

Abstract
Bottom-up structural analysis of disulfide-bond containing proteins usually involves time-consuming offline enzymatic digestion, chemical reduction and thiol protection prior to mass spectrometric detection, which takes many hours. This paper presents an expedited bottom-up approach, employing desorption electrospray ionization-mass spectrometry (DESI-MS) coupled with online pepsin digestion and online electrochemical reduction of disulfide bonds. Peptides are generated in high digestion yield as its precursor protein in acidic aqueous solution flows through a pepsin column, which can undergo direct electrolysis. The electrolytic behaviors of peptides, as online monitored by DESI-MS, suggest the presence or absence of disulfide bonds in the peptides, and also provide information to relate disulfide bond-containing peptide precursors to their corresponding reduced products. Furthermore, selective electrolysis simply using different reduction potentials can be adopted to generate either partially or fully reduced peptides to assist disulfide bond mapping. In addition, it turns out that DESI is suitable for ionizing peptides in water without organic solvent additives (organic solvent additives would not be compatible with the use of pepsin column). The feasibility of this method was demonstrated using insulin, a protein carrying three pairs of disulfide-bonds as an example, in which all disulfide bond linkages and most of the protein sequence were successfully determined. Strikingly, this method shortens the sample digestion, reduction and MS detection from hours to less than 7 min, which could be of high value in high-throughput proteomics research.
Keywords: Mass spectrometry, Electrochemistry, Disulfide bond mapping, Protein sequencing, Selective reduction
1. Introduction
Redox-active disulfide bonds are one of the most common protein post-translational modifications (~19% proteins contain multiple disulfide bonds [1]), which provide reversible covalent cross-linkages in native proteins for maintaining protein three-dimensional structures and their biological activities [2,3]. However, the presence of disulfide linkages increases the complexity for protein structure determination by mass spectrometry (MS). The cleavage of disulfide bond is often essential for protein/peptide analysis as dissociation of a reduced protein/peptide ion can give rise to more structurally informative fragment ions than that of the intact counterpart [2,3]. The traditional protocol to break a disulfide bond is chemical reduction using an excess amount of reagents like dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP). However, the reduction usually takes one-half to several hours and the removal of the excess amount of reductant is time-consuming. In addition, the resulting protein/peptide thiols often need to be protected due to the possibility of being reoxidized prior to MS analysis. Besides chemical reduction, other novel approaches include the cleavage of disulfide bonds via laser-based ionization [4–7], ultraviolet photodissociation [8–10], negative ion dissociation [11–14], electron-capture dissociation (ECD)[15], electron-transfer dissociation (ETD)[16–20], plasma-induced oxidation [21], or using new ion chemistry [22–28]. An alternative way for reducing disulfide bonds without involving chemical reductants is electrolytic reduction [29,30].
For structural analysis of disulfide bond-containing proteins, bottom-up approach involving digestion of proteins into peptides is often adopted as it is easier to analyze smaller peptides than intact proteins. However, the conventional enzymatic digestion method is slow and requires overnight incubation, which can be another time-consuming step. Protocols for fast protein digestion have been introduced, such as digestion using modified enzymes [31], microwave digestion [32–34], ultrasonic-assisted protein digestion [35–38] and proteolysis accelerated by infrared radiation [38–40]. Results have been shown that protein digestion can be finished in minutes without reducing efficiency and accuracy. Pepsin packed column is another alternative method to minimize digestion time, due to high concentration of enzyme attached to the column [41]. The online digestion using pepsin column has proved to be very useful for the analysis of proteins following hydrogen-deuterium exchange reactions [42], because backbone H/D exchange can be quenched at the acidic pH (typically pH 2.5) used for pepsin digestion. Recently, in our laboratory, online electrolytic reduction was combined with desorption electrospray ionization mass spectrometry (DESI-MS) for fast analysis of disulfide-bond containing proteins/peptides [43,44]. DESI-MS [45,46,47,48] is a recent advance in the field, which has been applied successfully to analysis of vast different analytes from pharmaceuticals to tissue imaging with little or no sample preparation. Our goal of integrating electrochemistry (EC) with DESI-MS (i.e., EC/DESI-MS method) is twofold. One is to use DESI-MS to study electrochemical reaction mechanisms in consideration of high specificity of MS detection; for instance, by online DESI-MS monitoring, the short-lived intermediate of chlorpromazine radical cation from chlorpromazine oxidation in an electrochemical cell was captured and the electrochemical nitroreduction mechanism was elucidated [49]. The other one is to find applications of the EC/DESI-MS method in proteomics. It has been shown that the EC/DESI-MS is useful for the structural analysis of disulfide bond containing proteins/peptides in either top-down [44] or bottom-up approach [43]. In the top-down approach employing ECD for ion dissociation, the number of fragment ions of lysozyme and β-lactoglobulin A increased to 3–13 fold after electrolytic reduction in comparison to those from the intact proteins [44]. In our previous bottom-up analysis approach [43], peptides and proteins were digested and then underwent online electrolysis and DESI-MS detection. Several useful findings were uncovered, including: (i) the disulfide-containing peptides in the digest mixture can be quickly identified, simply based on the abrupt decrease in their relative ion abundances after electrolysis and (ii) based on the mass relationship, precursor ions and their corresponding reduced product ions can be recognized [43]. However, in such a study, overnight trypsin digestion was a time-consuming step. The EC/DESI-MS method would be further benefited if online enzymatic digestion could be adopted.
In this study, we present an expedited method for disulfide bond-containing protein structural analysis, using online digestion and online electrolytic reduction combined with online MS analysis. In the experiment, by using a pepsin packed column as an immobilized enzyme reactor, fast protein digestion is achieved in high yield without overnight incubation. Insulin was selected as a model protein to demonstrate the feasibility of this new protocol. It turns out that protein can be quickly digested just as the aqueous solution is flowed through the pepsin column. The resulting peptides in the solution without organic solvent additives can be well ionized by DESI-MS. In addition, as reported before [43], based on the intensity changes before and after electro-reduction and their mass relationships, precursor ions and their corresponding reduced product ions were recognized. Furthermore, selective electrolysis was adopted to generate either partially or fully reduced peptides to assist disulfide linkage assignments. Results show that most of the protein sequence structure as well as all three disulfide bond locations of the examined protein were successfully determined using this new method. The method is very fast and it takes <7 min from sample injection to MS detection of peptide signal. This method is also green as no chemical reductant was used.
