

Abstract
Immunodeficiency, centromeric instability and facial anomalies type I (ICF1) syndrome is a rare genetic disease caused by mutations in DNA methyltransferase (DNMT) 3B, a de novo DNA methyltransferase. However, the molecular basis of how DNMT3B deficiency leads to ICF1 pathogenesis is unclear. Induced pluripotent stem cell (iPSC) technology facilitates the study of early human developmental diseases via facile in vitro paradigms. Here, we generate iPSCs from ICF Type 1 syndrome patient fibroblasts followed by directed differentiation of ICF1-iPSCs to mesenchymal stem cells (MSCs). By performing genome-scale bisulfite sequencing, we find that DNMT3B-deficient iPSCs exhibit global loss of non-CG methylation and select CG hypomethylation at gene promoters and enhancers. Further unbiased scanning of ICF1-iPSC methylomes also identifies large megabase regions of CG hypomethylation typically localized in centromeric and subtelomeric regions. RNA sequencing of ICF1 and control iPSCs reveals abnormal gene expression in ICF1-iPSCs relevant to ICF syndrome phenotypes, some directly associated with promoter or enhancer hypomethylation. Upon differentiation of ICF1 iPSCs to MSCs, we find virtually all CG hypomethylated regions remained hypomethylated when compared with either wild-type iPSC-derived MSCs or primary bone-marrow MSCs. Collectively, our results show specific methylome and transcriptome defects in both ICF1-iPSCs and differentiated somatic cell lineages, providing a valuable stem cell system for further in vitro study of the molecular pathogenesis of ICF1 syndrome. GEO accession number: GSE46030.
INTRODUCTION
DNA cytosine methylation is a major epigenetic factor that regulates many developmental events including genomic imprinting, X-inactivation and developmental gene regulation (1). DNA methylation is established and maintained by a family of DNA methyltransferases (DNMTs) including DNMT1, DNMT3A and DNMT3B (2). Mutations in these enzymes have been associated with a number of human diseases such as cancer, immunodeficiency and neurological disorders (3). For example, DNMT1 mutations are linked to hereditary sensory and autonomic neuropathy type 1 disease in which patients develop dementia and hearing loss in adulthood (4). DNMT3A mutations are coupled with subtypes of leukemia (5,6), and DNMT3B mutations cause a rare genetic disorder namely ICF syndrome (immunodeficiency, centromere instability and facial anomalies) (7,8). Relevant to the loss of DNMT3B activity, lymphoblasts from ICF patients exhibit hypomethylation of pericentromeric regions and selective demethylation of promoters associated with altered gene expression (3,9,10). Collectively, these results have highlighted the need for precise regulation of DNA methylation patterns for normal growth and development.
DNA methylation plays an important role in stem cell differentiation (11,12). We and others have demonstrated that DNA methylation acts in parallel with other known regulatory mechanisms such as transcriptional networks and histone modifications to control tissue- and cell-specific gene expression patterns (13–17). Upon differentiation, mouse and human embryonic stem cells (mESCs and hESCs) undergo a wave of de novo methylation which serves to silence pluripotency genes and establish tissue-specific methylation patterns (14,15,18,19). In the absence of Dnmts, demethylated mESCs undergo apoptosis soon after inducing in vitro differentiation (20,21). More recently, there has been increasing interest in elucidating the function of DNA methylation during cell reprogramming. It has been postulated that epigenetic barriers such as DNA methylation have to be overcome in order to achieve successful reprogramming of induced pluripotent stem cells (iPSCs). Indeed, selective promoter demethylation of pluripotent genes such as Oct4 and Nanog are associated with successful reprogramming of somatic cells to iPSCs (22–26). In addition, inhibiting DNMTs activities with 5-aza-cytidine, or DNMT1 knockdown, promotes partially reprogrammed cells into fully reprogrammed iPSCs (22,23,27). On the other hand, a wave of de novo methylation also occurs during late stage reprogramming (28). During this phase, tissue-specific genes and so-called partially methylated domains (PMDs) becomes hypermethylated (29,30), and hallmark enrichment of non-CG methylation becomes re-established in iPSCs (29,31,32). Together, these results indicate that major methylome alterations underlie cellular reprogramming.
To understand how DNMT3B contributes to methylation changes during cellular reprogramming and differentiation, we generated iPSCs from human Immunodeficiency, centromeric instability and facial anomalies type I (ICF1) syndrome patient fibroblasts carrying DNMT3B mutations, and profiled their global methylation levels and patterns through genome-wide bisulfite sequencing. We identified several unique targets of DNMT3B at both the large-scale megabase domains as well as at select gene promoters and enhancers. ICF1 iPSCs also exhibit dramatic loss of non-CG methylation, indicating that DNMT3B is the major enzyme for catalyzing this process. Through RNA sequencing, we identified altered gene expression in ICF1 iPSCs linked to the disease phenotype in ICF1 syndrome patients. Finally, upon differentiation of ICF1 iPSCs to mesenchymal stem cells (MSCs), we found many aberrations are preserved in ICF iPSC-derived MSCs. Overall, these data suggest that ICF iPSCs would be a valuable tool to study ICF pathogenesis in vitro.
RESULTS
Generation of human iPSCs from patient fibroblasts carrying DNMT3B mutations
To study the early molecular pathogenesis of ICF1 syndrome, we generated iPSCs derived from two fibroblast lines of ICF patients. These ICF1 fibroblasts carry compound heterozygous point mutations in the catalytic domain of DNMT3B, typical of type 1 ICF syndrome (Fig. 1A; see Material and Methods). Similar to Dnmt3b-deficient mouse somatic cells (33), ICF fibroblasts can also be reprogrammed with no obvious difference in reprogramming efficiency. Immunostaining and RT-PCR demonstrated ICF1-iPSCs expression of typical markers for pluripotency equivalent to normal iPSCs and hESCs (Supplementary Material, Fig. S1a and b). Incidentally, DNMT3A and DNMT3B transcript expressions are unaffected in ICF1 iPSCs (Supplementary Material, Fig. S1a). ICF1 iPSCs form teratomas in vivo, demonstrating that these reprogrammed cells can be differentiated into all three germ layer lineages (Supplementary Material, Fig. S1c). Interestingly, in contrast to major chromosomal instabilities seen in ICF1 patient B cells, ICF1-iPSCs karyotype appeared normal (Supplementary Material, Fig. S1d). However, hallmark hypomethylation at Satellite 2 repeats were consistently hypomethylated in ICF iPSCS (Supplementary Material, Fig. S1e). Overall, we conclude that DNMT3B is dispensable for reprogramming, but retains typical methylation patterns as previously observed in somatic cells.
