidoo A, Brooksbank R, Norton GR, Woodiwiss AJ. Cross-linking influences the impact of quantitative changes in myocardial collagen on cardiac stiffness and remodelling in hypertension in rats. Cardiovasc Res. 2003;57:632–641. doi: 10.1016/s0008-6363(02)00733-2. [DOI] [PubMed] [Google Scholar]
- 4.Baicu CF, Stroud JD, Livesay VA, Hapke E, Holder J, Spinale FG, Zile MR. Changes in extracellular collagen matrix alter myocardial systolic performance. Am J Physiol Heart Circ Physiol. 2003;284:H122–H132. doi: 10.1152/ajpheart.00233.2002. [DOI] [PubMed] [Google Scholar]
- 5.Burkhoff D, Maurer MS, Packer M. Heart failure with a normal ejection fraction. Is it really a disorder of diastolic function? [editorial] Circulation. 2003;107:656–658. doi: 10.1161/01.cir.0000053947.82595.03. [DOI] [PubMed] [Google Scholar]
- 6.Burlew BS, Weber KT. Connective tissue and the heart. Functional significance and regulatory mechanisms. Cardiol Clin. 2000;18:435–442. doi: 10.1016/s0733-8651(05)70154-5. [DOI] [PubMed] [Google Scholar]
- 7.Castoldi G, di Gioia CR, Bombardi C, Perego C, Perego L, Mancini M, Leopizzi M, Corradi B, Perlini S, Zerbini G, Stella A. Prevention of myocardial fibrosis by N-acetyl-seryl-aspartyl-lysyl-proline in diabetic rats. Clin Sci (Lond) 2010;118:211–220. doi: 10.1042/cs20090234. [DOI] [PubMed] [Google Scholar]
- 8.Cavasin MA. Therapeutic Potential of Thymosin-beta4 and its Derivative N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) in Cardiac Healing After Infarction. Am J Cardiovasc Drugs. 2006;6:305–311. doi: 10.2165/00129784-200606050-00003. [DOI] [PubMed] [Google Scholar]
- 9.Cavasin MA, Liao TD, Yang XP, Yang JJ, Carretero OA. Decreased endogenous levels of Ac-SDKP promote organ fibrosis. Hypertension. 2007;50:130–136. doi: 10.1161/HYPERTENSIONAHA.106.084103. [DOI] [PubMed] [Google Scholar]
- 10.Chen LJ, Zhao Y, Gao S, Chou IN, Toselli P, Stone P, Li W. Downregulation of lysyl oxidase and upregulation of cellular thiols in rat fetal lung fibroblasts treated with cigarette smoke condensate. Toxicol Sci. 2005;83:372–379. doi: 10.1093/toxsci/kfi019. [DOI] [PubMed] [Google Scholar]
- 11.Cingolani OH, Yang XP, Liu YH, Villanueva M, Rhaleb NE, Carretero OA. Reduction of cardiac fibrosis decreases systolic performance without affecting diastolic function in hypertensive rats. Hypertension. 2004;43:1067–1073. doi: 10.1161/01.HYP.0000125013.22494.c5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cleutjens JPM, Kandala JC, Guarda E, Guntaka RV, Weber KT. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995;27:1281–1292. doi: 10.1016/s0022-2828(05)82390-9. [DOI] [PubMed] [Google Scholar]
- 13.Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A. A critical role for NF-kappa B in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat Immunol. 2001;2:45–50. doi: 10.1038/83158. [DOI] [PubMed] [Google Scholar]
- 14.Fujihara CK, Antunes GR, Mattar AL, Malheiros DM, Vieira JM, Jr, Zatz R. Chronic inhibition of nuclear factor-kappaB attenuates renal injury in the 5/6 renal ablation model. Am J Physiol Renal Physiol. 2007;292:F92–F99. doi: 10.1152/ajprenal.00184.2006. [DOI] [PubMed] [Google Scholar]
- 15.Gingery A, Bradley EW, Pederson L, Ruan M, Horwood NJ, Oursler MJ. TGF-beta coordinately activates TAK1/MEK/AKT/NFkB and SMAD pathways to promote osteoclast survival. Exp Cell Res. 2008;314:2725–2738. doi: 10.1016/j.yexcr.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawaguchi M, Hay I, Fetics B, Kass DA. Combined ventricular systolic and arterial stiffening in patients with heart failure and preserved ejection fraction. Implications for systolic and diastolic reserve limitations. Circulation. 2003;107:714–720. doi: 10.1161/01.cir.0000048123.22359.a0. [DOI] [PubMed] [Google Scholar]
- 18.Kawano S, Kubota T, Monden Y, Tsutsumi T, Inoue T, Kawamura N, Tsutsui H, Sunagawa K. Blockade of NF-kappaB improves cardiac function and survival after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H1337–H1344. doi: 10.1152/ajpheart.01175.2005. [DOI] [PubMed] [Google Scholar]