2. Materials and methods
2.1. Materials
Bovine pancreas insulin was purchased from Sigma–Aldrich (St. Louis, MO). Formic acid was purchased from Spectrum Chemical Mfg. Corp (Gardena, CA). HPLC-grade methanol was purchased from Fisher Scientific (Fair Lawn, NJ). The de-ionized water used for sample preparation was obtained using a Nanopure Diamond Barnstead purification system (Barnstead International, Dubuque, IA).
2.2. Methods
The home-built apparatus for online coupling a thin-layer electrochemical flow cell with a Thermo Finnigan LCQ DECA ion trap mass spectrometer (San Jose, CA) by liquid sample DESI [50] was used and described before (Scheme 1). A pepsin packed column was added between the sample injection syringe and the electrochemical cell. The sample syringe, pepsin packed column and thin-layer flow cell were connected using two short pieces of PEEK tubes (length 4.5 cm, i.d. 0.25 mm). 5 µM intact insulin (MW: 5733.5 Da) in 1% formic acid, was flowed through the pepsin packed column at a rate of 5 µL/min [41]. A thin-layer µ-PrepCell™ electrochemical flow cell equipped with a magic diamond (MD) electrode (12 mm × 30 mm, Antec BV, Netherlands) as the working electrode (WE) was employed and a Roxy™ potentiostat (Antec BV, Netherlands) was used to apply a reduction potential to the cell for triggering electrolytic reduction. The reduced species flowed out of the thin-layer cell via a piece of fused silica capillary (length 17 cm, i.d. 0.1 mm) and then underwent ionization by DESI. The spray solvent for DESI was methanol/water (1:1 by volume) containing 1% formic acid at the injection rate of 5 µL/min. A high voltage of +5 kV was applied to the DESI spray probe. Collision induced dissociation (CID) was carried out to provide ion structural information.
Scheme 1.

Schematic showing the apparatus of the integrated online digestion, online electrolytic reduction and online MS analysis, for disulfide-bond containing protein structural analysis. WE: working electrode; AE: auxiliary electrode; and RE: reference electrode.
3. Results and discussion
Bovine pancreas insulin (MW 5733.5 Da, 51 amino acids, sequence is shown in Table 1) is known to have A and B chains linked by two inter-chain disulfide bonds, and the chain A of insulin has an additional intra-chain disulfide bond. Fig. 1a shows DESI-MS spectrum of the intact insulin in which +4, +5, and +6 of protein ions were observed at m/z 1434, 1147, and 957, respectively. Once the sample solution was flowed through the pepsin packed column at a flow rate of 5 µL/min, the protein was completely digested, as evidenced by the observation that all these protein peaks disappeared (Fig. 1b); at the same time, a series of new peptide peaks arose (denoted as ions 1–12), indicating a 100% digestion yield of the pepsin column. Note that, as the pepsin column cannot tolerate organic solvent due to possible enzyme denaturation issue, one has to use water as the sample solvent. However, proteins/peptides in water usually cannot be well ionized by regular ESI as organic solvent is typically required for effective micro-droplet evaporation to generate gaseous ions. It would reduce signal intensity without using any organic solvent. Interestingly, DESI offers a solution to this problem by using methanol/water (50:50, v/v) containing 1.0% formic acid as the spray solvent [51,52]. Indeed, in this case, the DESI signal of the digested insulin in water containing 1% formic acid was found to be about 4 times higher than the signal of electrosonic spray ionization (ESSI [53], a variant form of electrospray ionization) of the same sample (spectra not shown). For example, the intensity of ion 8 was 2.5e5 in ESSI-MS spectrum (data not shown) but increased to 1.1e6 in the DESI spectrum. In this regard, it is a benefit to use DESI to directly ionize peptides in aqueous solution following the pepsin column digestion.
Table 1.
Analysis results of insulin.
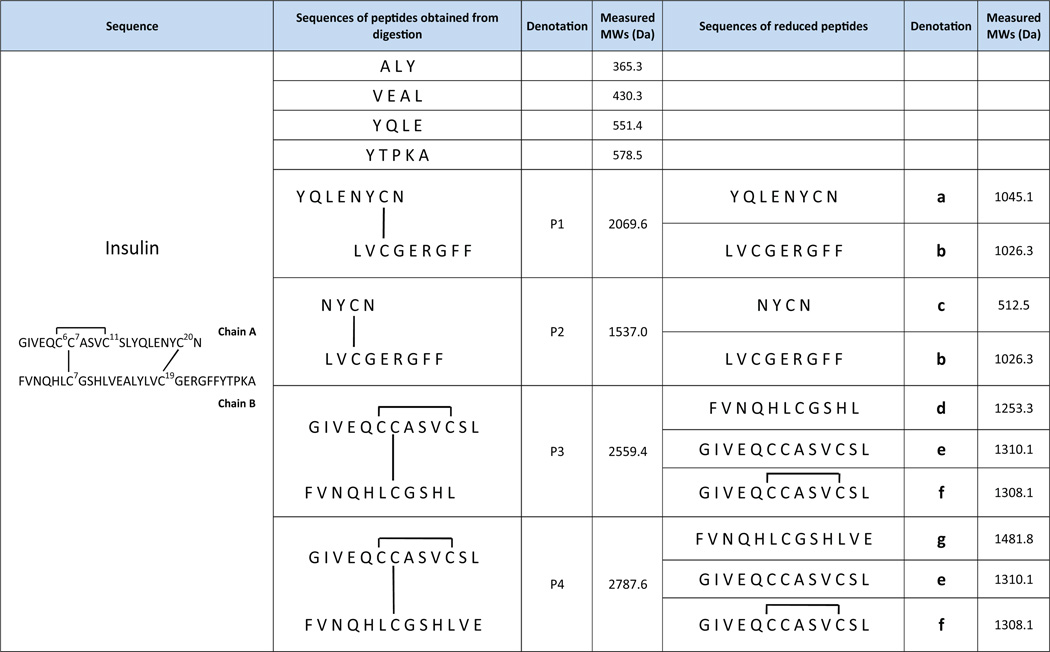 |
Fig. 1.