Figure 1.
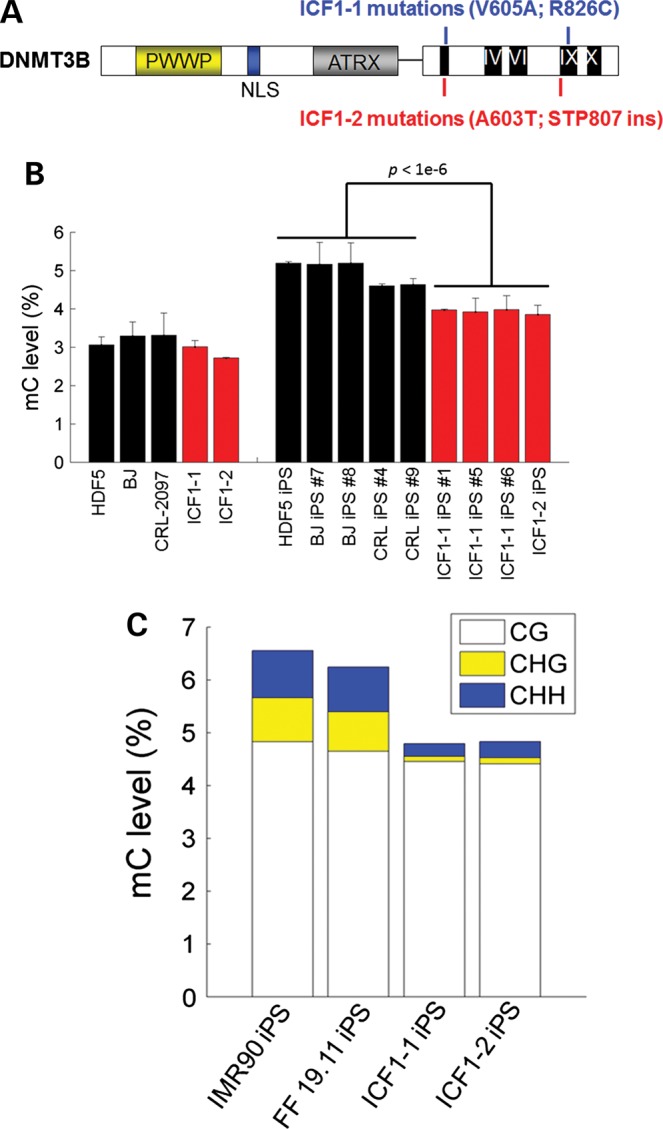
DNMT3B mutations lead to global DNA hypomethylation. (A). Schematic of the compound heterozygous point mutations in DNMT3B from two separate ICF1 syndrome patients. (B). Bar plot of LC–MS/MS-MRM measurements of total methylcytosine (5mC) levels in various cell types. 5mC levels are expressed as percentage of the total cytosine pool. Data represent mean and SDs from triplicate analyses. (C) Bar chart showing global methylation in CG, CHG and CHH motifs as measured by whole-genome shotgun bisulfite sequencing. Levels of CG (white), CHG (yellow) or CHH (blue) methylation are expressed as a percentage of total shared cytosine residues in all four cell lines (n = 9 766 941 CGs; n = 46 480 622 CHGs; and n = 117 356 835 CHHs).
DNMT3B contributes to de novo methylation during cellular reprogramming
iPSCs exhibit higher levels of methylation compared with somatic cells, suggesting that reprogramming involves a wave of de novo methylation. Because Dnmt3B is more dramatically upregulated in iPSCs when compared with levels of Dnmt1 and Dnmt3A (34), we hypothesized DNMT3B may play a major role in regulating methylation during cell reprogramming. To investigate the effect of DNMT3B deficiency on DNA methylation during reprogramming, we first measured global levels of 5mdC by combined liquid chromatography-tandem mass spectrometry with multiple reaction monitoring (LC–MS/MS-MRM) in a panel of somatic and iPS cells including ICF samples. Our analysis revealed that ICF iPSCs exhibit significantly reduced levels of global methylation compared with normal iPSCs (Student's t-test, P < 1 × 10−6) (Fig. 1B). In contrast, ICF1 fibroblasts have relatively similar levels of methylation compared with normal fibroblasts, which is consistent with previous literature (35). However, ICF1 iPSCs have higher methylation levels compared with both normal and ICF1 fibroblasts, indicating that DNMT3A also contributes to a wave of de novo methylation during cell reprogramming. To precisely map DNA methylation alterations in ICF1 iPSCs at base resolution, we performed whole-genome shotgun bisulfite sequencing in two separate ICF1 patient iPSCs and achieved an average coverage of 10× (ICF1-1 iPS) and 4× (ICF1-2 iPS) per strand. Consistent with the global levels of methylation as determined by LC–MS/MS-MRM, whole-genome bisulfite sequencing estimated a similar loss of global methylation in both ICF iPSC lines compared with control (Fig. 1C). Notably, our base pair resolution revealed non-CG methylation levels were drastically reduced, whereas CG methylation was only mildly diminished (Fig. 1C). Loss of non-CG methylation appeared to be uniform across chromosomes and genes (Supplementary Material, Fig. S2), which was confirmed at select loci using conventional bisulfite sequencing (Supplementary Material, Fig. S3).
Although the vast majority of non-CG methylation was abrogated in ICF iPSCs, some residual non-CG methylation was detected. Interestingly, both ICF1-1 and ICF1-2 iPSCs had greater levels of non-CG methylation compared with their parental fibroblast cell lines (Supplementary Material, Fig. S4a). Strikingly, the few remaining methylated non-CG sites were significantly conserved in both ICF1 iPSCs, suggesting that DNMT3A has a conserved minor role for targeting non-CG sites (Supplementary Material, Fig. S4b). Sequence motif analysis of the highly methylated non-CGs in control iPSCs reveals an enriched TACAG motif, consistent with previous reports (36) (Supplementary Material, Fig. S4b). However, the TACAG motif is abolished in the highly methylated non-CG sites in ICF iPSCs, though a strong CA motif is still present. Collectively, these data indicate that DNMT3A can redundantly compensate for DNMT3B deficiency at most CpG sites, but DNMT3B is the foremost enzyme in establishing non-CG methylation in human iPSCs.
