

Abstract
Mitochondrial dysfunction causes a range of early-onset neurological diseases and contributes to neurodegenerative conditions. The mechanisms of neurological damage however are poorly understood, as accessing relevant tissue from patients is difficult, and appropriate models are limited. Hence, we assessed mitochondrial function in neurologically relevant primary cell lines from a CI (complex I) deficient Ndufs4 KO (knockout) mouse (Ndufs4fky/fky) modelling aspects of the mitochondrial disease LS (Leigh syndrome), as well as MEFs (mouse embryonic fibroblasts). Although CI structure and function were compromised in all Ndufs4fky/fky cell types, the mitochondrial membrane potential was selectively impaired in the MEFs, correlating with decreased CI-dependent ATP synthesis. In addition, increased ROS (reactive oxygen species) generation and altered sensitivity to cell death were only observed in Ndufs4fky/fky primary MEFs. In contrast, Ndufs4fky/fky primary isocortical neurons and primary isocortical astrocytes displayed only impaired ATP generation without mitochondrial membrane potential changes. Therefore the neurological dysfunction in the Ndufs4fky/fky mouse may partly originate from a more severe ATP depletion in neurons and astrocytes, even at the expense of maintaining the mitochondrial membrane potential. This may provide protection from cell death, but would ultimately compromise cell functionality in neurons and astrocytes. Furthermore, RET (reverse electron transfer) from complex II to CI appears more prominent in neurons than MEFs or astrocytes, and is attenuated in Ndufs4fky/fky cells.
Keywords: metabolic stress, mitochondrial disease, mouse models, neuropathology, primary cells, reactive oxygen species
Abbreviations: BN-PAGE, blue native-PAGE; CI–CV, complex I–complex V; CS, citrate synthase; CV, complex V; DHE, dihydroethidium; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; H2O2, hydrogen peroxide; HBSS, Hanks buffered saline solution; KO, knockout; LS, Leigh syndrome; MEF, mouse embryonic fibroblast; NBM, neurobasal medium; OXPHOS, oxidative phosphorylation; PI, propidium iodide; RET, reverse electron transfer; ROS, reactive oxygen species; ΔΨm, mitochondrial membrane potential
Short abstract
We describe how the metabolic response in neurologically relevant primary astrocytes and neurons diverges from that of mouse embryonic fibroblasts isolated from the Ndufs4fky/fky mouse model of mitochondrial disease.
INTRODUCTION
Mitochondrial diseases are a collection of rare disorders, each featuring mitochondrial dysfunction, estimated to affect 1 in 5000 live births [1]. Presentation of the disease is diverse, potentially involving any tissue, with any severity and at any age [2]. Typically however, there is involvement of the more metabolically active tissues displaying high-energy demand, notably brain [3,4]. The genetic basis of the disease is also complex, with pathogenic mutations occurring in over 100 genes required to make a functional mitochondrion, including the OXPHOS (oxidative phosphorylation) system [4,5]. Although mutations in disease related OXPHOS genes are associated with a broad range of classical mitochondrial disorders, one of the most common of these is LS (Leigh syndrome) [4].
LS is a progressive neurodegenerative disorder where patients exhibit a broad range of neurologically based symptoms including ataxia, breathing abnormalities, optic atrophy, developmental retardation, hearing impairment and seizures [6–8]. Pathologically, the disease is characterized by symmetrical bilateral lesions in the basal ganglia, thalamus, or brain stem, increased lactate in the blood and/or cerebral spinal fluid, gliosis and neuronal demyelination [6–8]. Although the gross biochemical and clinical manifestations of LS are well characterized, the difficulty in obtaining fresh patient samples from neurological regions of interest has limited our understanding of the biochemical features at a cellular level.
To this end, a number of animal and cellular model systems of primary mitochondrial dysfunction have recently been generated (reviewed in [9,10]), including the Ndufs4fky/fky and Ndufs4 KO (knockout) mice lacking the OXPHOS CI (complex I) subunit NDUFS4 [11,12]. Like LS patients, the NDUFS4 deficient mice develop hyperintense bilateral lesions of the brain stem [13,14]. The mice also exhibit clinical manifestations similar to those observed in LS patients [11–14]. Moreover, the brain-specific Ndufs4 KO mouse is nearly biochemically and phenotypically indistinguishable from the whole animal KO, confirming the neurological basis of the disease and its clinical features [13]. As the NDUFS4 deficient mice have recapitulated many of the common features of LS, these mice provide the opportunity to examine how mitochondrial systems and dynamics are affected in a neurological setting.
So far, analysis of whole-brain preparations from NDUFS4-deficient mice [12,13] has revealed that, like NDUFS4-deficient patients and mouse fibroblasts [15,16], CI structure and function is disrupted. In each system, CI forms an ≈830 kDa crippled complex on BN-PAGE (blue native-PAGE). Furthermore, the CI defect was found to affect CI-dependent ATP synthesis in isolated brain mitochondria [12]. There is also evidence of oxidative damage to proteins in the olfactory bulb of NDUFS4-deficient mice, one of the areas of the brain affected by the disease [13]. The latter is of particular interest considering that NDUFS4-deficient patient and adult mouse fibroblasts produce elevated amounts of ROS (reactive oxygen species), including superoxide (O2•−) and H2O2 (hydrogen peroxide) [17–19]. It therefore seems reasonable to expect that disruption of CI in neurologically relevant cell types will also result in increased production of ROS, thus contributing to neuropathogenesis. However, in Ndufs4 KO mouse primary mesencephalic neurons, O2•− production in isolated mitochondria, and H2O2 production in whole cells was normal relative to controls [20,21].
Accordingly, characterizing cell-type-specific mechanisms of disease, such as ROS dynamics, will increase our understanding of the modes of disease pathogenesis in LS. To this end, we sought to isolate and characterize primary isocortical astrocytes and isocortical neurons from the Ndufs4fky/fky mouse. Despite astrocytes being the most abundant cell type in the mammalian brain [22,23], their role in mitochondrial disease is often overlooked. Astrocytes are metabolically intricately linked to neurons for proper cellular function through processes such as neurotransmitter recycling and extracellular potassium homoeostasis [23,24]. Furthermore, they have been implicated in disease progression in both LS patients and NDUFS4-deficient mice, where an infiltration of activated astrocytes into affected brain regions is a feature in both systems [7,13]. In conjunction, we also investigated mitochondrial function in Ndufs4fky/fky MEFs (mouse embryonic fibroblasts), which serve as a control cell type from a non-neurological tissue for our studies. Our examination of the OXPHOS system and associated mitochondrial processes in these primary Ndufs4fky/fky astrocytes, neurons and MEFs thus provided the opportunity to examine disease mechanisms at a cell-type-specific level.
EXPERIMENTAL
Mouse model
The Ndufs4fky/fky mouse was generated by the spontaneous insertion of the B2 SINE (Short Interspersed Nuclear Element) into the NADH dehydrogenase (ubiquinone) Fe–S protein 4 (Ndufs4) gene as previously described [12], and maintained on a BALB/c background. Heterozygous mice were bred for the generation of pups and embryos according to an approved protocol by the Murdoch Childrens Research Institute Animal Ethics Committee (A662). Mice were housed on 12 h light/dark cycles and provided ad libitum access to food and water.