(+)-DESI-MS spectra of (a) 5 µM intact insulin solution in water containing 1% formic acid; (b) 5 µM insulin solution after online pepsin digestion and before electrolytic reduction (applied potential: 0 V); and (c) 5 µM insulin solution after online pepsin digestion and online electrolytic reduction (applied potential: −1.5 V).
Tandem MS analysis was then applied to these new peptide ions 1–12. It was straightforward to analyze the sequences of peptide ions 1–5. For instance, based on the isotopic peak distribution, it is known that ion 1 at m/z 290.5 is doubly charged and ion 5 at m/z 579.5 is the corresponding singly charged ion. Upon CID (Fig. 1S–a, Supporting Information), ion 1 generated fragment ions of b2, b4-H2O, b4, y2, y3 and y4 (Table S3 in the Supporting Information summarizes the fragmentation results of all selected peptide ions). The appearance of y2,y3 and y4 tells this pentapeptide sequence to be either YTPKA or YTPAK. The presence of b4 points out that the 5th position residue is alanine. Thus the peptide corresponding to ions 1 and 5 is YTPKA, in agreement with insulin structure shown in Table 1. For the singly charged ion 2 at m/z 366.3, it dissociates into b2,y1, and y1-NH3 by CID (Fig. 1S–b, Supporting Information), suggesting its sequence as either ALY or LAY. Note that “L” in this sequence assignment could be its isobaric residue “I” as we could not distinguish leucine from isoleucine using CID data. This is also true for other assignments below. Ion 3 at m/z 431.3 is a singly charged peptide ion. Based on the formation of fragment ions y1, y2, and y3 by CID (Fig. 1S–c, Supporting Information), its sequence can be identified as VEAL. Singly charged ion 4 at m/z 552.4 produced fragment ions b2, b2-NH3, b3, b3-NH3,y2,y3, and y3-NH3 by CID (Fig. 1S–d, Supporting Information) and is thereby indicated to have the sequence of YQLE. Therefore, these ions 1–5 correspond to four peptides that do not carry disulfide bonds (i.e., the first four peptides from digestion shown in Table 1). Among these four peptides, YQLE is from insulin chain A and the other three are from the chain B. These peptides are small, probably due to low cleavage selectivity of pepsin. This is a characteristic of pepsin, which can lead to overlapping peptides such as VEAL and ALY mentioned above.
CID spectra for other peptide ions 6–12 (Fig. 1b) are also acquired but their interpretation are not as straightforward as those for ions 1–5. Part of the reason is that the fragment ions from backbone cleavages are limited (see further discussion below). We therefore first examined the peptide electrolytic reduction behaviors. Based on our previous observation [43], disulfide bond-containing peptides have decreased intensities after electrolysis while peptides without disulfide bonds do not. This would provide a simple way to differentiate these two groups of peptides generated from protein digestion. In this experiment, the digested insulin solution was flowed through the thin-layer electrochemical cell with an applied potential of −1.5 V for electrochemically cleaving disulfide bonds. Indeed, using the ion 1 (i.e., +2 ion of YTPKA) as the reference peak (the reason for that is that the base peak changed after electrolysis so this disulfide-free peptide ion is selected to serve as the reference), the relative intensities of peptide ions 6–12 decreased considerably after electrolysis. As shown in Table S1 (Supporting Information), the changes of their normalized intensities (based on 1) were not less than 69.5% and some of them (e.g., ions 7, 11, 12) completely disappeared (Fig. 1c). By contrast, the peaks of disulfide-free peptide ions 2–5 have much smaller changes varying from −2.3% to +18.8%. The small variation is probably from mass spectrometric signal fluctuation. Therefore it is very likely that the ions 6–12 correspond to disulfide bond-containing peptides. By examining the charges and m/z of 6–12 ions (Fig. 1b), they correspond to 4 disulfide bond-containing peptides detected from insulin peptic digest, which are denoted as P1 (MW 2069.6 Da; +3 ion: 6; +2 ion: 10), P2 (MW 1537.0 Da; +2 ion: 7), P3 (MW 2559.4 Da; +3 ion: 8; +2 ion: 11) and P4 (MW 2787.6 Da; +3 ion: 9; +2 ion: 12), respectively (refer to Table 1 for actual structures of P1–P4).
Following the online electrolytic reduction, new peaks (e.g., ions 13–25, Fig. 1c) appeared, corresponding to the reduced peptides. By examining the charges and m/z of 13–25 (Fig. 1c), these ions are from 7 peptides generated from electrolysis at −1.5 V, which are denoted as a (MW 1045.1 Da; +1 ion: 22), b (MW 1026.3 Da; +2 ion: 16; +1 ion: 21), c (MW 512.5 Da; +1 ion: 15), d (MW 1253.3 Da; +3 ion: 13; +2 ion: 17; +1 ion: 23), e (MW 1310.1 Da; +2 ion: 19; +1 ion: 25), f (MW 1308.1 Da; +2 ion: 18; +1 ion: 24) and g (MW 1481.8 Da; +3 ion: 14; +2 ion: 20), respectively (refer to Table 1 for actual structures of a–g).