Minor role for non-CG methylation in ICF1 iPSCs
Non-CG methylation was initially reported to have a positive association with gene expression in hESCs (31). To determine functional consequences for loss of non-CG methylation in ICF1 iPSCs, we performed mRNA-Seq on ICF1 and control iPSCs (Fig. 2A). Because average levels of non-CG methylation are relatively low (∼5%) compared with CG methylation (>80%), non-CG methylation may have subtle influence on gene expression. We therefore used relatively relaxed criterion to reduce the number of false negatives in differentially expressed genes (FDR <10% and >1.5-fold). Despite the loose cutoff, 270 and 195 significantly up- and downregulated genes in ICF iPSCs were identified (Fig. 2A; Supplementary Material, Table S1), indicating non-pervasive effects from loss of non-CG methylation. Indeed, non-CG methylation patterns across upregulated genes were no different from the genomic average in control hiPSCs (Supplementary Material, Fig. S5), suggesting that upregulated genes were probably not due to loss of non-CG methylation. Conversely, downregulated genes tended to have lower non-CG methylations in the gene-body compared with the genomic average. However, the reduced levels of non-CG methylation were also accompanied by reduced CG methylation levels, consistent with previous reports that non-CG methylation tend to flank methylated CGs (37,38). Gene ontology analysis revealed no functional enrichment for the set of upregulated genes, whereas the downregulated set of genes were significantly enriched for terms in gastrulation, neural development and cell adhesion (Fig. 2B).
Figure 2.
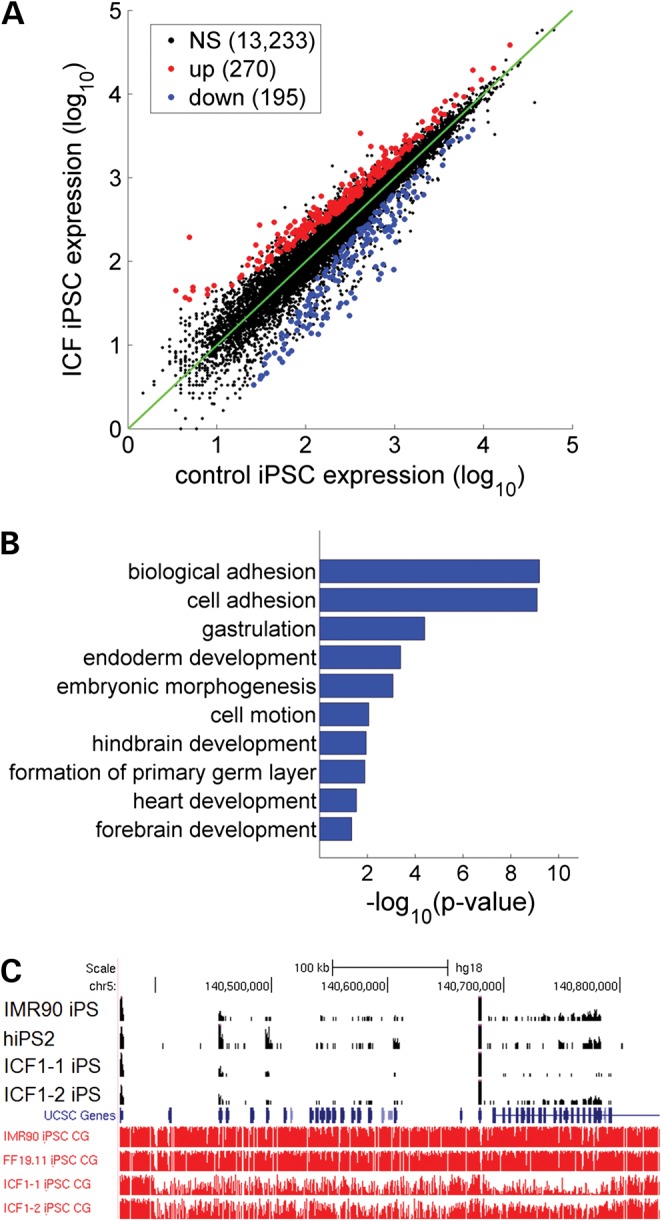
ICF1 iPSCs exhibit altered gene expression. (A). Scatterplot of average expression levels from ICF1-1 and ICF1-2 and control IMR90/FF1911 iPSCs are shown on X- and Y-axes, respectively. Significantly up- and downregulated genes (FDR <10% and fold-change >1.5) in ICF1 iPSCs are shown in red and blue, respectively. (B). Gene ontology terms of downregulated genes in ICF1 iPSCs. The abscissa shows Benjamani–Hochberg-adjusted P-values. (C). Genome-browser view of demethylated protocadherin gene clusters. Top four tracks (in black) are normalized RNA-seq reads (RPM) and bottom four tracks are CG methylation levels (in red) as measured by bisulfite sequencing (Y-axis limits between 0 and 1).
A previous report identified genes that appeared differentially regulated by non-CG methylation in mouse glial and neuronal cells (39). For example, 174 genes were uniquely CH hypermethylated in glial cells but not neurons. We asked whether these genes potentially share similar non-CG regulation in hiPSCs. Strikingly, we found a small yet significant overlap of 7 (out of 134 expressed in hiPSCs) neuronal genes that are mCH hypermethylated in control hiPSCs, but are downregulated in ICF1 hiPSCs (P = 6.6 × 10−4, hypergeometric test). These genes included FOXG1, PLXNA2, SPRY2, TUBB2A and most notably, the gamma protocadherin cluster (PCDHG-3, -4 and -5). With exception of the PCDH gene cluster, all other genes were not differentially methylated in the CG context and therefore may be downregulated due to loss of non-CG methylation. By visual inspection of the protocadherin clusters, we found dramatic CG demethylation across the entire cluster (>30%) associated with gene repression (Fig. 2C). Collectively, our findings are consistent with the previously reported positive correlation between non-CG methylation and gene expression (31), though it is still unclear whether non-CG methylation directly regulates these genes. Nevertheless, many downregulated genes involved in the neural development pathway may contribute to mental defects observed in ICF syndrome patients.