- 19.Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, Rahn HP, Plehm R, Wellner M, Elitok S, Gratze P, Dechend R, Luft FC, Muller DN. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119:2904–2912. doi: 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- 20.Liao TD, Yang XP, D’Ambrosio M, Zhang Y, Rhaleb NE, Carretero OA. N-acetyl-seryl-aspartyl-lysyl-proline attenuates renal injury and dysfunction in hypertensive rats with reduced renal mass: Council for High Blood Pressure Research. Hypertension. 2010;55:459–467. doi: 10.1161/HYPERTENSIONAHA.109.144568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin CX, Rhaleb NE, Yang XP, Liao TD, D’Ambrosio MA, Carretero OA. Prevention of aortic fibrosis by N-acetyl-seryl-aspartyl-lysyl-proline in angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol. 2008;295:H1253–H1261. doi: 10.1152/ajpheart.00481.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu YH, D’Ambrosio M, Liao TD, Peng H, Rhaleb NE, Sharma U, Andre S, Gabius HJ, Carretero OA. N-acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling and dysfunction induced by galectin-3, a mammalian adhesion/growth-regulatory lectin. Am J Physiol Heart Circ Physiol. 2009;296:H404–H412. doi: 10.1152/ajpheart.00747.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez B, Gonzalez A, Hermida N, Valencia F, de Teresa E, Diez J. Role of lysyl oxidase in myocardial fibrosis. From basic science to clinical aspects. Am J Physiol Heart Circ Physiol. 2010;299:H1–H9. doi: 10.1152/ajpheart.00335.2010. [DOI] [PubMed] [Google Scholar]
- 24.Mann DL, Spinale FG. Activation of matrix metalloproteinases in the failing human heart: breaking the tie that binds. Circulation. 1998;98:1699–1702. doi: 10.1161/01.cir.98.17.1699. [DOI] [PubMed] [Google Scholar]
- 25.Marfella R, D’Amico M, Di Filippo C, Baldi A, Siniscalchi M, Sasso FC, Portoghese M, Carbonara O, Crescenzi B, Sangiuolo P, Nicoletti GF, Rossiello R, Ferraraccio F, Cacciapuoti F, Verza M, Coppola L, Rossi F, Paolisso G. Increased activity of the ubiquitin-proteasome system in patients with symptomatic carotid disease is associated with enhanced inflammation and may destabilize the atherosclerotic plaque: effects of rosiglitazone treatment. J Am Coll Cardiol. 2006;47:2444–2455. doi: 10.1016/j.jacc.2006.01.073. [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee D, Sen S. Collagen phenotypes during development and regression of myocardial hypertrophy in spontaneously hypertensive rats. Circ Res. 1990;67:1474–1480. doi: 10.1161/01.res.67.6.1474. [DOI] [PubMed] [Google Scholar]
- 27.Muller DN, Dechend R, Mervaala EMA, Park JK, Schmidt F, Fiebeler A, Theuer J, Breu V, Ganten D, Haller H, Luft FC. NF-κB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension. 2000;35:193–201. doi: 10.1161/01.hyp.35.1.193. [DOI] [PubMed] [Google Scholar]
- 28.Norton GR, Tsotetsi J, Trifunovic B, Hartford C, Candy GP, Woodiwiss AJ. Myocardial stiffness is attributed to alterations in cross-linked collagen rather than total collagen or phenotypes in spontaneously hypertensive rats. Circulation. 1997;96:1991–1998. doi: 10.1161/01.cir.96.6.1991. [DOI] [PubMed] [Google Scholar]
- 29.Omata M, Taniguchi H, Koya D, Kanasaki K, Sho R, Kato Y, Kojima R, Haneda M, Inomata N. N-acetyl-seryl-aspartyl-lysyl-proline ameliorates the progression of renal dysfunction and fibrosis in WKY rats with established anti-glomerular basement membrane nephritis. J Am Soc Nephrol. 2006;17:674–685. doi: 10.1681/ASN.2005040385. [DOI] [PubMed] [Google Scholar]
- 30.Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol. 2007;292:F330–F339. doi: 10.1152/ajprenal.00059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papachroni KK, Piperi C, Levidou G, Korkolopoulou P, Pawelczyk L, Diamanti-Kandarakis E, Papavassiliou AG. Lysyl oxidase interacts with AGEs signaling to modulate collagen synthesis in polycystic ovarian tissue. J Cell Mol Med. 2010;14:2460–2469. doi: 10.1111/j.1582-4934.2009.00841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng H, Carretero OA, Liao TD, Peterson EL, Rhaleb NE. Role of N-acetyl-seryl-aspartyl-lysyl-proline in the antifibrotic and anti-inflammatory effects of the angiotensin-converting enzyme inhibitor Captopril in hypertension. Hypertension. 2007;49:695–703. doi: 10.1161/01.HYP.0000258406.66954.4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng H, Carretero OA, Peterson EL, Rhaleb NE. Ac-SDKP inhibits transforming growth factor-betal-induced differentiation of human cardiac fibroblasts into myofibroblasts. Am J Physiol Heart Circ Physiol. 