

Abstract
Gain-of-function mutations in the olfactomedin domain of the MYOC gene facilitate the toxic accumulation of amyloid-containing myocilin aggregates, hastening the onset of the prevalent ocular disorder primary open-angle glaucoma. Aggregation of wild-type myocilin has been reported in other glaucoma subtypes, suggesting broader relevance of misfolded myocilin across the disease spectrum, but the absence of myocilin does not cause disease. Thus, strategies aimed at eliminating myocilin could be therapeutically relevant for glaucoma. Here, a novel and selective Grp94 inhibitor reduced the levels of several mutant myocilin proteins as well as wild-type myocilin when forced to misfold in cells. This inhibitor rescued mutant myocilin toxicity in primary human trabecular meshwork cells. Mechanistically, in vitro kinetics studies demonstrate that Grp94 recognizes on-pathway aggregates of the myocilin olfactomedin domain (myoc-OLF), accelerates rates of aggregation and co-precipitates with myoc-OLF. These results indicate that aberrant myocilin quaternary structure drives Grp94 recognition, rather than peptide motifs exposed by unfolded protein. Inhibition of Grp94 ameliorates the effects of Grp94-accelerated myoc-OLF aggregation, and Grp94 remains in solution. In cells, when wild-type myocilin is driven to misfold and aggregate, it becomes a client of Grp94 and sensitive to Grp94 inhibition. Taken together, the interaction of Grp94 with myocilin aggregates can be manipulated by cellular environment and genetics; this process can be exploited with Grp94 inhibitors to promote the clearance of toxic forms of myocilin.
INTRODUCTION
Primary open-angle glaucoma (POAG) is a degenerative eye disease characterized by retinal ganglion cell loss and optic nerve head damage. This is often preceded by increased intraocular pressure (IOP) due to impaired aqueous humor outflow through the anterior anatomical segment called the trabecular meshwork (TM), leading to irreversible vision loss (1–4). Mutations in the MYOC gene, which encodes the myocilin protein, cause ∼5% of hereditary POAG in populations throughout the world, including ∼100 000 people in the US (5,6). As a result, the pathogenic mechanisms of mutant myocilin and its corresponding native biological function(s) have been subjects of intensive investigation over the past decade (7–10). Myocilin, composed of an N-terminal coiled-coil and 31 kDa C-terminal olfactomedin domain (myoc-OLF), is generally believed to be a secreted protein (11). The majority of the non-synonymous MYOC gene lesions known to cause POAG are found within myoc-OLF (12,13), leading to intracellular sequestration and aggregation (14–17). These observations suggest a functional link between this domain and pathogenicity. The vast majority of work in the field indicates that mutations in myocilin do in fact cause a toxic gain-of-function for the protein (18,19).
Our team has recently provided a molecular basis for this toxicity: mutations in the OLF domain of myocilin promote the toxic aggregation of the myocilin protein into amyloid aggregates (13,20). This non-native structure is known to be nearly impossible to disaggregate, likely explaining why, unlike other proteins, mutant myocilin cannot be efficiently cleared by endoplasmic reticulum-associated degradation (ERAD) and instead accumulates in the ER, leading to activation of ER stress pathways and TM cell death. Indeed, we recently showed that knocking down or inhibiting the ER chaperone Grp94 facilitates the degradation of mutant myocilin in cells via autophagy. Grp94 preserves mutant myocilin in the ER by attempting to force it through the ERAD pathway that involves the valosin containing protein (VCP/p97) and the proteasome.
Here, we have identified a potent and selective Grp94 inhibitor that lowers the levels of several mutant myocilin species. Grp94 inhibition reduces the toxicity of mutant myocilin in human primary TM cells. The mechanism for Grp94 association with myocilin is through its recognition of myocilin aggregates, not the folded mutant protein as originally hypothesized. Based on this observation, we show that even wild-type (WT) myocilin, if misfolded and aggregated, can become a Grp94 client, and therefore can be successfully degraded by inhibiting Grp94. Since ocular hypertension in glaucoma is thought to be a result of the overexpression of WT myocilin (21,22), Grp94 inhibition could be more broadly applicable to glaucoma treatment than just for individuals with inherited POAG.