We then focus on the structural analysis of the peptides P1–P4 and the reduced peptides a–g by tandem mass analysis. We start from the P1 identification. Among the “pool” of the reduced peptides a–g, the only possible pair of peptides that come from P1 reduction are peptides a and b. Table S2 (Supporting Information) lists all possible combinations of electrochemically generated peptides a–g. Only the mass sum of a and b (MW 2071.4 Da) is close to that of P1 (MW 2069.6 Da) and the former value is higher than the latter by 1.8 Da. The mass difference suggests that P1 has one inter-chain disulfide bond linking its two chains, a and b. The theoretical mass difference should be 2.0 Da; due to the low resolution of the ion trap instrument, there is small difference between measured and theoretical masses. Upon CID, the ion 22 of m/z 1046.1 (i.e., +1 ion of peptide a) generates fragment ions of b4, b5, b5-NH3, b6, b6-NH3, b7, b7-NH3,y6, and y6-NH3 (Fig. 2b; fragment ions are also listed in Table S3 in the Supporting Information). Since b4, b5, b6 and b7 from successive backbone cleavage are observed, it tells the C-terminal sequence of peptide a, which can be assigned as XXXXNYCN (i.e., the chain A of P1; X represents an unidentified residue) and the cysteine residue is at the 7th position. Likewise, upon CID, ion 16 of m/z 514.5 (i.e., +2 ion of peptide b) yields b2, b3, b4, b7, b8,y1,y3,y4,y6,y7,y72+, and y8 (Fig.2c).These fragment ions cover all amide backbones and suggest the peptide b sequence to be LVCGERGFF (i.e., the chain B of P1), in which there is one cysteine residue located in the 3rd position. Therefore, P1 has sequence of XXXXNYCN/LVCGERGFF (the slash “/” represents the disulfide bond linkage between two chains) and the disulfide bond connects the 7th residue of chain A to the 3rd residue of chain B. This sequence is in agreement with the CID MS/MS spectrum of +3 ion of P1 at m/z 691.6 (i.e., ion 6) shown in Fig. 2a. Upon CID, ion 6 gives rise to fragment ions A(b2), A(b3), A(b4), A/B(b8)3+, B(y6), B/A(y4)2+, B/A(y5)2+, B/A(y6)2+, A/B(y7)2+, A/B(b8)2+, and B/A(b7)2+. In terms of notation, A(b2) refers to the b2 fragment ion of A chain and B/A(y4)2+ refers to y42+ ion of A chain which is linked with B chain by a disulfide bond. The formation of A(b2), A(b3), A(b4), B/A(Y4)2+, B/A(Y5)2+ and B/A(Y6)2+ fragment ions further shows the 3rd and 4th residues of chain A to be leucine and glutamic acid, respectively. Thereby, P1 can be further identified as XXLENYCN/LVCGERGFF, agreeing with the insulin sequence shown in Table 1. Clearly, just based on the CID of P1 ions, only 7 backbone cleavages are observed, which are not enough to determine either disulfide bond location or the peptide sequence. In contrast, by combining electrolytic reduction results, both the linkage of disulfide bond and most of sequence of P1 can be determined.
Fig. 2.

CID MS/MS spectra of (a) [P1+3H]3+(m/z 692); (b) singly charged peptide a (m/z 1046) and (c) doubly charged peptide b (m/z 514). Note that peptides a and b arose from electrolytic reduction of peptide P1.
Similar analysis can be done with P2, another disulfide bond-containing peptide. Among reduced peptides a–g, only the mass sum of peptide b and c (MW 1538.8 Da) is 1.8 Da higher than that of P2 (MW 1537.0 Da, Table S2, Supporting Information), revealing that b and c are linked by one inter-chain disulfide bond and they are the reduced products from P2. Upon CID, the ion 15 of m/z 513.5 (i.e., +1 ion of peptide c) yields b2, b3, and y2 (Fig. 2S–b, Supporting Information). Thus, the cysteine residue of c is pinpointed at the 3rd position due to the observation of b2 and b3, and the sequence of peptide c is suggested to be NYCN or YNCN. Since peptide b is known to be LVCGERGFF, P2 sequence should be either NYCN/LVCGERGFF or YNCN/LVCGERGFF and the disulfide bond connects the 3rd residue of chain A to the 3rd residue of chain B. One can see that P2 is a peptide overlapping with P1, which is due to the low cleavage selectivity of pepsin. The determined sequence of P2 also agrees with the CID MS/MS spectrum of P2 ion (i.e., ion 7 at m/z 769.5), which shows the fragment ions of A(b2), A/B(b7)2+, A/B(b8)2+, B/A(b3)2+, A/B(b3), A/B(b5), A/B(b6), B(Y2), B(Y4), B(Y2)2+,A/B(Y7)2+, B(y6), B/A(Y2), and A/B(y7) Fig. 2S–a, Supporting Information).
By electrolytic reduction, the relative intensity of +3 ion of P3 (i.e., ion 8) decreases by 96.3% (Table S1, Supporting Information), apparently suggesting the presence of disulfide bonds in P3. Among all possible combinations shown in Table S2 (Supporting Information), the mass sum of peptides d and e (MW 2563.4 Da) is 4.0 Da more than that of P3 (MW 2559.4 Da), which indicates that peptides d and e are the reduced products from P3 and there are two disulfide bonds in P3. CID of ion 17 (i.e., +2 ion of peptide d) produces fragment ions b2, b3, b5, b6, b7, b8, b9, b10, b102+, y2, y4, y5, y6, y7, y8,y8-NH3,y9, and y10 (Fig. 3b). The sequence of d can be therefore assigned as FVNQHLCGSHL in which there is one cysteine residue is at the 7th residue. In addition, CID of ion 19 (i.e., +2 ion of peptide e) yields b3, b4, b5, b6, b7, b8, b9, b10, b10-H2O, b11, b11-NH3, b12, b12-H2O, y3,y4,y5,y6,y7,y8,y9,y10, and y11 (Fig. 3c).The observation of successive b3–b12 and y3–y11 points out that there are three cysteine residues, located at 6th, 7th and 11th residues, respectively. Also, the fragmentation pattern shows the sequence of e to be XXVEQCCASVCSL. Because there are two disulfide bonds in P3, the peptide e should have one intra-chain disulfide and another interchain disulfide linkage with d for constituting P3. Since e has three cysteine residues, there are three possibilities for the intra-chain disulfide bond location, between Cys6 and Cys7, between Cys6 and Cys11, or between Cys7 and Cys11. Based on the formation of a series of A(b3), A(b4), A(b5), B/A(b11)2+ and B/A(b12)2+ from CID of +3 ion of P3 at m/z 854.8 (i.e., ion 8, Fig. 3a) and no backbone cleavage of the chain A (i.e., peptide e) between Cys6 and Cys11, it infers that Cys6 and Cys11 are bridged by an intra-disulfide bond linkage. The missing cleavage region between Cys6 and Cys11 of A chain is due to the protection by the intra-disulfide bond linkage. Therefore, the remaining Cys7 of A chain should be linked with the Cys7 of B chain via an inter-chain disulfide bond and the sequence of P3 is 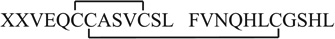 , agreeing with the insulin structure.