Cell culture methods
Primary isocortical astrocytes and isocortical neurons were isolated from individual 3–5-day-old pups and 17–18-day post-fertilization embryo isocortices, respectively, as previously described [25,26]. Briefly, tissues were collected in HBSS (Hanks buffered saline solution) without Mg2+ and Ca2+ (Gibco), and digested in 0.6% (w/v) trypsin with 2.78 mM glucose [30 min, 37°C, 5% (v/v) CO2]. Following trypsin neutralization with 1 mg/ml soya trypsin inhibitor (Invitrogen) containing 100 units/ml DNAse I (Invitrogen), cells were mechanically dissociated and diluted in HBSS containing Mg2+ and Ca2+. Isolated astrocytes were diluted in DMEM containing 25 mM glucose (abbreviated in text as ‘glucose’ medium; Thermo scientific) supplemented with 10% (v/v) FCS, and plated in flasks pre-coated with 50 μg/ml poly D-lysine. At confluency, astrocyte cultures were shaken at 200 rpm, 37°C for 14–16 h to remove contaminating cell types. Isolated neurons were diluted in NBM (neurobasal medium; Invitrogen) supplemented with B-27 (Invitrogen), 292 mg/l L-glutamine, and plated in flasks pre-coated with 50 μg/ml poly D-lysine, or coverslips pre-coated with both 50 μg/ml poly D-lysine and 10 μg/ml laminin. The purity of neuronal and astrocyte cultures was assessed by immunocytochemistry (Supplemental Materials and methods section and Supplementary Figure S1).
Primary MEFs were isolated from day 12.5–14.5 post-fertilization embryos. After the removal of the head and visceral organs, tissues were disrupted mechanically and subsequently digested with 0.125% trypsin (30 min, 37°C, 5% CO2). Cells were then mechanically dissociated, pelleted, and plated in glucose medium in culture flasks coated with 0.1% gelatin.
Cells were maintained at 37°C with 5% CO2 in a humidified chamber. All cell isolation buffers (HBSS) and culture media contained 60.3 mg/l penicillin and 100 mg/l streptomycin. mtDNA (mitochondrial DNA) deficient (ρ°) and control (ρ+) mouse LM(TK−) cells [27] were maintained on glucose medium (described above) with 50 μg/ml uridine. Where described, astrocytes and MEFs were alternatively cultured in DMEM containing 5 mM galactose (abbreviated in text as ‘galactose’ medium; Gibco) supplemented with 10% dialysed FCS and 292 mg/l L-glutamine.
Mitochondrial isolation
Mitochondria were isolated from primary cells as previously described [28].
CI and CS (citrate synthase) activity
CI and CS activities in primary cell cultures were determined relative to total protein at 30°C as previously described [29]. Briefly, cells were homogenized in buffer (200 mM mannitol, 70 mM sucrose, 5 mM HEPES free acid, 1 mM EGTA, pH 7.2) with a Teflon-glass homogenizer in combination with a benchtop drill at 1000 rpm. Debris was removed by a low speed spin at 600 g. For CI activity measurements, the samples were diluted in buffer containing CoenzymeQ1, NADH, potassium cyanide and antimycin A in the presence or absence of rotenone. The activity of CI was measured in a spectrophotometer as the rotenone-sensitive rate of NADH oxidation at 340 nm over 3 min. For CS activity measurements, sample was diluted in buffer containing 5,5′-dithio-bis-(2-nitrobenzoic acid), oxaloacetate and acetyl CoA. The rate of thionitrobenzoate anion production was monitored in a spectrophotometer over 3 min at 412 nm.
Western blot analysis
Primary cell pellets were solubilized in RIPA buffer [50 mM Tris/HCl, 150 mM NaCl, 1% (v/v) Triton X-100, 1% (w/v) Na deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA, pH to 7.4] containing protease inhibitor (Roche), diluted in LDS (lithium dodecyl sulfate) sample buffer (Invitrogen), heat denatured (10 min, 60°C) and separated on 10% Novex bis-tris gels (Invitrogen). Gels were transferred to 0.45 μm PVDF membrane (Millipore) for immunodecoration. Membranes were probed with anti-NDUFS4 (MitoSciences, MS104) or anti-SDHA (Molecular Probes, A-11142).
BN-PAGE
Cell pellets were solubilized in either 1% digitonin or 1% Triton X-100 and protein complexes separated by BN-PAGE as described [30]. Briefly, 50–80 μg of whole cell lysate was separated per lane on 4–10 or 4–13% BN-PAGE gels (Invitrogen). Gels were transferred to PVDF membrane (Millipore) for immunodecoration. Membranes were probed with antibodies against the CI subunit NDUFA9 [31], CII subunit SDHA (Invitrogen) or complex III subunit Core1 (Invitrogen).
ATP synthesis assay
ATP synthesis rates were measured in technical duplicates in primary cell cultures and isolated mitochondria as described [32]. Briefly, 10 μg of cells, or 2 μg isolated mitochondria were diluted 10-fold in ATP synthesis buffer containing 40–80 μg/ml digitonin (whole cells only) and substrates with/without inhibitors [succinate (10 mM); malonate (1 mM); glutamate (10 mM); pyruvate (10 mM); malate (10 mM); rotenone (2.5 μM)] and incubated at 37°C for 20 min. Reactions were stopped with perchloric acid on ice, neutralized with potassium hydroxide and MOPS. ATP concentration in samples was determined using a luciferase-based assay (Roche, 11699695001) in a microplate reader (BMG Labtech, FLUOstar OPTIMA). Rates are reported as the ratio to measurements made with succinate+rotenone.
Membrane potential measurements
Cells were cultured in black 96-well plates (2×104 cells/well for primary astrocytes and MEFs, 5×104 cells/well for ρ° and ρ+ LM(TK−) cells) and incubated with media containing 75 nM tetramethylrhodamine methyl ester (TMRM; Sigma) and 2 μg/ml Hoechst-33258 (Sigma) (40 min, 37°C, 5% CO2). Subsequently, cells were washed with PBS before the ΔΨm (mitochondrial membrane potential) was determined in a minimum of three wells as the ratio of TMRM to Hoechst fluorescence (BMG Labtech, FLUOstar OPTIMA). Cells treated with the CI inhibitor rotenone (2.5 μM) or the protonophore FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone); Sigma; 20 μM) were used as controls.
Alternatively, the ΔΨm was determined by microscopy in cells cultured in glass bottom dishes (WPI, FD-35-100). Cells were incubated in HBSS with Mg2+ and Ca2+ containing 37.5 nM TMRM (30 min, 37°C, 5% CO2). Cells were visualized at 37°C on a DeltaVision OMX V3 Imaging System (Applied Precision) using the TRITC (tetramethylrhodamine β-isothiocyanate) filter set with an oil-immersion objective (Olympus, IX71), fitted with a CoolSNAP HQ2 camera (Photometrics). After initial imaging, either 20 μM FCCP or 2.5 μM rotenone was added to dishes and regions of interest re-captured. Images were de-convoluted, cropped and projected with maximum intensity (Applied Precision, SoftWoRx v5.5). Mitochondrial rich perinuclear regions were selected in greater than 60 cells per dish and average TMRM fluorescence determined pre- and post-FCCP or rotenone application.
Superoxide (O2•−) detection
Cells were cultured in black 96-well plates (2×104 cells/well) and incubated with media containing 10 μg/ml DHE (dihydroethidium; Sigma) and 2 μg/ml Hoechst-33258 (40 min, 37°C, 5% CO2). Cells were washed in PBS, and the rate of O2•− production determined in a minimum of three wells as the ratio of DHE to Hoechst fluorescence (BMG Labtech, FLUOstar OPTIMA). Cells treated with the CI inhibitor rotenone (2.5 μM) were used as a control.
H2O2 detection
The rate of H2O2 production was measured as previously described [33] in 5 μg isolated mitochondria using the fluorescent probe Amplex Red with the substrate and inhibitor combinations detailed in the text [succinate (10 mM); malonate (1 mM); glutamate (10 mM); pyruvate (10 mM); malate (10 mM); rotenone (2.5 μM)]. H2O2 production was measured fluorometrically over a 20 min period (BMG Labtech, FLUOstar OPTIMA).