Select regions of CG demethylation in ICF1 iPSCs
Although global CG methylation patterns appeared normal across chromosomes, we observed select regions of hypomethylation in ICF1 iPSCs in both megabase and kilobase regions. For example, megabase regions were dramatically demethylated in pericentromeric and subtelomeric regions (Supplementary Material, Fig. S2). Satellite 2 repeats (typically found at pericentromeric regions) were confirmed to be selectively demethylated but not other repeat motifs (Supplementary Material, Fig. S6). In total, we identified 110 Mb demethylated regions that showed at least 30% reduced CG methylation (Supplementary Material, Table S2). Overall, these regions represent <10% of all analyzed CpGs and are also hypomethylated in fibroblast cells, suggesting impaired de novo methylation during reprogramming (Supplementary Material, Fig. S7). Intriguingly, analysis of DNMT binding in these regions did not show particular enrichment for DNMT3B compared with DNMT3A or DNMT1 (Supplementary Material, Fig. S8).
Unexpectedly, megabase regions of CpG demethylation in ICF-iPSCs had a significant overlap (∼80%) and share well-defined borders with previously identified non-CG DMRs (29) (Fig. 3A). In normal hiPSCs, non-CG DMRs are megabase domains with similar levels of CG methylation, but lower levels of non-CG methylation when compared with hESCs. Importantly, somatic cells also show low levels of CpG methylation in these genomic regions (29) (Fig. 3B). Therefore, the partial reduction of CpG methylation in ICF-iPSCs supports the notion that DNMT3B is involved in de novo CpG methylation at non-CG DMRs in normal iPSCs (Fig. 3A-B). Strikingly, in normal hESCs, non-CG DMR regions are depleted of 5hmC, though the boundaries are clearly demarked by 5hmC enrichment (Supplementary Material, Fig. S9). This is reminiscent of DNA methylation canyons recently characterized in mouse hematopoietic stem cells (40). Altogether, our analysis shows non-CG DMRs represent a unique epigenetic context that involves specialized targeting of DNMT3B for maintaining CG but not non-CG methylation.
Figure 3.

ICF iPSCs have aberrant megabase domains of hypomethylation. (A). Genome browser view of methylation level in a representative normal iPSCs and ICF1-1 iPSCs. Note coincidence of reduced non-CG methylation in normal iPSCs and reduced CG methylation in ICF1 iPSCs. CG methylation tracks are colored in red (y-axis limits between 0 and 1) and non-CG methylation tracks are colored in blue (y-axis limits between 0 and 0.05 for CHG methylation and 0–0.02 for CHH methylation). (B). Metaplot analysis of CG methylation in non-CG DMRs for control and ICF1 iPSCs as well as somatic cells. (C). Methylation profile of X-chromosome in ICF1 (red) and control (black) iPSCs. Loci of upregulated genes are shown in red dots and known loci of X-chromosome inactivation escape genes are shown in blue dots. (D). Average methylation profile across LADs in control and ICF iPSCs and somatic cells.
Next, we examined the methylation status at other large-scale regions of reduced CpG methylation in somatic cells, termed PMDs, which normally become fully methylated in iPSCs (29). However, PMDs in ICF iPSCs showed a range of methylation levels, indicating some PMDs do not fully reacquire DNA methylation in the absence of DNMT3B, whereas others can be compensated by DNMT3A in ICF iPSCs (Supplementary Material, Fig. S10). We also looked into nuclear lamina-associated domains (or LADs). Although LAD methylation levels are comparable between normal and ICF iPSCs, we found dramatic LAD hypomethylation in ICF lymphoblasts (Fig. 3C). These observations are consistent with the normal karyotype of ICF1 iPSCs, but atypical of the karyotype observed in ICF lymphoblasts. Thus, LAD hypomethylation appears specific to the hematopoietic lineage, and may contribute to chromosome aberrations observed uniquely in ICF B cells.
Although ICF lymphoblasts displayed globally hypomethylated genes compared with control lymphoblasts, they are most severely demethylated in LADs and the X-chromosome. However, unlike LADs, ICF iPSCs also exhibit chromosome-wide demethylation on the X-chromosome (Fig. 3D). Strikingly, X-chromosome demethylation was associated with a large number of upregulated genes, many of which were found on the X-chromosome short arm. Further analysis showed that many of these genes (∼25%) are previously characterized XCI-escape genes, while the remaining are associated with inactive genes. Thus, our current study indicates a greater number of deregulated X-linked genes upon X-chromosome demethylation than was previously appreciated.
Select gene promoter and enhancer hypomethylation in ICF1 iPSCs
We next examined whether DNMT3B could directly regulate a subset of gene promoters or enhancers. We found that 19 (15%) upregulated genes showed at least 20% promoter demethylation, indicating that these genes may be uniquely regulated by DNMT3B (Fig. 4A). For example, TCERG1L and FAM19A5 showed localized proximal promoter demethylation concomitant with increased gene expression (Fig. 4B). DNMT3B targeting did not appear to depend on promoter CG density. TCERG1L and FAM19A5, RBMXL2 and ZNF560 gene promoters contain CpG islands, whereas gene promoters for NLRP2 and CXorf61 have low CG density. Some downregulated genes also showed significant promoter demethylation, but many of these can be explained by broad demethylation of the entire protocadherin cluster (Fig. 2C). These genes are likely downregulated by some indirect mechanism.
Figure 4.

Selective promoter and enhancer demethylation in ICF1 iPSCs. (A). Scatterplot showing the relative fold change in gene expression (y-axis) and absolute changes in proximal promoter methylation (x-axis) for the subset of differentially expressed genes as shown in Figure 2A. Genes with significant promoter demethylation (delta-beta >30%) and transcript up- or downregulated are colored in red and blue, respectively. (B). Genome browser view of the FAM19A5 gene locus. Top four tracks (in black) are normalized RNA-seq reads (RPM) and bottom four tracks are CG methylation levels (in red) as measured by bisulfite sequencing. Both alternative promoters are selectively demethylated (highlighted in blue bar). (C). Bar graph showing number of hypomethylated enhancers in various cell types that are also hypomethylated in ICF1 iPSCs compared with control iPSCs. (D). Genome browser example of a specifically hypomethylated enhancer that only exhibit H3K27ac occupancy in GM12878 lymphoblasts but not H1 hESCs. Red tracks are methylation levels measured by bisulfite sequencing (Y-axis limits between 0 and 1). Blue tracks are normalized tags of H3K27ac from ENCODE.