, agreeing with the insulin structure.
Fig. 3.

CID MS/MS spectra of (a) [P3+3H]3+ (m/z 855); (b) doubly charged peptide d(m/z 628); (c) doubly charged peptide e (m/z 656); and (d) [peptide f+H]+(m/z 1309). Note that e arose from full reduction of peptide P3 while f resulted from partial reduction of peptide P3.
As shown in Table S2 (Supporting Information), the mass sum of peptides d and another peptide f (MW 2561.4 Da) is 2.0 Da higher than that of P3 (MW 2559.4 Da), which indicates that f is a partially reduced chain A of P3. Indeed, the measured mass of peptide f (MW 1308.1 Da) is 2.0 Da less than that of peptide e (MW 1310.1 Da), based on the detection of their corresponding +1 ions, 24 (m/z 1309.1) and 25 (m/z 1311.1, Fig. 1c). We further testify this hypothesis by electrolytic reduction at different potentials. When the reduction potential was reduced from −1.5 V (Fig. 4b) to −1.2 V (Fig. 4a), peptide f was a major reduction product as +1 ion of f is much higher than +1 of e. When the potential was increased to −2.0 V, only peptide e was detected (Fig. 4c). These results clearly show that stepwise selective reduction can be achieved simply using different reduction potentials. The partially reduced product generated at lower reduction potential is structurally useful. In this case, CID of +1 ion of f gives rise to fragment ions b11, b12, y8, y9, y10 and y11 (Fig. 3d). Again, the backbone cleavages between Cys6 and Cys11 are not seen, confirming that an intra-chain disulfide bond bridges these two cysteines. This result agrees with the sequence of f of 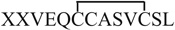 , as the partially reduced chain A of P3. It is also indicative that, upon electrochemical reduction, the interchain disulfide bond is preferentially reduced than the intra-chain disulfide bond. This observation is consistent with previous reports of electrochemistry research [30,54], probably due to the higher stress of the inter-chain disulfide bonds in comparison to the intra-chain ones. Using polarography, Cecil et al. showed that, at −1.35 V only the inter-chain disulfide bonds of insulin were reduced but all three bonds were reduced at−1.8 V [54]. Bard and co-workers [30] also reported that, for intact insulin molecules, the two inter-chain disulfide bonds were more rapidly reduced than the intra-chain disulfide bond. Partial reduction of peptides containing multiple disulfide bonds is often necessary for disulfide bond mapping. This is usually achieved by using different amount of chemical reductants for reduction in different time periods. In this study, we show that it can be achieved simply using electroreduction at different potentials.
, as the partially reduced chain A of P3. It is also indicative that, upon electrochemical reduction, the interchain disulfide bond is preferentially reduced than the intra-chain disulfide bond. This observation is consistent with previous reports of electrochemistry research [30,54], probably due to the higher stress of the inter-chain disulfide bonds in comparison to the intra-chain ones. Using polarography, Cecil et al. showed that, at −1.35 V only the inter-chain disulfide bonds of insulin were reduced but all three bonds were reduced at−1.8 V [54]. Bard and co-workers [30] also reported that, for intact insulin molecules, the two inter-chain disulfide bonds were more rapidly reduced than the intra-chain disulfide bond. Partial reduction of peptides containing multiple disulfide bonds is often necessary for disulfide bond mapping. This is usually achieved by using different amount of chemical reductants for reduction in different time periods. In this study, we show that it can be achieved simply using electroreduction at different potentials.
Fig. 4.

(+)-DESI-MS spectra showing the stepwise reduction of P3 at (a) applied potential of − 1.2 V; (b) applied potential of −1.5 V; and (c) applied potential of −2.0 V.
In addition, peptide P4 is the combination of peptides e and g (Table S2, Supporting Information), which also has two disulfide bonds. Upon CID (Fig. 3S–b), ion 20 at m/z 741.9 (i.e., +2 ion of peptide g) generates fragment ions b5–b10, b102+,y3,y4, and y8–12 (Table S3, Supporting Information). The fragmentation behavior suggests the sequence of g to be FVNQHLCGSHXXX and that one cysteine residue is located at its 7th residue position. Therefore, P4 is determined as 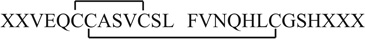 , which is an overlapping peptide with P3 and has a longer chain B. Upon CID of +3 ion of P4 at m/z 930.7 (i.e., ion 9, Fig. 3S–a in the Supporting Information), it generates fragment ions of B/A(b11)3+, B/A(b12)3+, A/B(b7)2+, B/A(b11)2+, B/A(b12)2+, B(y6), A/B(y11)3+, B/A(y11)3+, A/B(y12)3+, A/B(y8)2+, B/A(y8)2+, B/A(y9)2+, B/A(y10)2+, A/B(y11)2+, B/A(y11)2+, A/B(y12)2+, A/B(y10)2+, and A/B(y9)2+. Missing cleavage between Cys6 and Cys11 of the chain A further supports that the intra-chain disulfide bond connects the cysteine residues of 6th and 11th of the P4 chain A.