Cell death assays
Where described, cells were treated with 50 μM H2O2 in culture media for 6 h. Cells were harvested using trypsin, and diluted to 2×105 cells/ml in annexin-V binding buffer (10 mM Hepes, 140 mM NaCl, 2.5 mM CaCl2, pH to 7.4). An aliquot of 500 μl was co-incubated with 5 μl annexin-V (Invitrogen) and 1 μg/ml PI (propidium iodide, Sigma) for 10 min at room temperature then analysed by flow cytometry (Becton Dickinson, LSR II flow cytometer) with the FITC-A and PI-488-695/40-A lasers. Viable cells were distinguished from dead cells using the forward- and side-scatter parameters (De Novo, FCS express V4). Healthy cells were considered as annexin-V and PI double negative, apoptotic cells as annexin-V positive, and necrotic cells as annexin-V and PI double positive. Gating strategy is depicted in Supplementary Figures S2(A) and S2(B).
Statistics
Unless otherwise described, error bars represent standard error of the mean (S.E.M.) and P values are generated from two-tailed unpaired t tests (GraphPad, PRISM V6.0b) and reported when ≤0.05.
RESULTS
CI activity is equally reduced in Ndufs4fky/fky cell lines
The NDUFS4 subunit was undetectable in all Ndufs4fky/fky primary cell types examined (Figure 1A), consistent with the systemic disruption of NDUFS4 in the mouse and in keeping with previous reports on all tissues studied [12]. This was not changed by culturing Ndufs4fky/fky primary MEFs and astrocytes under conditions that favour ATP generation by the OXPHOS system (on galactose). CI activity was severely impaired in Ndufs4fky/fky primary MEFs (21% of +/+), astrocytes (23% of +/+) and neurons (42% of +/+) under standard culture conditions (on glucose) (Figure 1B), similar to reports on mouse tissues [12]. To account for alterations in mitochondrial volume and/or number, CI activity was normalized to CS activity, which was not affected by loss of NDUFS4 (Figure 1C). It is worth noting that while the activity of CI relative to CS was similar in each of the Ndufs4fky/fky cell types, the activity of CI relative to CS in +/+ cells was lower in neurons than in astrocytes and MEFs (Figure 1B).
Figure 1. CI activity in Ndufs4fky/fky cell lines.

Expression of the CI subunit NDUFS4 was examined in cell lysates that were prepared from primary cell cultures from +/+ (white bars) and Ndufs4fky/fky (grey bars) mice that were grown in either glucose or galactose medium (primary MEFs, primary astrocytes), or NBM (primary neurons) by Western blot analysis using antibodies against the CI subunit NDUFS4 and the CII subunit SDHA as a loading control (A). Images have been digitally cropped for clarity. CI activity relative to CS activity (B) and CS activity (C) were measured in primary cell culture lysates from +/+ and Ndufs4fky/fky mice maintained on either glucose medium (primary MEFs, primary astrocytes) or NBM (primary neurons) by spectrophotometry. Error bars=S.E.M., n≥7, P values generated from two-tailed unpaired t tests. CI, complex I; CII, complex II; CS, citrate synthase.
Taken together with previous data obtained from the Ndufs4fky/fky mice [12], it appears unlikely that a tissue-specific difference in CI activity is the basis for the predominant neurological dysfunction in the Ndufs4fky/fky mouse.
CI appears as a crippled complex in Ndufs4fky/fky cell lines
We next investigated the assembly status of CI in the primary cell lines. Consistent with previous reports, using BN-PAGE we observed that CI lacking NDUFS4 forms a crippled assembly intermediate (Ci) of ≈830 kDA and is reduced in abundance when cells were solubilized in Triton X-100 to isolate individual OXPHOS complexes (Figure 2A: +/+, odd lanes; Ndufs4fky/fky, even lanes) [12,15,16,34]. This was independent of culture conditions for all primary cells (Figure 2A). Furthermore, when Ndufs4fky/fky cell pellets were solubilized in the milder detergent digitonin to retain supramolecular interactions with other OXPHOS complexes (Figures 2B and 2D: +/+, odd lanes; Ndufs4fky/fky, even lanes), supercomplexes appeared at smaller molecular weights corresponding to supercomplexes containing the crippled CI. Again, this was observed independent of culture conditions for all primary cells (Figures 2B and 2D).
Figure 2. BN-PAGE analyses of respiratory complexes in Ndufs4fky/fky cell lines.
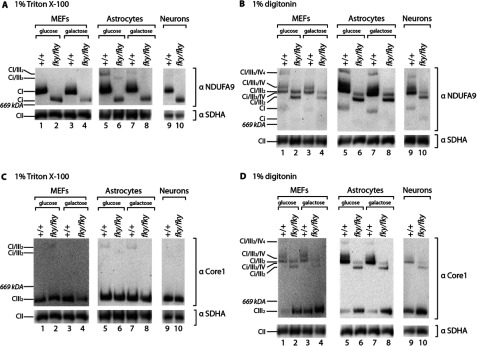
Cells derived from +/+ and Ndufs4fky/fky mice maintained on either glucose or galactose medium (primary MEFs, primary astrocytes), or NBM (primary neurons), were solubilized in buffer containing either 1% Triton X-100 (A and C) or 1% digitonin (B, D), and protein complexes separated by BN-PAGE. Gels were transferred to membrane and immunodecorated with antibody targeted to the CI subunit NDUFA9 (A, B), or targeted to the CIII subunit Core1 (C, D). CII subunit SDHA was used as a loading control. n=1–3. Images have been digitally cropped for clarity. CI–IV, complexes I–IV; Ci, crippled CI formed in the absence of NDUFS4.
In addition, the level of mature complex III (CIII2) was similar in all cells and under all culture conditions examined (Figure 2C). However the abundance of free CIII2 not bound in supercomplexes appeared to be increased in Ndufs4fky/fky cells (Figure 2D), consistent with reduced abundance of supercomplex species containing crippled CI.
Together these data suggest that the assembly of CI itself and supercomplexes containing crippled CI is equally disrupted in detergent solubilized cell pellets across all primary Ndufs4fky/fky cell types and culture conditions. Accordingly, these data do not provide any further insight into the mechanisms behind the tissue-specific nature of the disease, nor suggest any adaptive changes in MEFs and astrocytes on galactose in response to a greater dependence on OXPHOS for cellular ATP production.
The ΔΨm is selectively impaired in Ndufs4fky/fky primary MEFs
As noted above, CI activity and assembly were severely impaired in all Ndufs4fky/fky primary cell types studied. Considering the integral nature of CI to proton pumping across the IMM, it might be expected that this could affect the ΔΨm. Moreover, culturing cells on galactose increases the dependence on OXPHOS, which could further impact the ΔΨm. Nonetheless, under basal conditions, the ΔΨm was significantly impaired only in Ndufs4fky/fky primary MEFs (Figures 3A and 3B), while it appeared normal in astrocytes (Figures 3C and 3D) and neurons (Figures 3E and 3F). More specifically, the ΔΨm at rest in Ndufs4fky/fky primary MEFs on glucose medium was depolarized (more electroneutral; Figure 3A; 75% of +/+), and appeared further depolarized on galactose medium (Figure 3B; 60% of +/+). As control experiments, we found that the ΔΨm was equally depolarized in all cell types under all culture conditions following application of the CI inhibitor rotenone, or the protonophore FCCP (Figures 3A–3F). In addition, methodological approaches were validated using mouse fibroblast ρ° cells, which do not possess a functional OXPHOS system [27]. In the ρ° cells at rest, the ΔΨm was clearly depolarized when measured by both plate reader (Supplementary Figure S3A; 62% of ρ+) and microscopy (Supplementary Figure S3B and S3C; 48% of ρ+), consistent with previous observations in similar cells [35,36].