We next examined whether deregulation of enhancers could affect some of the observed gene expression changes. Globally, enhancers showed significant demethylation in ICF1 iPSCs (P < 0.01, Kolmogorov–Smirnov test). In total, we identified, 39 (∼0.8%) class I enhancers and 54 (2.4%) class II enhancers that were demethylated by >50% compared with control iPSCs (Supplementary Material, Fig. S11). Class I enhancers are characterized with H3K4me1 and H3K27ac marks, whereas class II enhancers are associated with H3K4me1 and H3K27me3 marks (41). To gain greater perspective for these enhancers, we leveraged publically available methylomes to determine whether ICF iPSCs share enhancer methylation patterns with other cell types. Our analysis indicated enhancer demethylation was associated with several different cell types (Fig. 4C). These cell-type-specific enhancers also gain H3K27ac, consistent with newly acquired enhancer activity (Fig. 4D). Together, our data suggest multiple modes for gene deregulation by DNMT3B.
We compared our methylome and gene expression dataset to publically available data on ICF1 patient lymphoblasts (9,42), and found some deregulated genes observed in ICF1 iPSCs were also seen in lymphoblasts. For example, previous studies have found deregulated genes involved in immune function and neurogenesis in ICF1 patient lymphoblasts (9,42). By cross-referencing our set of deregulated genes in ICF1 iPSCs with previous studies, we identified 11 commonly upregulated genes between ICF1 iPSCs and ICF1 lymphoblastoids (P = 7.7 × 10−4, hypergeometric test) including ADCY1, BEX2, CASK, EIF2S3, FRAS1, GLT1D1, KCNN2, PDPN, PLS3, TBL1X and WWC3. Of these, five have been implicated in neural function, and another three are involved in cell adhesion. Notably, GLTD1 showed selective promoter demethylation, indicating GLTD1 may be a direct target of DNMT3B (Supplementary Material, Fig. S12). Commonly downregulated genes include COL5A1, JAKMIPI, RHOBTB3, S100A11 and a cluster of protocadherin (PCDH) genes (Fig. 2C). Deregulation of the PCDH gene cluster may be associated with the neural deficiency observed in ICF syndrome patients. Together, our results demonstrated that ICF iPSCs exhibit characteristic features of gene expression associated with disease phenotypes of ICF syndrome.
Differentiation of ICF1 iPSCs into MSCs
To demonstrate that ICF iPSCs provide a valuable resource for modeling ICF pathogenesis, we differentiated ICF iPSCs into MSCs following a previously established protocol (43) (Supplementary Material, Fig. S13). LC–MS/MS-MRM analysis showed that ICF-derived MSCs exhibited slightly reduced global methylation compared with ES- or iPSC-derived MSCs (Fig. 5A). We next performed reduced representation bisulfite sequencing (RRBS) for ICF1-iPSCs-derived MSCs and a subset of control MSCs including primary bone marrow cells. Our RRBS data consistently showed globally minor changes in CG methylation (Fig. 5B). Since RRBS does not comprehensively cover the genome, we were primarily able to assess differential methylation at CG islands and gene promoters. In total, we identified 181 gene promoters demethylated by >30% in ICF1 MSCs when compared with hESC-derived MSCs and primary bone marrow. Strikingly, 87 (48%) of demethylated promoters in ICF1-MSCs overlapped with demethylated promoters identified in ICF1 iPSCs (P < 10−16, hypergeometric test) (Fig. 5C). These shared gene promoters included FAM19A5 and the protocadherin cluster of genes (Fig. 5D). As expected, gene ontology analysis of demethylated promoters in ICF1-MSCs revealed enrichment in cell-adhesion terms, again similar to what was observed in ICF1 iPSCs. In summary, ICF1-iPSC differentiated MSCs retain aberrant DNA methylation patterns similar to what was observed in ICF1 iPSCs. Thus, our MSC differentiation paradigm provides proof of principle that ICF1 iPSCs would serve as a useful tool for further studies of ICF1 molecular pathogenesis in various tissues.
Figure 5.

Methylation profiling of ICF1-iPSCs-derived MSCs. (A). Results of LC–MS/MS-MRM measurements of total methylcytosine levels in various MSC cell types as well as primary bone marrow. 5mC levels are expressed as percentage of the total cytosine pool. Data represents mean and standard deviations from triplicate analyses. (B). Bar chart showing global methylation in shared CG sites as measured by RRBS. Values represent average methylation of all measurable CpGs (n = 738 952). (C). Venn diagram representing number of demethylated gene promoters in ICF1 cells compared with respective controls. (D). Genome browser view of DNA methylation levels in the protocadherin cluster. Methylation level of individual CpGs is represented in vertical red bars (between 0 and 1), with the horizontal black line representing methylation level of 0.5 for comparison.
DISCUSSION
Through reprogramming ICF patient fibroblasts carrying DNMT3B mutations, we discovered that DNMT3B is necessary for many de novo methylation signatures, but is dispensable for overall iPSC derivation. Our finding reinforces the findings from a recent study that showed mouse iPSCs can be readily generated from embryonic fibroblasts deficient of either Dnmt3a or Dnmt3b, or both (33). Thus, although a wave of de novo methylation in cell reprogramming contributes to iPSC-specific methylation, demethylation in pluripotency genes is essential for successful cell reprogramming. Of note, mouse ESCs tolerate total demethylation while maintaining stem cell self-renewal (21,44).
Our finding that DNMT3B is the predominant enzyme for catalyzing non-CG methylation has support in the literature. For example, Suetake et al. (45) demonstrated that recombinant Dnmt3b has greater activity toward CpA and CpT compared with Dnmt3a. Therefore, the greater contribution for DNMT3B-mediated non-CG methylation may be due to biochemical differences in enzyme specificities. However, recombinant proteins may not accurately represent non-CG activity in vitro. We have performed genome-wide bisulfite sequencing in Dnmt3a−/− and Dnmt3b−/− mESCs (Huang et al., unpublished), and found that non-CG methylation is equivalently and mildly reduced in both cell lines, suggesting that Dnmt3a and Dnmt3b are functionally redundant for non-CG methylation in mESCs. Interestingly, Ziller et al. (46) demonstrated a strong correlation between non-CpG methylation and DNMT3 expression levels through siRNA knockdown experiments in human ESCs. Their study also showed knockdown of DNMT3B led to a greater loss of non-CG methylation. However, their study may also be confounded by knockdown efficiencies. In the current study, we showed that DNMT3B mutations lead to the loss of over 90% of non-CG methylation, indicating DNMT3B is the predominant de novo methyltransferase for non-CG methylation in hiPSCs. We have also examined the localization of residual highly methylated non-CGs (>30% methylation) in ICF1 iPSCs. Consistent with previous reports in germ cells that non-CG methylation tend to accumulate around methylated CGs (37,38), remnant non-CGs in ICF1 iPSCs also tend to cluster around highly methylated CGs compared with unmethylated non-CpG sites (data not shown). These data support that notion that DNMT3A has some minor non-CG activity that localizes around highly methylated regions.