, which is an overlapping peptide with P3 and has a longer chain B. Upon CID of +3 ion of P4 at m/z 930.7 (i.e., ion 9, Fig. 3S–a in the Supporting Information), it generates fragment ions of B/A(b11)3+, B/A(b12)3+, A/B(b7)2+, B/A(b11)2+, B/A(b12)2+, B(y6), A/B(y11)3+, B/A(y11)3+, A/B(y12)3+, A/B(y8)2+, B/A(y8)2+, B/A(y9)2+, B/A(y10)2+, A/B(y11)2+, B/A(y11)2+, A/B(y12)2+, A/B(y10)2+, and A/B(y9)2+. Missing cleavage between Cys6 and Cys11 of the chain A further supports that the intra-chain disulfide bond connects the cysteine residues of 6th and 11th of the P4 chain A.
In addition, by coupling online digestion/online reduction/online mass analysis, it greatly shortens the time for sequencing and disulfide bond mapping experiments. In this study, it takes less than 7 min from the start of the protein sample injection to the detection of MS signal of digested and reduced peptides, which otherwise could take several hours. The time delay of 7 min is mainly due to the dead volumes of the pepsin packed column and the electrochemical cell and is also sample infusion rate dependent.
4. Conclusions
EC/DESI-MS in conjunction with online enzymatic digestion has been shown to be powerful in the structural analysis of disulfide bond-containing proteins. The most striking feature of the method is its speed. Besides, the employment of electrochemical reduction avoids the use of chemical reductants and the reduction behaviors of peptides in protein digests allow rapid identification of disulfide bond-containing peptides as well as their corresponding reduced products. Subsequent tandem MS analysis provides sequence information plus disulfide bond linkage patterns. Selective reduction of disulfide bonds using different potentials provides partially or fully reduced peptides which can be helpful in the structure determination of the precursor peptides. In summary, the proposed EC/DESI-MS method with online enzymatic digestion appears to be of value for improving traditional bottom-up analysis methods for the structure determination of proteins carrying disulfide bonds. In the future, fast enzymatic technique with high digestion specificity such as trypsin would be possible to further promote the method.
Supplementary Material
Acknowledgements
Financial support from NSF Career Award (CHE-1149367) and research support from Antec BV, Dr. Agnieszka Kraj and Mr. Martin Eysberg are gratefully acknowledged. We also thank support from NIH NIGMS (8 P41 GM103422-35 to Professor Michael L. Gross of Washington University in St. Louis).
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ijms.2013.04.009.
Contributor Information
Hao Zhang, Email: zhanghao@wuchem.wustl.edu.
Hao Chen, Email: chenh2@ohio.edu.
References
- 1.Alegre-Cebollada J, Kosuri P, Rivas-Pardo JA, Fernandez JM. Direct observation of disulfide isomerization in a single protein. Nature Chemistry. 2011;3:882–887. doi: 10.1038/nchem.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bilusich D, Bowie JH. Fragmentations of (M–H)- anions of underivatised peptides. Part 2: characteristic cleavages of Ser and Cys and of disulfides and other post-translational modifications, together with some unusual internal processes. Mass Spectrometry Reviews. 2009;28:20–34. doi: 10.1002/mas.20206. [DOI] [PubMed] [Google Scholar]
- 3.Gorman JJ, Wallis TP, Pitt JJ. Protein disulfide bond determination by mass spectrometry. Mass Spectrometry Reviews. 2002;21:183–216. doi: 10.1002/mas.10025. [DOI] [PubMed] [Google Scholar]
- 4.Jones MD, Patterson SD, Lu HS. Determination of disulfide bonds in highly bridged disulfide-linked peptides by matrix-assisted laser desorption/ionization mass spectrometry with post source decay. Analytical Chemistry. 1998;70:136–143. doi: 10.1021/ac9707693. [DOI] [PubMed] [Google Scholar]
- 5.Qiu X, Cui M, Li H, Liu Z, Liu S. Prompt disulfide fragmentations of disulfide-containing proteins in a matrix-assisted laser desorption/ionization source. Rapid Communications in Mass Spectrometry. 2007;21:3520–3525. doi: 10.1002/rcm.3230. [DOI] [PubMed] [Google Scholar]
- 6.Peng IX, Loo RO, Shiea J, Loo JA. Reactive-electrospray-assisted laser desorption/ionization for characterization of peptides and proteins. Analytical Chemistry. 2008;80:6995–7003. doi: 10.1021/ac800870c. [DOI] [PubMed] [Google Scholar]
- 7.Love CB, Tan L, Francisco JS, Xia Y. Competition of charge- vs. radical-directed fragmentation of gas-phase protonated cysteine sulfinyl radicals. Journal of the American Chemical Society. 2013 doi: 10.1021/ja4008744. [DOI] [PubMed] [Google Scholar]
- 8.Shaw JB, Ledvina AR, Zhang X, Julian RR, Brodbelt JS. Tyrosine deprotonation yields abundant and selective backbone cleavage in peptide anions upon negative electron transfer dissociation and ultraviolet photodissociation. Journal of the American Chemical Society. 2012;134:15624–15627. doi: 10.1021/ja3032086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parthasarathi R, He Y, Reilly JP, Raghavachari K. New insights into the vacuum UV photodissociation of peptides. Journal of the American Chemical Society. 2010;132:1606–1610. doi: 10.1021/ja907975v. [DOI] [PubMed] [Google Scholar]
- 10.Stinson CA, Xia Y. Radical induced disulfide bond cleavage within peptides via ultraviolet irradiation of an electrospray plume. Analyst. 2013;138:2840–2846. doi: 10.1039/c3an00303e. [DOI] [PubMed] [Google Scholar]
- 11.Calabrese A, Good N, Wang T, He J, Bowie J, Pukala T. A negative ion mass spectrometry approach to identify cross-linked peptides utilizing characteristic disulfide fragmentations. Journal of American Society for Mass Spectrometry. 2012;23:1364–1375. doi: 10.1007/s13361-012-0407-x. [DOI] [PubMed] [Google Scholar]