Figure 3. Analysis of the ΔΨm in Ndufs4fky/fky cell lines.

The ΔΨm was measured by the accumulation of TMRM in mitochondria and made relative to cell number in primary MEFs and astrocytes from +/+ (white bars) and Ndufs4fky/fky (grey bars) mice, maintained on glucose (A, C) or galactose (B, D) medium. The ΔΨm was reduced with the CI inhibitor rotenone or the protonophore FCCP, or modulated by the CV inhibitor oligomycin, as indicated. The ΔΨm was assessed by microscopy in primary neurons (E) pre- and post-incubation with rotenone, by measuring the accumulation of the cationic dye TMRM in the mitochondrial-rich perinuclear region. Greater than 60 cells per dish were analysed using ≥2 cell cultures. Representative images are shown for primary neurons in (F), pre- and post-incubation with FCCP. Error bars=S.E.M., n≥3 (A–D), P values generated from two-tailed unpaired t tests. CI and CV, complexes I and V; ΔΨm, mitochondrial membrane potential.
Primary MEFs and astrocytes did however respond differently to the application of the CV (complex V) inhibitor oligomycin. Inhibition of CV blocks the flow of electrons back into the mitochondrial matrix, which should result in hyperpolarization (more electronegative) of the ΔΨm when OXPHOS is in action. In primary MEFs cultured on glucose medium (Figure 3A), oligomycin had no effect on the ΔΨm, yet it hyperpolarized the ΔΨm when cells were cultured on galactose (Figure 3B). In contrast, in primary astrocytes the ΔΨm was depolarized after applying oligomycin irrespective of culture media (Figures 3C and 3D). The phenomenon was not genotype specific though, and could reflect toxicity of the oligomycin to primary astrocytes.
CI-dependent ATP synthesis capacity is reduced in Ndufs4fky/fky primary astrocytes and neurons, but only impaired in primary MEFs when cultured on galactose
The ΔΨm is utilized to drive a number of mitochondrial processes. One of the most critical of these, and considered to be the core function of mitochondria, is ATP synthesis via CV [37]. In this context, we sought to determine the maximal capacity of CI-dependent ATP synthesis in permeabilized Ndufs4fky/fky primary cells under optimal substrate conditions. These measurements were obtained in digitonin permeabilized Ndufs4fky/fky primary cells provided with the CI-dependent substrates glutamate+malate or pyruvate+malate, with or without the CI inhibitor rotenone. All rates were made relative to the CII-dependent rate determined using succinate+rotenone, since these should not be affected by CI deficiency. Importantly, no statistically significant differences were detected between genotypes for rates of CII-dependent ATP synthesis (Table 1).
Table 1. CII-dependent rate of ATP synthesis in isolated mitochondria and permeabilized cells from Ndufs4fky/fky primary cell lines.
*Rate of ATP synthesis measured using the substrate succinate with the complex I inhibitor rotenone±S.E.M. (n). No statistically significant differences were detected (P<0.050 level) between +/+ and fky/fky samples, measured by unpaired t tests, correcting for multiple comparisons using the Holm–Sidak method (GraphPad, PRISM V6.0b).
| ATP (μmol/g/min)* | ||||
|---|---|---|---|---|
| Primary cell line | Medium | Preparation | +/+ | fky/fky |
| MEFs | Glucose | Permeabilzed cells | 36±5.1 (9) | 37±5.0 (13) |
| Galactose | Permeabilized cells | 26±4.1 (7) | 19±4.6 (8) | |
| Glucose | Isolated mitochondria | 61±12 (6) | 65±11 (6) | |
| Astrocytes | Glucose | Permeabilized cells | 61±6.1 (13) | 68±7.6 (11) |
| Galactose | Permeabilized cells | 48±3.9 (7) | 45±1.4 (5) | |
| Glucose | Isolated mitochondria | 84±11 (3) | 89±22 (4) | |
| Neurons | NBM | Permeabilized cells | 24±1.7 (17) | 29±2.3 (21) |
In Ndufs4fky/fky primary MEFs, the CI-dependent ATP synthesis rates were almost indistinguishable from controls on glucose medium (Figures 4A and 4E; 82–105% of +/+) but reduced on galactose (Figure 4C; 55–60% of +/+). In contrast, in Ndufs4fky/fky primary astrocytes the CI-dependent rate was reduced with glutamate+malate (Figures 4B, 4D and 4F; 67–79% of +/+ on glucose and 77% of +/+ on galactose), but normal with pyruvate+malate irrespective of culture media (Figures 4B, 4D and 4F). In Ndufs4fky/fky primary neurons the rate of ATP synthesis was reduced with all CI-dependent substrates (Figure 4G; 63–74% of +/+). To confirm the specificity of the substrates, we determined that the CI-dependent rate of ATP synthesis was almost completely abolished in the presence of the CI inhibitor rotenone in all Ndufs4fky/fky primary cell types under all culture conditions (Figures 4A–4G).
Figure 4. Rates of ATP synthesis in Ndufs4fky/fky cell lines, expressed relative to the CII-dependent rate determined using succinate+rotenone (S+R).

The CII- (S) and CI-dependent (G+M, P+M) rates of ATP synthesis were measured in permeabilized cells or isolated mitochondria from MEFs and astrocytes maintained on glucose (A, B, E, F) or galactose (C, D), and neurons maintained on NBM medium (G) from +/+ (white bars) and Ndufs4fky/fky (grey bars) mice. Where indicated, rotenone was added to inhibit CI. Error bars=S.E.M. Replicates: S (n≥3); G+M (n≥3); G+M+R (n≥2); P+M (n≥3); and P+M+R (n≥2). P values generated from two-tailed unpaired t tests. CI, complex I; G, glutamate; M, malate; P, pyruvate; R, rotenone; S, succinate.
We also noted in permeabilized primary neurons (Figure 4G) that the rate of ATP synthesis with succinate alone relative to the rate with succinate+rotenone was lower (<1), compared with permeabilized MEFs or astrocytes (Figures 4A–4D). Moreover, this relative rate was more reduced in +/+ than Ndufs4fky/fky neurons (Figure 4G; 0.60 versus 0.74). In addition, a relative rate of <1 is observed in isolated mitochondria from +/+ MEFs and astrocytes, but not Ndufs4fky/fky cells (Figures 4E and 4F). Together these data imply that RET (reverse electron transfer) may occur in all primary cell types examined. The term RET describes the process in which ubiquinol generated by CII donates electrons to a reverse CI reaction rather than to the forward CIII reaction [38]. RET is blocked by rotenone inhibition of CI. The data also suggest that RET is partially blocked in the Ndufs4fky/fky primary cells, most prominently in neurons. This is presumably because the CI defect blocks both forward and reverse electron transfer by CI. In addition, these observations indicate that a small amount of residual CI-dependent substrate in our permeabilized cells that is not present in isolated mitochondria may artificially increase the CII-dependent rate of ATP synthesis. Indeed, while the general rates of ATP synthesis were very low when no substrate was provided (Supplementary Figures S4A–S4G), a slightly higher rate was observed in the permeabilized cells compared with the isolated mitochondria and neurons. As a control, we determined that the rates of CII-dependent ATP synthesis in all cell types were sensitive to the CII inhibitor malonate (Supplementary Figures S4A–S4G; rate relative to succinate+rotenone<1).