2010;298:H1357–H1364. doi: 10.1152/ajpheart.00464.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng H, Carretero OA, Raij L, Yang F, Kapke A, Rhaleb NE. Antifibrotic effects of N-acetyl-seryl-aspartyl-lysyl-proline on the heart and kidney in aldosterone-salt hypertensive rats. Hypertension. 2001;37:794–800. doi: 10.1161/01.hyp.37.2.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng H, Carretero OA, Vuljaj N, Liao TD, Motivala A, Peterson EL, Rhaleb NE. Angiotensin-Converting enzyme inhibitors: a new mechanism of action. Circulation. 2005;112:2436–2445. doi: 10.1161/CIRCULATIONAHA.104.528695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhaleb NE, Peng H, Harding P, Tayeh M, LaPointe MC, Carretero OA. Effect of N-acetyl-seryl-aspartyl-lysyl-proline on DNA and collagen synthesis in rat cardiac fibroblasts. Hypertension. 2001;37:827–832. doi: 10.1161/01.hyp.37.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhaleb NE, Peng H, Yang XP, Liu YH, Mehta D, Ezan E, Carretero OA. Long-term effect of N-acetyl-seryl-aspartyl-lysyl-proline on left ventricular collagen deposition in rats with 2-kidney, 1-clip hypertension. Circulation. 2001;103:3136–3141. doi: 10.1161/01.cir.103.25.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rhaleb NE, Pokharel S, Sharma U, Carretero OA. Renal protective effects of N-acetyl-Ser-Asp-Lys-Pro in deoxycorticosterone acetate-salt hypertensive mice. J Hypertens. 2011;29:330–338. doi: 10.1097/HJH.0b013e32834103ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Z, Wakil AE, Rockey DC. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc Natl Acad Sci U S A. 1997;94:10663–10668. doi: 10.1073/pnas.94.20.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith-Mungo LI, Kagan HM. Lysyl oxidase: properties, regulation and multiple functions in biology. Matrix Biol. 1998;16:387–398. doi: 10.1016/s0945-053x(98)90012-9. [DOI] [PubMed] [Google Scholar]
- 41.Tobar N, Villar V, Santibanez JF. ROS-NFkappaB mediates TGF-beta1-induced expression of urokinase-type plasminogen activator, matrix metalloproteinase-9 and cell invasion. Mol Cell Biochem. 2010;340:195–202. doi: 10.1007/s11010-010-0418-5. [DOI] [PubMed] [Google Scholar]
- 42.Trackman PC. Diverse biological functions of extracellular collagen processing enzymes. J Cell Biochem. 2005;96:927–937. doi: 10.1002/jcb.20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weber KT, Sun Y, Tyagi SC, Cleutjens JP. Collagen network of the myocardium: function, structural remodeling and regulatory mechanisms. J Mol Cell Cardiol. 1994;26:279–292. doi: 10.1006/jmcc.1994.1036. [DOI] [PubMed] [Google Scholar]
- 44.Wei L. Immunological aspect of cardiac remodeling: T lymphocyte subsets in inflammation-mediated cardiac fibrosis. Exp Mol Pathol. 2011;90:74–78. doi: 10.1016/j.yexmp.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 45.Woodiwiss AJ, Tsotetsi OJ, Sprott S, Lancaster EJ, Mela T, Chung ES, Meyer TE, Norton GR. Reduction in myocardial collagen cross-linking parallels left ventricular dilatation in rat models of systolic chamber dysfunction. Circulation. 2001;103:155–160. doi: 10.1161/01.cir.103.1.155. [DOI] [PubMed] [Google Scholar]
- 46.Yang F, Yang XP, Liu YH, Xu J, Cingolani O, Rhaleb NE, Carretero OA. Ac-SDKP reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension. 2004;43:229–236. doi: 10.1161/01.HYP.0000107777.91185.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu Q, Horak K, Larson DF. Role of T lymphocytes in hypertension-induced cardiac extracellular matrix remodeling. Hypertension. 2006;48:98–104. doi: 10.1161/01.HYP.0000227247.27111.b2. [DOI] [PubMed] [Google Scholar]
- 48.Yu Q, Vazquez R, Zabadi S, Watson RR, Larson DF. T-lymphocytes mediate left ventricular fibrillar collagen cross-linking and diastolic dysfunction in mice. Matrix Biol. 2010;29:511–518. doi: 10.1016/j.matbio.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zibadi S, Vazquez R, Moore D, Larson DF, Watson RR. Myocardial lysyl oxidase regulation of cardiac remodeling in a murine model of diet-induced metabolic syndrome. Am J Physiol Heart Circ Physiol. 2009;297:H976–H982. doi: 10.1152/ajpheart.00398.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.