- 12.Thakur SS, Balaram P. Fragmentation of peptide disulfides under conditions of negative ion mass spectrometry: studies of oxidized glutathione and contryphan. Journal of American Society for Mass Spectrometry. 2008;19:358–366. doi: 10.1016/j.jasms.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Zhang M, Kaltashov IA. Mapping of protein disulfide bonds using negative ion fragmentation with a broadband precursor selection. Analytical Chemistry. 2006;78:4820–4829. doi: 10.1021/ac060132w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bilusich D, Bowie JH. Identification of intermolecular disulfide linkages in underivatised peptides using negative ion electrospray mass spectrometry. A joint experimental and theoretical study. Rapid Communications in Mass Spectrometry. 2007;21:619–628. doi: 10.1002/rcm.2872. [DOI] [PubMed] [Google Scholar]
- 15.Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM, Carpenter BK, McLafferty FW. Electron capture dissociation of gaseous multiply-charged proteins is favored at disulfide bonds and other sites of high hydrogen atom affinity. Journal of the American Chemical Society. 1999;121:2857–2862. [Google Scholar]
- 16.Gunawardena HP, Gorenstein L, Erickson DE, Xia Y, McLuckey SA. Electron transfer dissociation of multiply protonated and fixed charge disulfide linked polypeptides. International Journal of Mass Spectrometry. 2007;265:130–138. [Google Scholar]
- 17.Cole S, Ma X, Zhang X, Xia Y. Electron transfer dissociation (ETD) of peptides containing intrachain disulfide bonds. Journal of American Society for Mass Spectrometry. 2012;23:310–320. doi: 10.1007/s13361-011-0300-z. [DOI] [PubMed] [Google Scholar]
- 18.Ni W, Lin M, Salinas P, Savickas P, Wu S-L, Karger B. Complete mapping of a cystine knot and nested disulfides of recombinant human arylsulfatase a by multi-enzyme digestion and LC–MS analysis using CID and ETD. Journal of American Society for Mass Spectrometry. 2013;24:125–133. doi: 10.1007/s13361-012-0510-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu S-L, Jiang H, Hancock WS, Karger BL. Identification of the unpaired cysteine status and complete mapping of the 17 disulfides of recombinant tissue plasminogen activator using LC–MS with electron transfer dissociation/collision induced dissociation. Analytical Chemistry. 2010;82:5296–5303. doi: 10.1021/ac100766r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu S-L, Jiang H, Lu Q, Dai S, Hancock WS, Karger BL. Mass spectrometric determination of disulfide linkages in recombinant therapeutic proteins using online LC–MS with electron-transfer dissociation. Analytical Chemistry. 2008;81:112–122. doi: 10.1021/ac801560k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia Y, Cooks RG. Plasma induced oxidative cleavage of disulfide bonds in polypeptides during nanoelectrospray ionization. Analytical Chemistry. 2010;82:2856–2864. doi: 10.1021/ac9028328. [DOI] [PubMed] [Google Scholar]
- 22.Barlow CK, O'Hair RAJ. Gas-phase peptide fragmentation: how understanding the fundamentals provides a springboard to developing new chemistry and novel proteomic tools. Journal of Mass Spectrometry. 2008;43:1301–1319. doi: 10.1002/jms.1469. [DOI] [PubMed] [Google Scholar]
- 23.Chrisman PA, McLuckey SA. Dissociations of disulfide-linked gaseous polypeptide/protein anions: ion chemistry with implications for protein identification and characterization. Journal of Proteome Research. 2002;1:549–557. doi: 10.1021/pr025561z. [DOI] [PubMed] [Google Scholar]
- 24.Lioe H, Duan M, O'Hair RAJ. Can metal ions be used as gas-phase disulfide bond cleavage reagents? A survey of coinage metal complexes of model peptides containing an intermolecular disulfide bond. Rapid Communications in Mass Spectrometry. 2007;21:2727–2733. doi: 10.1002/rcm.3122. [DOI] [PubMed] [Google Scholar]
- 25.Kim HI, Beauchamp JL. Identifying the presence of a disulfide linkage in peptides by the selective elimination of hydrogen disulfide from collisionally activated alkali and alkaline earth metal complexes. Journal of the American Chemical Society. 2008;130:1245–1257. doi: 10.1021/ja075698w. [DOI] [PubMed] [Google Scholar]
- 26.Gunawardena HP, O'Hair RAJ, McLuckey SA. Selective disulfide bond cleavage in gold(I) cationized polypeptide ions formed via gas-phase ion/ion cation switching. Journal of Proteome Research. 2006;5:2087–2092. doi: 10.1021/pr0602794. [DOI] [PubMed] [Google Scholar]
- 27.Mihalca R, van der Burgt YEM, Heck AJR, Heeren RMA. Disulfide bond cleavages observed in SORI-CID of three nonapeptides complexed with divalent transition-metal cations. Journal of Mass Spectrometry. 2007;42:450–458. doi: 10.1002/jms.1175. [DOI] [PubMed] [Google Scholar]
- 28.Teesch LM, Orlando RC, Adams J. Location of the alkali metal ion in gas-phase peptide complexes. Journal of the American Chemical Society. 1991;113:3668–3675. [Google Scholar]
- 29.Honeychurch MJ. The reduction of disulfide bonds in proteins at mercury electrodes. Bioelectrochemistry and Bioenergetics. 1997;44:13–21. [Google Scholar]
- 30.Stankovich MT, Bard AJ. The electrochemistry of proteins and related substances: part II. Insulin. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry. 1977;85:173–183. [Google Scholar]
- 31.Havlis J, Thomas H, Sebela M, Shevchenko A. Fast-response proteomics by accelerated in-gel digestion of proteins. Analytical Chemistry. 2003;75:1300–1306. doi: 10.1021/ac026136s. [DOI] [PubMed] [Google Scholar]
- 32.Juan H-F, Chang S-C, Huang H-C, Chen S-T. A new application of microwave technology to proteomics. Proteomics. 2005;5:840–842. doi: 10.1002/pmic.200401056. [DOI] [PubMed] [Google Scholar]