Rates of ROS generation are selectively elevated in Ndufs4fky/fky primary MEFs
Dysfunction of CI may affect not only the ΔΨm and associated ATP synthesis, but may also be associated with increased ROS production. ROS have the potential to cause oxidative damage to cellular structures, including DNA, proteins and lipids, if not rapidly detoxified by cells [39,40]. As such, we sought to quantify the amount of O2•− produced by Ndufs4fky/fky primary MEFs and astrocytes cultured on either glucose or galactose media, using the O2•− specific probe DHE. Additionally, CI- and CII-dependent H2O2 production was measured in isolated mitochondria from MEFs and astrocytes grown on the glucose medium using Amplex Red. Owing to limitations of sample availability, we were unable to perform comparable measurements of O2•− and H2O2 production in Ndufs4fky/fky primary neurons.
Compared with control cells, Ndufs4fky/fky primary MEFs generated increased amounts of O2•− at rest on glucose medium (Figure 5A; 150% of +/+), but not on galactose medium (Figure 5B). In comparison, the rates of O2•− production at rest in Ndufs4fky/fky primary astrocytes cultured on either glucose (Figure 5D) or galactose (Figure 5E) medium were indistinguishable from controls. Rotenone treatment stimulated O2•− in all cell types, independent of genotype or culture conditions (Figures 5A, 5B, 5D and 5E). However, the rotenone dependent increase in O2•− production was less for both Ndufs4fky/fky primary MEFs (Figure 5C) and astrocytes (Figure 5F) compared with +/+, regardless of the culture media.
Figure 5. O2•− and H2O2 production in Ndufs4fky/fky cell lines.

The rate of O2•− production was measured by the cellular accumulation of DHE and made relative to the total cell number in primary MEFs and astrocytes from +/+ (white bars) and Ndufs4fky/fky (grey bars) mice, maintained on glucose (A, D), or galactose (B, E) medium. The rate of O2•− production was modulated with the CI inhibitor rotenone, as indicated. The ratio of O2•− production from rotenone treated cells to non-treated cells (+ rotenone/- rotenone) is presented in (C) and (F). Rates of H2O2 production were measured with Amplex Red in isolated mitochondria from glucose-grown primary MEFs and astrocytes from +/+ and Ndufs4fky/fky mice (G, H) and results are displayed either relative to succinate+rotenone (left y-axis) or glutamate+malate (right y-axis). To maintain statistical power, comparisons have only been made between genotypes with the exception of between groups comparing succinate with or without rotenone, given this difference is of particular interest (G, H). Error bars=S.E.M., n≥3, P values generated from two-tailed unpaired t tests.
In contrast, the rates of H2O2 production were equivalent to controls in isolated mitochondria from Ndufs4fky/fky MEFs (Figure 5G) and astrocytes (Figure 5H) with both CI- (glutamate+malate) and CII-dependent substrates (succinate). As well, the rotenone induced increase in H2O2 production linked to CI was equivalent in mitochondria from both +/+ and Ndufs4fky/fky primary MEFs and astrocytes (Figures 5G and 5H). Notably though, the CII-dependent rate of H2O2 production in both MEF and astrocyte mitochondria was lower when incubated with the CI-inhibitor rotenone than without. Moreover, although not significant, this trend appears to be more pronounced in both Ndufs4fky/fky MEFs and astrocytes compared to controls. This further suggests the occurrence of RET in our primary cells, and that the process is attenuated in Ndufs4fky/fky cells.
Ndufs4fky/fky primary MEFs are more susceptible to entering cell death on galactose
Our results suggest that alterations to mitochondrial function are cell type specific, whereby under standard culture conditions on glucose medium, Ndufs4fky/fky primary MEFs manifest a reduced ΔΨm and increased ROS production, while the astrocytes and neurons exhibit a reduced CI-dependent ATP synthesis capacity. Moreover, Ndufs4fky/fky primary MEFs, but not astrocytes, appeared on average to be more sensitive to maintenance on galactose, as evidenced by a further depolarization of the ΔΨm and a corresponding drop in ATP synthesis. All these processes have the potential to affect initiation of cell death [41]. Accordingly, we examined cell death in Ndufs4fky/fky primary cells. These investigations revealed that Ndufs4fky/fky primary MEFs (Figures 6A, 6C and 6E), but not astrocytes (Figures 6B, 6D and 6F), were more sensitive to cell death (see Supplementary Table S1 for statistics on analyses of viable cells). However, this increased sensitivity to cell death was only observed in Ndufs4fky/fky primary MEFs when maintained on galactose for an extended period of time, followed by the application of the acute stressor H2O2. Not only was there an increased percentage of dead cells at the time of analysis (Figure 6A; 1.8 times +/+), but an increased percentage of viable cells that were entering into what appeared to be necrotic cell death (Figure 6E; 13 times +/+). In contrast to Ndufs4fky/fky primary MEFs, Ndufs4fky/fky astrocytes (Figures 6B, 6D and 6F) displayed no marked change in sensitivity to cell death compared to +/+ under all culture conditions tested.
Figure 6. Cell death in Ndufs4fky/fky cell lines.

The propensity of primary MEFs and astrocytes from +/+ and Ndufs4fky/fky mice to enter cell death was measured by flow cytometry. The ratio of dead cells relative to +/+ (on glucose medium) for primary MEFs is presented in (A) and astrocytes (B), either cultured on glucose medium for 1 day or galactose medium for 1 day or 4 days, with or without acute H2O2 treatment for 6 h prior to analysis. The percentage of viable single-celled primary MEFs is presented in (C, E) and astrocytes (D, F), either cultured on glucose for 1 day or galactose for 1 day or 4 days, with (E, F) or without (C, D) acute H2O2 treatment for 6 h prior to analysis. Error bars=S.E.M., n≥4, P values generated from two-tailed unpaired t tests correcting for multiple comparisons using the Holm–Sidak method (GraphPad, PRISM V6.0b).*=P≤0.050, **=P≤0.005. PI, propidium iodide.
DISCUSSION
It is generally accepted that OXPHOS mutations likely cause cell-specific effects, due to the differential energy requirements and metabolic pathways utilized by various cell and tissue types [9,42]. Here, we sought to characterize this further by analysing mitochondrial function in neurological and non-neurological primary cell types from the Ndufs4fky/fky mouse model mimicking aspects of LS [12]. In conjunction, we used culture conditions (galactose) that forced Ndufs4fky/fky primary MEFs and astrocytes to rely less on glycolytic ATP generation and more on OXPHOS linked ATP production to emphasize any underlying mitochondrial defects. Astrocytes in particular have been shown to up-regulate glycolysis in response to respiratory inhibition [43,44]. This study significantly makes direct comparisons between primary cells derived from the same animal model of OXPHOS deficiency, the Ndufs4fky/fky mouse. As well, it encompasses one of the first comprehensive analyses of astrocytes from a mammalian model of mitochondrial CI deficiency.
Ndufs4fky/fky MEFs were chosen as a control non-neurological cell type, since OXPHOS-deficient patient fibroblasts are the source of much of our current knowledge regarding the cellular biochemistry of mitochondrial diseases [45]. The biochemical profile under standard culture conditions of Ndufs4fky/fky primary MEFs described in the present study closely matches studies conducted in NDUFS4-deficient human and mouse fibroblast model systems. Notably, CI activity is reduced, CI assembly is impaired, the ΔΨm is slightly depolarized, and ROS production is typically increased [15–17,46,47]. These data suggest that disease pathogenesis could partly result from a compromised ΔΨm, which is essential for maintaining basic cellular functions such as ATP synthesis, mitochondrial protein import, cellular Ca2+ homoeostasis, and ultimately cell survival. Furthermore, ROS are also implicated in disease progression, causing oxidative damage to cellular structures. However, since LS is primarily a neurological disorder, fibroblasts are not well suited for the study of mechanisms contributing to the neuropathology.