The gene expression analysis of ICF-iPSCs allowed us to identify several genes that are deregulated both in ICF patient somatic cells and iPSCs. In our study, we used a relatively relaxed criterion for calling significantly deregulated genes, and still found small differences in transcriptomes between normal and ICF1 iPSCs. It is likely that DNMT3A compensates for a vast majority of DNMT3B deficiency. Indeed, CG methylation is largely unchanged with exception at select loci in ICF1 iPSCs. In addition, mutations in DNMT3B do not affect transcript and protein levels, so non-catalytic functions of DNMT3B may still be preserved (10). Nevertheless, some genes were consistently deregulated between normal and ICF1 iPSCs. Most notably, loss of cell adhesion appears to be a recurrent theme underlying ICF transcriptome in all cell types. In addition, we found that several genes required for lymphocyte differentiation and genome stability are already deregulated in ICF iPSCs, suggesting that altered lymphocyte cell development in ICF patients can be predicted from altered gene expression in mutant ICF-iPSCs. Thus, the derivation of ICF-iPSCs also provides a valuable system to study ICF syndrome pathogenesis through in vitro differentiation into a variety of somatic cell types relevant to ICF syndrome phenotypes such as hematopoietic and neural crest lineage cells.
MATERIAL AND METHODS
Derivation and cultures of human iPSCs and hESCs
The production of human iPSCs follows the protocol previously described (47,48) using retroviruse expression of OCT4, SOX2, KLF4 and c-MYC. One line of ICF1 patient fibroblasts (ICF1-1 as designated in this paper) was purchased from Coriell Institute of Medical Research (GM08747), and the other line of ICF1 patient fibroblasts (ICF1-2) was from Great North Children's Hospital in Newcastle, UK (Dr. Andrew Gennery) (49). Mutations were confirmed via Sanger sequencing (Supplementary Material, Fig. S14). Control ESC and iPSC lines have been described previously (32). Human ESC and hiPSCs were maintained in DME supplemented with 20% KSR, nonessential amino acids (Invitrogen), l-glutamine (Mediatech), Penn/Strep, 2-mercaptoethanol with a feeder layer of MEFs as previously described (50). For both gene expression and methylation analysis, the hESCs and hiPSCs were passaged onto feeder-free gelatin-coated plates twice before harvesting RNA and DNA. RNA was isolated using Trizol (Invitrogen) while DNA was isolated using PureLink™ genomic DNA purification kit (Invitrogen).
Southern blot analysis
Genomic DNAs were digested with HpaII enzymes overnight and fractionated through agarose gel electrophoresis then transferred to a nitrocellulose membrane. Satellite 2 and 3 probes were end-labeled with P32 and were hybridized with Quickhyb (Stratagene, San Diego), following the manufacturer's protocol. Radioautography was performed with hybridized membranes and developed with a Kodak film developer.
Bisulfite conversion and sequencing
Bisulfite conversion was performed as described (50). Briefly, genomic DNA was overnight digested with BglII, then incubated with a sodium bisulfite for 16 h. Bisulfite-treated DNA was desalted and precipitated and 10% of the precipitated material was used for each PCR. Nested primers were used, and products were gel purified and used for Topo Cloning (Invitrogen).
Whole-genome bisulfite sequencing analysis
Whole-genome shotgun bisulfite sequencing libraries (BS-seq) were prepared as previously described (29) and sequenced on the Illumina HiSeq2000 machine following manufacturer instructions. BS-seq raw reads were mapped to the hg18 genome using default parameters in BS-Seeker (51). Methylation calling was performed using in-house scripts following previously described methods (31). Metaplot figures for DMRs were constructed by dividing DMRs into 10 equally sized bins flanked both up- and down-stream with 10 equally sized bins that were normalized by the DMR length. For chromosome wide plots, a smoothed average of 10 kb windows was used. Regions of differential methylation was calculated by pooling cytosine methylation levels for control or ICF1 iPSCs, then using cutoff of FDR <1% (based on the Kolmogorv–Smirnov test) and delta-beta >30%. Proximal promoters were defined as 500 bp sequences up- and down-stream of the transcription start site (TSS). Enhancers were defined as previously described (41,52).
RNA-sequencing analysis
RNA isolation and library construction was performed as previously described (53). Reads were mapped to the hg18 genome using default parameters from BWA aligner (54); unmapped reads were further mapped to hg18 reference mRNA sequences. Uniquely aligned transcript reads were normalized to RPKM and low or not expressed transcripts (RPKM <0.5) were filtered out. The remaining gene list was used as input for differential expression analysis by running default parameters in DEseq (55). Genes with Benjamini and Hochberg-adjusted P-values (FDR) <10% and mean fold-change >1.5 were considered differentially expressed. Gene ontology analysis was performed using DAVID Bioinformatics Resource (56).
Quantitative real-time PCR
RNA was DNase I treated (Invitrogen) and then quantified. cDNA conversion was completed using the iScript kit (Bio-Rad). Quantitative PCR was done on a MyIQ Thermocycler (Bio-Rad) using the Sybr Green Supermix (Bio-Rad). Relative expression levels were normalized to 18S amplicons.
Mesenchymal stem cell differentiation
MSC differentiation was performed as previously described (43). Briefly, human ESC/iPSC colonies were detached with 1 mg/ml collagenase IV (Invitrogen) for 10–15 min. Detached hESC colonies were incubated in bFGF-depleted hESC medium suspension in bacterial dishes for 7 days to form EBs. Media was changed every other day. EBs were re-attached on a tissue culture dish after selecting for well-rounded EBs under the microscope. Attached EBs were cultured in DMEM high glucose (Gibco) containing 15% fetal bovine serum (FBS) (Gibco), non-essential amino acid (NEAA) (Gibco), antibodic (Gibco) and 10 ng/ml bFGF (RD). Cell outgrowths from EBs were sorted by flow cytometry (Aria II, BD Biosciences).
Flow cytometry was performed by dissociating cells with 0.25% trypsin ethylenediaminetetraacetic acid (Invitrogen) at 37° for 1 min, washed with PBS containing 2% FBS and incubated for 30 min with antibodies. Antibodies for hMSC are as follows: CD73 (APC, Biolegend) and CD146 (PE, Biolegend).