- 33.Pramanik BN, Mirza UA, Ing YH, Liu Y-H, Bartner PL, Weber PC, Bose AK. Microwave-enhanced enzyme reaction for protein mapping by mass spectrometry: a new approach to protein digestion in minutes. Protein Science. 2002;11:2676–2687. doi: 10.1110/ps.0213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun W, Gao S, Wang L, Chen Y, Wu S, Wang X, Zheng D, Gao Y. Microwave-assisted protein preparation and enzymatic digestion in proteomics. Molecular & Cellular Proteomics. 2006;5:769–776. doi: 10.1074/mcp.T500022-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.López-Ferrer D, Capelo JL, Vázquez J. Ultra fast trypsin digestion of proteins by high intensity focused ultrasound. Journal of Proteome Research. 2005;4:1569–1574. doi: 10.1021/pr050112v. [DOI] [PubMed] [Google Scholar]
- 36.Carreira RJ, Cordeiro FM, Moro AJ, Rivas MG, Rial-Otero R, Gaspar EM, Moura I, Capelo JL. New findings for in-gel digestion accelerated by high-intensity focused ultrasound for protein identification by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Journal of Chromatography A. 2007;1153:291–299. doi: 10.1016/j.chroma.2006.09.078. [DOI] [PubMed] [Google Scholar]
- 37.Rial-Otero R, Carreira RJ, Cordeiro FM, Moro AJ, Santos HM, Vale G, Moura I, Capelo JL. Ultrasonic assisted protein enzymatic digestion for fast protein identification by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry: sonoreactor versus ultrasonic probe. Journal of Chromatography A. 2007;1166:101–107. doi: 10.1016/j.chroma.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 38.Dycka F, Bobal P, Mazanec K, Bobalova J. Rapid and efficient protein enzymatic digestion: an experimental comparison. Electrophoresis. 2012;33:288–295. doi: 10.1002/elps.201100123. [DOI] [PubMed] [Google Scholar]
- 39.Bao H, Liu T, Chen X, Chen G. Efficient in-gel proteolysis accelerated by infrared radiation for protein identification. Journal of Proteome Research. 2008;7:5339–5344. doi: 10.1021/pr800572e. [DOI] [PubMed] [Google Scholar]
- 40.Wang S, Bao H, Zhang L, Yang P, Chen G. Infrared-assisted on-plate proteolysis for MALDI-TOF-MS peptide mapping. Analytical Chemistry. 2008;80:5640–5647. doi: 10.1021/ac800349u. [DOI] [PubMed] [Google Scholar]
- 41.Wang L, Pan H, Smith DL. Hydrogen exchange-mass spectrometry: optimization of digestion conditions. Molecular & Cellular Proteomics. 2002;1:132–138. doi: 10.1074/mcp.m100009-mcp200. [DOI] [PubMed] [Google Scholar]
- 42.Ahn J, CJung M, Wyndham K, Yu YQ, Engen JR. Pepsin immobilized on high-strength hybrid particles for continuous flow online digestion at 10000 psi. Analytical Chemistry. 2012;84:7256–7262. doi: 10.1021/ac301749h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Dewald HD, Chen H. Online mass spectrometric analysis of proteins/peptides following electrolytic cleavage of disulfide bonds. Journal of Proteome Research. 2011;10:1293–1304. doi: 10.1021/pr101053q. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Cui W, Zhang H, Dewald HD, Chen H. Electrochemistry-assisted top-down characterization of disulfide-containing proteins. Analytical Chemistry. 2012;84:3838–3842. doi: 10.1021/ac300106y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takats ZWJM, Gologan B, Cooks RG. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science. 2004;306:471–473. doi: 10.1126/science.1104404. [DOI] [PubMed] [Google Scholar]
- 46.Venter A, Nefliu M, Graham Cooks R. Ambient desorption ionization mass spectrometry. Trends in Analytical Chemistry. 2008;27:284–290. [Google Scholar]
- 47.Cooks RG, Ouyang Z, Takats Z, Wiseman JM. Ambient Mass Spectrometry. Science. 2006;311:1566–1570. doi: 10.1126/science.1119426. [DOI] [PubMed] [Google Scholar]
- 48.Wiseman JM, Puolitaival SM, Takáts Z, Cooks RG, Caprioli RM. Mass spectrometric profiling of intact biological tissue by using desorption electrospray ionization. Angewandte Chemie International Edition. 2005;44:7094–7097. doi: 10.1002/anie.200502362. [DOI] [PubMed] [Google Scholar]
- 49.Lu M, Wolff C, Cui W, Chen H. Investigation of some biologically relevant redox reactions using electrochemical mass spectrometry interfaced by desorption electrospray ionization. Analytical and Bioanalytical Chemistry. 2012;403:355–365. doi: 10.1007/s00216-011-5679-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miao Z, Chen H. Direct analysis of liquid samples by desorption electrospray ionization-mass spectrometry (DESI-MS) Journal of American Society for Mass Spectrometry. 2009;20:10–19. doi: 10.1016/j.jasms.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 51.Miao Z, Wu S, Chen H. The study of protein conformation in solution via direct sampling by desorption electrospray ionization mass spectrometry. Journal of American Society for Mass Spectrometry. 2010;21:1730–1736. doi: 10.1016/j.jasms.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moore BN, Hamdy O, Julian RR. Protein structure evolution in liquid DESI as revealed by selective noncovalent adduct protein probing. International Journal of Mass Spectrometry. 2012;330–332:220–225. doi: 10.1016/j.ijms.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takáts Z, Wiseman JM, Gologan B, Cooks RG. Electrosonic spray ionization. A gentle technique for generating folded proteins and protein complexes in the gas phase and for studying ion-molecule reactions at atmospheric pressure. Analytical Chemistry. 2004;76:4050–4058. doi: 10.1021/ac049848m. [DOI] [PubMed] [Google Scholar]
- 54.Cecil R, Weitzman PDJ. The electroreduction of the disulfide bonds of insulin and other proteins. Biochemical Journal. 1964;93:1–11. doi: 10.1042/bj0930001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.