We find then that the biochemical profile of Ndufs4fky/fky primary MEFs is divergent from that of the neurologically relevant astrocytes and neurons. That is, despite both neurological and non-neurological Ndufs4fky/fky primary cell types manifesting reduced CI activity and disrupted CI formation, the effect on mitochondrial function differs. In particular, Ndufs4fky/fky primary astrocytes and neurons have a remarkably normal ΔΨm under standard culture conditions. Nonetheless, our data indicate that the CI-dependent ATP synthesis rates in Ndufs4fky/fky primary astrocytes and neurons is reduced, paralleling previous studies in brain from the Ndufs4fky/fky mouse also reporting reduced CI-dependent ATP synthesis (65% of +/+) [12]. This represents the opposite trend to the Ndufs4fky/fky primary MEFs, perhaps alluding to the maintenance of the ΔΨm as an adaptive response by the Ndufs4fky/fky neurological cell types to protect against cell death. Such a response might result from modulation of CV activity, perhaps via altering levels of the IF1 regulatory protein [48] or reverse action of the complex [49,50]. It should be noted though that we have reported maximal rates of ATP synthesis, while the ΔΨm was monitored under steady state conditions. Also, neurons themselves may be more sensitive to the disruption of CI activity than other cell types as indicated by our results that neurons have a reduced CI capacity relative to mitochondrial volume and/or number in comparison to astrocytes and MEFs.
Moreover, while increased ROS production was a feature in NDUFS4 deficient patient and mouse fibroblasts [17,46], there was little evidence of this in our Ndufs4fky/fky primary astrocytes, nor the previously described NDUFS4 deficient mesencephalic neurons [20,21]. Previous cellular studies have indicated correlations between the degree of CI inhibition and the level of ROS production [18,51]. In contrast, we observed variations in ROS production between different cell types from the same mouse model with the same level of CI inhibition. This variation could partly result from differing levels of cellular anti-oxidants, such as the decreased glutathione observed in neurons versus astrocytes [52,53]. Again, this emphasizes the pathogenic relevance of investigating these parameters in appropriate cells. As well, it should be noted that both Ndufs4fky/fky primary MEFs and astrocytes showed a smaller increase in ROS generation after addition of rotenone. This may reflect an adaptation to the loss of NDUFS4 and the unstable CI observed in vitro in these cells, such that the basal rate of ROS generation in CI-deficient cells is closer to the maximal rate than in +/+ cells.
Nonetheless, oxidative damage has been observed in the brain of Ndufs4 KO mice [13]. Although oxidative damage may be a major contributing factor to disease, the source of ROS is likely to be external from the affected cell types. Cavanagh and Harding [7] suggested that oxidative damage to neurological tissue in LS patients is mediated by the effect of pH changes to the neurovasculature (hyperlacticacidaemia). Moreover, these pH changes could be more pronounced at a local level in the brain due to its high rate of metabolic activity [7,54]. Combined with the highly oxygenated environment of the mammalian brain, conditions may be conducive to the catalysis of ROS formation, thus mediating oxidative damage in affected brain regions [7]. Alternatively, oxidative damage in the brain may result from gliosis, an infiltration of activated glial cells, including astrocytes. Gliosis is a feature of LS [7,8], and was also observed in Ndufs4 KO mice [13,14]. These activated glial cells may release pro-inflammatory cytokines, further recruiting immune cells to the affected brain region, producing a burst of ROS and driving oxidative damage [7,13,14,55]. Furthermore, aberrant activation of astrocytes could cause tissue damage by promoting inflammation, contributing to scarring and preventing axonal regrowth from neurons [55,56]. Indeed with few exceptions, there is little correlation between the ROS production and oxidative damage observed in neurologically relevant models of mitochondrial disease [9].
This study also finds evidence to suggest that electrons from ubiquinol (QH2) generated by succinate oxidation at CII can be transferred in both the forward direction to CIII and the reverse direction to CI (RET) [38,57]. This was indicated by both the increased efficiency of the CII-dependent rate of ATP synthesis and the higher rate of H2O2 production, when measured in the presence of the CI inhibitor rotenone. Notably, the trend appears to be more pronounced in neurons than in primary MEFs or astrocytes, and is attenuated in Ndufs4fky/fky primary cells compared with controls. The physiological relevance of this phenomenon to disease is unclear, although RET-induced changes in redox balance or generation of ROS may influence cell signalling. It is therefore of interest that neurons have higher rates of RET than other cells. Nonetheless, as these experiments measured maximal rates of activity, they likely do not reflect the in vivo activity of the system. Moreover, the measurements describing RET were performed in the absence of CI-dependent substrates, unlike in the in vivo environment where CI substrates may influence forward electron transport through CI.
When grown with galactose instead of glucose, to force OXPHOS dependent respiration, Ndufs4fky/fky primary MEFs again phenotypically deviated from astrocytes. While Ndufs4fky/fky primary MEFs responded by further depolarizing their ΔΨm with a corresponding reduction in CI-dependent ATP synthesis capacity, Ndufs4fky/fky primary astrocytes responded no differently than when maintained under standard culture conditions. Moreover, Ndufs4fky/fky primary MEFs on galactose have an increased propensity for cell death, while Ndufs4fky/fky primary astrocytes do not. This suggests that Ndufs4fky/fky primary MEFs have insufficient reserve OXPHOS capacity to adapt to maintenance on galactose. In contrast, Ndufs4fky/fky astrocytes may have a larger spare OXPHOS capacity, or the ability to reduce their metabolic demand more readily, thus preferentially maintaining their ΔΨm. This difference however was only significant with the acute application of H2O2, again perhaps indicative of underlying differences in the antioxidant defence systems in these two cell types that were not investigated in the present study.
Ndufs4fky/fky primary neurological cell types may preferentially maintain some mitochondrial processes over others, such as the ΔΨm over ATP synthesis, to avoid sensitization to cell death. This would be consistent with the phenomenon described in LS patients known as relative neuronal sparing, characterized by the preservation of neurons in localized regions of cell death [7,8]. Likewise, it was observed in CI-deficient mt-Nd5/6 transgenic cybrid-derived neurons and astrocytes that the ΔΨm was elevated by up to 40% under resting conditions [53], yet collapsed with application of the CV inhibitor oligomycin. Furthermore, neurons are known to possess other mechanisms to evade cell death, such as their reduced sensitivity to cytochrome c-mediated apoptosis [58].
Considering that the CI-dependent ATP synthesis capacity of Ndufs4fky/fky astrocytes and neurons is greatly reduced, energy-dependent processes in these cells, such as the capacity to generate action potentials, may be compromised. Likewise, this may impact the astrocytes’ ability to recycle glutamine and potassium released as neurotransmitters by neurons, maintain the blood–brain barrier and propagate action potentials [23,24,56,59]. The effect of the CI deficiency on the capacity of astrocytes to recycle neurotransmitters released by neurons is of particular importance, given that this process alone likely accounts for a large proportion of the brain's energy budget, and may be hypersensitive to an ATP deficiency [54].
Collectively, we show that Ndufs4fky/fky primary MEFs respond differently to the CI deficiency compared with the astrocytes and neurons, which may reflect that the tissue from which the MEFs are derived is less metabolically active than brain. Furthermore, measurements obtained from the primary astrocytes and neurons were conducted at rest and it remains to be determined how robust mitochondrial function is in stimulated cells. Ultimately, these results highlight the need to study disease mechanisms in relevant cells and tissues, since studies in less relevant samples, such as fibroblasts, may be misleading.