LC–MS/MS-MRM for measuring global methylation levels
This was performed as previously described (57). Briefly, genomic DNA was digested using DNA Degradase Plus (Zymo Research) and injected into a reverse phase ultra-high-performance liquid chromatography column (Eclipse C18, 2.1 × 10 mm, 1.8 μm particle size, Agilent) and eluted with a methanol gradient. The effluent was passed through a triple quadruple mass spectrometer (Agilent 6460 QQQ) via an electrospray ion source (Agilent Jet Stream). The spectrometer was run in positive MRM mode, and 5mC levels for each sample were quantified by dividing the 5mC MRM peak area by the combined peak areas of 5mC, 5hmC, and C (unmodified cytosine). Calibration curves were generated using known 5mC standards (Zymo Research).
The 5mdC peak in BJ fibroblasts and IPSCs from BJ fibroblasts is 3 and 5%, while similar measurements for the same cell types have reported levels of 5 and 8%, respectively (57). We attribute these variations to small differences in homogeneity/heterogeneity of the cell types in different batches prepared at different times by different people, and to improvements in the assay where we have made efforts to minimize ion suppression which is particularly worrisome for poorly retained analytes such as dC and 5mdC. Nevertheless, reproducibility of relative peak intensities within an experiment is very good, as noted by the small error bars.
SUPPLEMENTARY MATERIAL
FUNDING
This project is supported by NIH DE022928 and UCLA CTSI Grant UL1TR000124 (G.F.). K.H. is supported by the Eli & Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA. Additional support is from 973 Basic Research Program in China (2011CB965102).
Supplementary Material
Acknowledgments
Conflict of Interest statement. None declared.
REFERENCES
- 1.Jaenisch R., Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33(suppl.):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 2.Goll M.G., Bestor T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 3.Feng J., Fan G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int. Rev. Neurobiol. 2009;89:67–84. doi: 10.1016/S0074-7742(09)89004-1. [DOI] [PubMed] [Google Scholar]
- 4.Klein C.J., Botuyan M.V., Wu Y., Ward C.J., Nicholson G.A., Hammans S., Hojo K., Yamanishi H., Karpf A.R., Wallace D.C., et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 2011;43:595–600. doi: 10.1038/ng.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shah M.Y., Licht J.D. DNMT3A mutations in acute myeloid leukemia. Nat. Genet. 2011;43:289–290. doi: 10.1038/ng0411-289. [DOI] [PubMed] [Google Scholar]
- 6.Ley T.J., Ding L., Walter M.J., McLellan M.D., Lamprecht T., Larson D.E., Kandoth C., Payton J.E., Baty J., Welch J., et al. DNMT3A mutations in acute myeloid leukemia. New Engl. J. Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu G.L., Bestor T.H., Bourc'his D., Hsieh C.L., Tommerup N., Bugge M., Hulten M., Qu X., Russo J.J., Viegas-Pequignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 8.Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 9.Jin B., Tao Q., Peng J., Soo H.M., Wu W., Ying J., Fields C.R., Delmas A.L., Liu X., Qiu J., et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum. Mol. Genet. 2008;17:690–709. doi: 10.1093/hmg/ddm341. [DOI] [PubMed] [Google Scholar]
- 10.Ehrlich M., Sanchez C., Shao C., Nishiyama R., Kehrl J., Kuick R., Kubota T., Hanash S.M. ICF, an immunodeficiency syndrome: DNA methyltransferase 3B involvement, chromosome anomalies, and gene dysregulation. Autoimmunity. 2008;41:253–271. doi: 10.1080/08916930802024202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bibikova M., Laurent L.C., Ren B., Loring J.F., Fan J.B. Unraveling epigenetic regulation in embryonic stem cells. Cell Stem Cell. 2008;2:123–134. doi: 10.1016/j.stem.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Huang K., Fan G. DNA methylation in cell differentiation and reprogramming: an emerging systematic view. Regen. Med. 2010;5:531–544. doi: 10.2217/rme.10.35. [DOI] [PubMed] [Google Scholar]
- 13.Fouse S.D., Shen Y., Pellegrini M., Cole S., Meissner A., Van Neste L., Jaenisch R., Fan G. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell. 2008;2:160–169. doi: 10.1016/j.stem.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meissner A., Mikkelsen T.S., Gu H., Wernig M., Hanna J., Sivachenko A., Zhang X., Bernstein B.E., Nusbaum C., Jaffe D.B., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawkins R.D., Hon G.C., Lee L.K., Ngo Q., Lister R., Pelizzola M., Edsall L.E., Kuan S., Luu Y., Klugman S., et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M., et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young R.A. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohn F., Weber M., Rebhan M., Roloff T.C., Richter J., Stadler M.B., Bibel M., Schubeler D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell. 2008;30:755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Stadler M.B., Murr R., Burger L., Ivanek R., Lienert F., Scholer A., Wirbelauer C., Oakeley E.J., Gaidatzis D., Tiwari V.K., et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 20.Farthing C.R., Ficz G., Ng R.K., Chan C.F., Andrews S., Dean W., Hemberger M., Reik W. Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genet. 2008;4:e1000116. doi: 10.1371/journal.pgen.1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsumura A., Hayakawa T., Kumaki Y., Takebayashi S., Sakaue M., Matsuoka C., Shimotohno K., Ishikawa F., Li E., Ueda H.R., et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells. 2006;11:805–814. doi: 10.1111/j.1365-2443.2006.00984.x. [DOI] [PubMed] [Google Scholar]
- 22.Mikkelsen T.S., Hanna J., Zhang X., Ku M., Wernig M., Schorderet P., Bernstein B.E., Jaenisch R., Lander E.S., Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huangfu D., Osafune K., Maehr R., Guo W., Eijkelenboom A., Chen S., Muhlestein W., Melton D.A. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 2008;26:1269–1275. doi: 10.1038/nbt.1502. [DOI] [PubMed] [Google Scholar]
- 24.Bhutani N., Brady J.J., Damian M., Sacco A., Corbel S.Y., Blau H.M. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan E.M., Ratanasirintrawoot S., Park I.H., Manos P.D., Loh Y.H., Huo H., Miller J.D., Hartung O., Rho J., Ince T.A., et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat. Biotechnol. 2009;27:1033–1037. doi: 10.1038/nbt.1580. [DOI] [PubMed] [Google Scholar]