Online data
ACKNOWLEDGEMENTS
We kindly thank Tomris Mustafa (The Florey Institute of Neuroscience and Mental Health) for assistance with neuronal and astrocyte culturing procedures, as well as Ian Trounce (Centre for Eye Research Australia) and Doug Wallace (Center for Mitochondrial and Epigenomic Medicine, Children's Hospital of Philadelphia, U.S.A.) for the kind gift of the ρ° cells. We also thank Matthew Burton (Murdoch Childrens Research Institute, Australia) for assistance in performing and analysing data collected by flow cytometry and live cell imaging.
AUTHOR CONTRIBUTION
The following authors contributed to the concept and design of the study: Matthew J. Bird; Xiaonan W. Wijeyeratne; Jasper C. Komen; Michael T. Ryan; David R. Thorburn; and Ann E. Frazier. The collection and/or assembly of the data was performed by Matthew J. Bird, Xiaonan W. Wijeyeratne, Adrienne Laskowski and Ann E. Frazier. Analysis and interpretation of the data were performed by Matthew J. Bird, Xiaonan W. Wijeyeratne, Michael T. Ryan, David R. Thorburn and Ann E. Frazier. The manuscript was written by Matthew J. Bird, Jasper C. Komen, Michael T. Ryan, David R. Thorburn and Ann E. Frazier.
FUNDING
The authors’ research was supported by grants from the Australian National Health and Medical Research Council (NHMRC) [grant number APP102222] and the Muscular Dystrophy Association (MDA) [grant number MDA113072], an NHMRC Principal Research Fellowship to David Thorburn [grant number 1022896], an NHMRC Career Development Award to Ann Frazier [grant number 541920], and an Australian Postgraduate Award to Matthew Bird [grant number APA2011]. Additional support was provided by the Australian Mitochondrial Disease Foundation and the Victorian Government's Operational Infrastructure Support Programme.
References
- 1.Skladal D., Halliday J., Thorburn D. R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- 2.Munnich A., Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am. J. Med. Genet. 2001;106:4–17. doi: 10.1002/ajmg.1391. [DOI] [PubMed] [Google Scholar]
- 3.Pfeffer G., Chinnery P. F. Diagnosis and treatment of mitochondrial myopathies. Ann. Med. 2013;45:4–16. doi: 10.3109/07853890.2011.605389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koopman W. J. H., Distelmaier F., Smeitink J. A. M., Willems P. H. G. M. OXPHOS mutations and neurodegeneration. EMBO J. 2013;32:9–29. doi: 10.1038/emboj.2012.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DiMauro S., Schon E. A. Mitochondrial disorders in the nervous system. Annu. Rev. Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- 6.Baertling F., Rodenburg R. J., Schaper J., Smeitink J. A., Koopman W. J. H., Mayatepek E., Morava E., Distelmaier F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry. 2014;85:257–265. doi: 10.1136/jnnp-2012-304426. [DOI] [PubMed] [Google Scholar]
- 7.Cavanagh J. B., Harding B. N. Pathogenic factors underlying the lesions in Leigh's disease. Tissue responses to cellular energy deprivation and their clinico-pathological consequences. Brain. 1994;117:1357–1376. doi: 10.1093/brain/117.6.1357. [DOI] [PubMed] [Google Scholar]
- 8.Rahman S., Blok R. B., Dahl H. H., Danks D. M., Kirby D. M., Chow C. W., Christodoulou J., Thorburn D. R. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996;39:343–351. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- 9.Bird M. J., Thorburn D. R., Frazier A. E. Modelling biochemical features of mitochondrial neuropathology. Biochim. Biophys. Acta. 2014;1840:1380–1392. doi: 10.1016/j.bbagen.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 10.Wallace D. C., Fan W. The pathophysiology of mitochondrial disease as modeled in the mouse. Genes Dev. 2009;23:1714–1736. doi: 10.1101/gad.1784909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kruse S. E., Watt W. C., Marcinek D. J., Kapur R. P., Schenkman K. A., Palmiter R. D. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008;7:312–320. doi: 10.1016/j.cmet.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leong D. W., Komen J. C., Hewitt C. A., Arnaud E., McKenzie M., Phipson B., Bahlo M., Laskowski A., Kinkel S. A., Davey G. M., et al. Proteomic and metabolomic analyses of mitochondrial complex I-deficient mouse model generated by spontaneous B2 short interspersed nuclear element (SINE) insertion into NADH dehydrogenase (ubiquinone) Fe–S protein 4 (Ndufs4) gene. J. Biol. Chem. 2012;287:20652–20663. doi: 10.1074/jbc.M111.327601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quintana A., Kruse S. E., Kapur R. P., Sanz E., Palmiter R. D. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. U. S. A. 2010;107:10996–11001. doi: 10.1073/pnas.1006214107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quintana A., Zanella S., Koch H., Kruse S. E., Lee D., Ramirez J. M., Palmiter R. D. Fatal breathing dysfunction in a mouse model of Leigh syndrome. J. Clin. Invest. 2012;122:2359. doi: 10.1172/JCI62923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Assouline Z., Jambou M., Rio M., Bole-Feysot C., de Lonlay P., Barnerias C., Desguerre I., Bonnemains C., Guillermet C., Steffann J., et al. A constant and similar assembly defect of mitochondrial respiratory chain complex I allows rapid identification of NDUFS4 mutations in patients with Leigh syndrome. Biochim. Biophys. Acta. 2012;1822:1062–1069. doi: 10.1016/j.bbadis.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Valsecchi F., Monge C., Forkink M., de Groof A. J. C., Benard G., Rossignol R., Swarts H. G., van Emst-de Vries S. E., Rodenburg R. J., Calvaruso M. A., et al. Metabolic consequences of NDUFS4 gene deletion in immortalized mouse embryonic fibroblasts. Biochim. Biophys. Acta. 2012;1817:1925–1936. doi: 10.1016/j.bbabio.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Valsecchi F., Grefte S., Roestenberg P., Joosten-Wagenaars J., Smeitink J. A. M., Willems P. H. G. M., Koopman W. J. H. Primary fibroblasts of NDUFS4−/− mice display increased ROS levels and aberrant mitochondrial morphology. Mitochondrion. 2013;13:436–443. doi: 10.1016/j.mito.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 18.Verkaart S., Koopman W. J. H., van Emst-de Vries S. E., Nijtmans L. G. J., van den Heuvel L. W. P. J., Smeitink J. A. M., Willems P. H. G. M. Superoxide production is inversely related to complex I activity in inherited complex I deficiency. Biochim. Biophys. Acta. 2007;1772:373–381. doi: 10.1016/j.bbadis.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Verkaart S., Koopman W. J. H., Cheek J., van Emst-de Vries S. E., van den Heuvel L. W. P. J., Smeitink J. A. M., Willems P. H. G. M. Mitochondrial and cytosolic thiol redox state are not detectably altered in isolated human NADH: ubiquinone oxidoreductase deficiency. Biochim. Biophys. Acta. 2007;1772:1041–1051. doi: 10.1016/j.bbadis.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Choi W.-S., Kruse S. E., Palmiter R. D., Xia Z. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc. Natl. Acad. Sci. U. S. A. 2008;105:15136–15141. doi: 10.1073/pnas.0807581105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi W.-S., Palmiter R. D., Xia Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson's disease model. J. Cell Biol. 2011;192:873–882. doi: 10.1083/jcb.201009132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freeman M. R. Specification and morphogenesis of astrocytes. Science. 2010;330:774–778. doi: 10.1126/science.1190928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barres B. A. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 24.Dienel G. A. Astrocytic energetics during excitatory neurotransmission: What are contributions of glutamate oxidation and glycolysis? Neurochem. Int. 2013;63:244–258. doi: 10.1016/j.neuint.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du F., Qian Z. M., Zhu L., Wu X. M., Qian C., Chan R., Ke Y. Purity, cell viability, expression of GFAP and bystin in astrocytes cultured by different procedures. J. Cell. Biochem. 2010;109:30–37. doi: 10.1002/jcb.22375. [DOI] [PubMed] [Google Scholar]
- 26.Meberg P. J., Miller M. W. Culturing hippocampal and cortical neurons. Methods Cell Biol. 2003;71:111–127. doi: 10.1016/S0091-679X(03)01007-0. [DOI] [PubMed] [Google Scholar]
- 27.Trounce I. A., Schmiedel J., Yen H. C., Hosseini S., Brown M. D., Olson J. J., Wallace D. C. Cloning of neuronal mtDNA variants in cultured cells by synaptosome fusion with mtDNA-less cells. Nucleic Acids Res. 2000;28:2164–2170. doi: 10.1093/nar/28.10.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKenzie M., Lazarou M., Thorburn D. R., Ryan M. T. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J. Mol. Biol. 2006;361:462–469. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 29.Frazier A. E., Thorburn D. R. Biochemical analyses of the electron transport chain complexes by spectrophotometry. Methods Mol. Biol. 2012;837:49–62. doi: 10.1007/978-1-61779-504-6. [DOI] [PubMed] [Google Scholar]
- 30.McKenzie M., Lazarou M., Thorburn D. R., Ryan M. T. Analysis of mitochondrial subunit assembly into respiratory chain complexes using Blue Native polyacrylamide gel electrophoresis. Anal. Biochem. 2007;364:128–137. doi: 10.1016/j.ab.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 31.Dunning C. J., McKenzie M., Sugiana C., Lazarou M., Silke J., Connelly A., Fletcher J. M., Kirby D. M., Thorburn D. R., Ryan M. T. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. EMBO J. 2007;26:3227–3237. doi: 10.1038/sj.emboj.7601748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wanders R. J., Ruiter J. P., Wijburg F. A. Studies on mitochondrial oxidative phosphorylation in permeabilized human skin fibroblasts: application to mitochondrial encephalomyopathies. Biochim. Biophys. Acta. 1993;1181:219–222. doi: 10.1016/0925-4439(93)90024-U. [DOI] [PubMed] [Google Scholar]
- 33.Morten K. J., Ackrell B. A., Melov S. Mitochondrial reactive oxygen species in mice lacking superoxide dismutase 2: attenuation via antioxidant treatment. J. Biol. Chem. 2006;281:3354–3359. doi: 10.1074/jbc.M509261200. [DOI] [PubMed] [Google Scholar]
- 34.Calvaruso M. A., Willems P., van den Brand M., Valsecchi F., Kruse S., Palmiter R., Smeitink J., Nijtmans L. Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum. Mol. Genet. 2012;21:115–120. doi: 10.1093/hmg/ddr446. [DOI] [PubMed] [Google Scholar]
- 35.Appleby R. D., Porteous W. K., Hughes G., James A. M., Shannon D., Wei Y.-H., Murphy M. P. Quantitation and origin of the mitochondrial membrane potential in human cells lacking mitochondrial DNA. Eur. J. Biochem. 1999;262:108–116. doi: 10.1046/j.1432-1327.1999.00350.x. [DOI] [PubMed] [Google Scholar]
- 36.Buchet K., Godinot C. Functional F1-ATPase essential in maintaining growth and membrane potential of human mitochondrial DNA-depleted ρ° cells. J. Biol. Chem. 1998;273:22983–22989. doi: 10.1074/jbc.273.36.22983. [DOI] [PubMed] [Google Scholar]
- 37.Boyer P. D. The ATP synthase–a splendid molecular machine. Annu. Rev. Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]
- 38.Dröse S., Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012;748:145–169. doi: 10.1007/978-1-4614-3573-0. [DOI] [PubMed] [Google Scholar]
- 39.Balaban R. S., Nemoto S., Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 40.Venditti P., Di Stefano L., Di Meo S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion. 2013;13:71–82. doi: 10.1016/j.mito.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 41.Galluzzi L., Kepp O., Kroemer G. Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012;13:780–788. doi: 10.1038/nrm3479. [DOI] [PubMed] [Google Scholar]
- 42.Lombès A., Auré K., Bellanné-Chantelot C., Gilleron M., Jardel C. Unsolved issues related to human mitochondrial diseases. Biochimie. 2014;100:171–176. doi: 10.1016/j.biochi.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 43.Bolaños J. P., Almeida A., Moncada S. Glycolysis: a bioenergetic or a survival pathway? Trends Biochem. Sci. 2010;35:145–149. doi: 10.1016/j.tibs.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 44.Bouzier-Sore A. K., Pellerin L. Unraveling the complex metabolic nature of astrocytes. Front. Cell. Neurosci. 2013;7:179. doi: 10.3389/fncel.2013.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saada A. Mitochondria: mitochondrial OXPHOS (dys) function ex vivo–The use of primary fibroblasts. Int. J. Biochem. Cell Biol. 2014;48:60–65. doi: 10.1016/j.biocel.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 46.Distelmaier F., Koopman W. J. H., Van Den Heuvel L. P., Rodenburg R. J., Mayatepek E., Willems P. H. G. M., Smeitink J. A. M. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132:833–842. doi: 10.1093/brain/awp058. [DOI] [PubMed] [Google Scholar]
- 47.Lazarou M., McKenzie M., Ohtake A., Thorburn D. R., Ryan M. T. Analysis of the assembly profiles for mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol. Cell. Biol. 2007;27:4228–4237. doi: 10.1128/MCB.00074-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campanella M., Casswell E., Chong S., Farah Z., Wieckowski M. R., Abramov A. Y., Tinker A., Duchen M. R. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008;8:13–25. doi: 10.1016/j.cmet.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 49.McKenzie M., Liolitsa D., Akinshina N., Campanella M., Sisodiya S., Hargreaves I., Nirmalananthan N., Sweeney M. G., Abou-Sleiman P. M., Wood N. W., et al. Mitochondrial ND5 gene variation associated with encephalomyopathy and mitochondrial ATP consumption. J. Biol. Chem. 2007;282:36845–36852. doi: 10.1074/jbc.M704158200. [DOI] [PubMed] [Google Scholar]
- 50.Chinopoulos C., Adam-Vizi V. Mitochondria as ATP consumers in cellular pathology. Biochim. Biophys. Acta. 2010;1802:221–227. doi: 10.1016/j.bbadis.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 51.Barrientos A., Moraes C. T. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J. Biol. Chem. 1999;274:16188–16197. doi: 10.1074/jbc.274.23.16188. [DOI] [PubMed] [Google Scholar]
- 52.Dringen R., Hirrlinger J. Glutathione pathways in the brain. Biol. Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- 53.Abramov A. Y., Smulders-Srinivasan T. K., Kirby D. M., Acin-Perez R., Enriquez J. A., Lightowlers R. N., Duchen M. R., Turnbull D. M. Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations. Brain. 2010;133:797–807. doi: 10.1093/brain/awq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mergenthaler P., Lindauer U., Dienel G. A., Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 2013;36:587–597. doi: 10.1016/j.tins.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singh S., Swarnkar S., Goswami P., Nath C. Astrocytes and microglia: responses to neuropathological conditions. Int. J. Neurosci. 2011;121:589–597. doi: 10.3109/00207454.2011.598981. [DOI] [PubMed] [Google Scholar]
- 56.Sofroniew M. V. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- 57.Pryde K. R., Hirst J. Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011;286:18056–18065. doi: 10.1074/jbc.M110.186841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vaughn A. E., Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 2008;10:1477–1483. doi: 10.1038/ncb1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int. 2000;36:291–300. doi: 10.1016/S0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.