- 26.Wernig M., Lengner C.J., Hanna J., Lodato M.A., Steine E., Foreman R., Staerk J., Markoulaki S., Jaenisch R. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat. Biotechnol. 2008;26:916–924. doi: 10.1038/nbt1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi Y., Desponts C., Do J.T., Hahm H.S., Scholer H.R., Ding S. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell. 2008;3:568–574. doi: 10.1016/j.stem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Polo J.M., Anderssen E., Walsh R.M., Schwarz B.A., Nefzger C.M., Lim S.M., Borkent M., Apostolou E., Alaei S., Cloutier J., et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lister R., Pelizzola M., Kida Y.S., Hawkins R.D., Nery J.R., Hon G., Antosiewicz-Bourget J., O'Malley R., Castanon R., Klugman S., et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doi A., Park I.H., Wen B., Murakami P., Aryee M.J., Irizarry R., Herb B., Ladd-Acosta C., Rho J., Loewer S., et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lister R., Pelizzola M., Dowen R.H., Hawkins R.D., Hon G., Tonti-Filippini J., Nery J.R., Lee L., Ye Z., Ngo Q.M., et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang K., Shen Y., Xue Z., Bibikova M., April C., Liu Z., Cheng L., Nagy A., Pellegrini M., Fan J.B., et al. A panel of CpG methylation sites distinguishes human embryonic stem cells and induced pluripotent stem cells. Stem Cell Reports. 2014;2:36–43. doi: 10.1016/j.stemcr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pawlak M., Jaenisch R. De novo DNA methylation by Dnmt3a and Dnmt3b is dispensable for nuclear reprogramming of somatic cells to a pluripotent state. Genes Dev. 2011;25:1035–1040. doi: 10.1101/gad.2039011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stadtfeld M., Maherali N., Breault D.T., Hochedlinger K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell. 2008;2:230–240. doi: 10.1016/j.stem.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ji W., Hernandez R., Zhang X.Y., Qu G.Z., Frady A., Varela M., Ehrlich M. DNA demethylation and pericentromeric rearrangements of chromosome 1. Mutat. Res. 1997;379:33–41. doi: 10.1016/s0027-5107(97)00088-2. [DOI] [PubMed] [Google Scholar]
- 36.Chen P.Y., Feng S., Joo J.W., Jacobsen S.E., Pellegrini M. A comparative analysis of DNA methylation across human embryonic stem cell lines. Genome Biol. 2011;12:R62. doi: 10.1186/gb-2011-12-7-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ichiyanagi T., Ichiyanagi K., Miyake M., Sasaki H. Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Res. 2013;41:738–745. doi: 10.1093/nar/gks1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shirane K., Toh H., Kobayashi H., Miura F., Chiba H., Ito T., Kono T., Sasaki H. Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet. 2013;9:e1003439. doi: 10.1371/journal.pgen.1003439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lister R., Mukamel E.A., Nery J.R., Urich M., Puddifoot C.A., Johnson N.D., Lucero J., Huang Y., Dwork A.J., Schultz M.D., et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeong M., Sun D., Luo M., Huang Y., Challen G.A., Rodriguez B., Zhang X., Chavez L., Wang H., Hannah R., et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Genet. 2014;46:17–23. doi: 10.1038/ng.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S.A., Flynn R.A., Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ehrlich M., Buchanan K.L., Tsien F., Jiang G., Sun B., Uicker W., Weemaes C.M., Smeets D., Sperling K., Belohradsky B.H., et al. DNA methyltransferase 3B mutations linked to the ICF syndrome cause dysregulation of lymphogenesis genes. Hum. Mol. Genet. 2001;10:2917–2931. doi: 10.1093/hmg/10.25.2917. [DOI] [PubMed] [Google Scholar]
- 43.Ye L., Fan Z., Yu B., Chang J., Al Hezaimi K., Zhou X., Park N.H., Wang C.Y. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell. 2012;11:50–61. doi: 10.1016/j.stem.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hutnick L.K., Huang X., Loo T.C., Ma Z., Fan G. Repression of retrotransposal elements in mouse embryonic stem cells is primarily mediated by a DNA methylation-independent mechanism. J. Biol. Chem. 2010;285:21082–21091. doi: 10.1074/jbc.M110.125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suetake I., Miyazaki J., Murakami C., Takeshima H., Tajima S. Distinct enzymatic properties of recombinant mouse DNA methyltransferases Dnmt3a and Dnmt3b. J. Biochem. 2003;133:737–744. doi: 10.1093/jb/mvg095. [DOI] [PubMed] [Google Scholar]
- 46.Ziller M.J., Muller F., Liao J., Zhang Y., Gu H., Bock C., Boyle P., Epstein C.B., Bernstein B.E., Lengauer T., et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7:e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R., et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 49.Gennery A.R., Slatter M.A., Bredius R.G., Hagleitner M.M., Weemaes C., Cant A.J., Lankester A.C. Hematopoietic stem cell transplantation corrects the immunologic abnormalities associated with immunodeficiency-centromeric instability-facial dysmorphism syndrome. Pediatrics. 2007;120:e1341–e1344. doi: 10.1542/peds.2007-0640. [DOI] [PubMed] [Google Scholar]
- 50.Shen Y., Chow J., Wang Z., Fan G. Abnormal CpG island methylation occurs during in vitro differentiation of human embryonic stem cells. Hum. Mol. Genet. 2006;15:2623–2635. doi: 10.1093/hmg/ddl188. [DOI] [PubMed] [Google Scholar]
- 51.Chen P.Y., Cokus S.J., Pellegrini M. BS Seeker: precise mapping for bisulfite sequencing. BMC Bioinformatics. 2010;11:203. doi: 10.1186/1471-2105-11-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ziller M.J., Gu H., Muller F., Donaghey J., Tsai L.T., Kohlbacher O., De Jager P.L., Rosen E.D., Bennett D.A., Bernstein B.E., et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Y., Huang K., Nakatsu M.N., Xue Z., Deng S.X., Fan G. Identification of novel molecular markers through transcriptomic analysis in human fetal and adult corneal endothelial cells. Hum. Mol. Genet. 2013;22:1271–1279. doi: 10.1093/hmg/dds527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 57.Le T., Kim K.P., Fan G., Faull K.F. A sensitive mass spectrometry method for simultaneous quantification of DNA methylation and hydroxymethylation levels in biological samples. Anal. Biochem. 2011;412:203–209. doi: 10.1016/j.ab.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.