RESULTS
Intracellular mutant myocilin levels and toxicity are reduced following Grp94 knockdown using siRNA
We previously showed that Grp94 could regulate the levels of I477N-mutant myocilin, but we sought to determine if modulating Grp94 levels could be generalized to other disease-associated variants. HEK cells were transfected with myocilin harboring I477N, Y437H, P370L, W286R or N480K substitutions and the effects of Grp94 overexpression (Fig. 1A) or siRNA knockdown (Fig. 1B) on their intracellular levels were evaluated by western blot. Overexpression of Grp94 preserved mutant myocilin in cells. Conversely, Grp94 knockdown dramatically reduced the levels of each mutant myocilin species. Optimal Grp94 knockdown of >60% was achieved with one of three siRNAs tested. We then fractionated lysates from cells transfected with each myocilin mutant and either control or Grp94 siRNA to determine the impact of Grp94 suppression on mutant myocilin solubility, as previously described (17,23). Briefly, cells were lysed in a buffer containing 0.5% Triton, centrifuged at 16 000 g for 10 min and supernatant was recovered for the ‘soluble’ fraction. The pellet from the spin was re-suspended in loading buffer and 9 M urea to produce the ‘insoluble’ fraction. Grp94 knockdown reduced both soluble and insoluble myocilin species, suggesting, as expected, that Grp94 suppression was facilitating myocilin clearance, not forcing it into an insoluble state (Fig. 1C and D). Quantification was achieved from four independent experiments, including those shown in Figure 1B and C. These findings further imply that Grp94 modulation is broadly applicable to POAG cases resulting from different myocilin mutations.
Figure 1.

Consequences of Grp94 knockdown or overexpression on levels of intracellular and aggregated mutant myocilin. (A) HEK cells were co-transfected with each indicated myocilin mutant and either Grp94 or Control 6TR cDNA. Western blot analyses of cell lysates were performed 48 h after cDNA transfection. (B) Prior to transfection with each myocilin mutant, cells were transfected with either Grp94 or Control siRNA. Western blot analysis of cell lysates were performed 72 h following siRNA transfection and 48 h after cDNA transfection. (C) Prior to transfection with each myocilin mutant, cells were transfected with either Grp94 or Control siRNA. Triton solubility fractions of cell lysates were analyzed by western blot. Insoluble myocilin remaining in the supernatant after centrifugation cannot migrate through the stacking gel and is considered insoluble supernatant, while insoluble myocilin from the post-centrifugation pellet is suspended in 9 m urea and therefore runs at the standard 55 kDa MW. (D) Quantification of (C) shows significant reductions in each myocilin mutant caused by Grp94 suppression by siRNA as a percent of control siRNA following normalization to loading control. *P < 0.05.
Improvement of Grp94 inhibitor efficacy through structure-based design
We previously determined that the Grp94 inhibitor, BnIm 01, could reduce mutant myocilin levels (23). Based on molecular modeling studies of the pan-Hsp90 inhibitor radamide bound to Grp94, we proposed the imidazole ring as a bioisosteric replacement for the cis-amide moiety (Fig. 2A and B). Consistent with our computational predictions, the first compound synthesized, BnIm 01, exhibited ∼100:1 selectivity for Grp94 versus cytoplasmic Hsp90α/β, as determined by inhibition of Grp94-mediated Toll-like receptor protein presentation at the cell surface versus western blot analyses of Hsp90-dependent proteins found in the cytoplasm (24). On the basis of our computational model, we speculated that creating a molecule with increased hydrophobicity at the C4-position of the N-benzyl group would improve drug interaction with Grp94 and further prevent its interaction with Hsp90 proteins, thereby enhancing selectivity (25).
Figure 2.
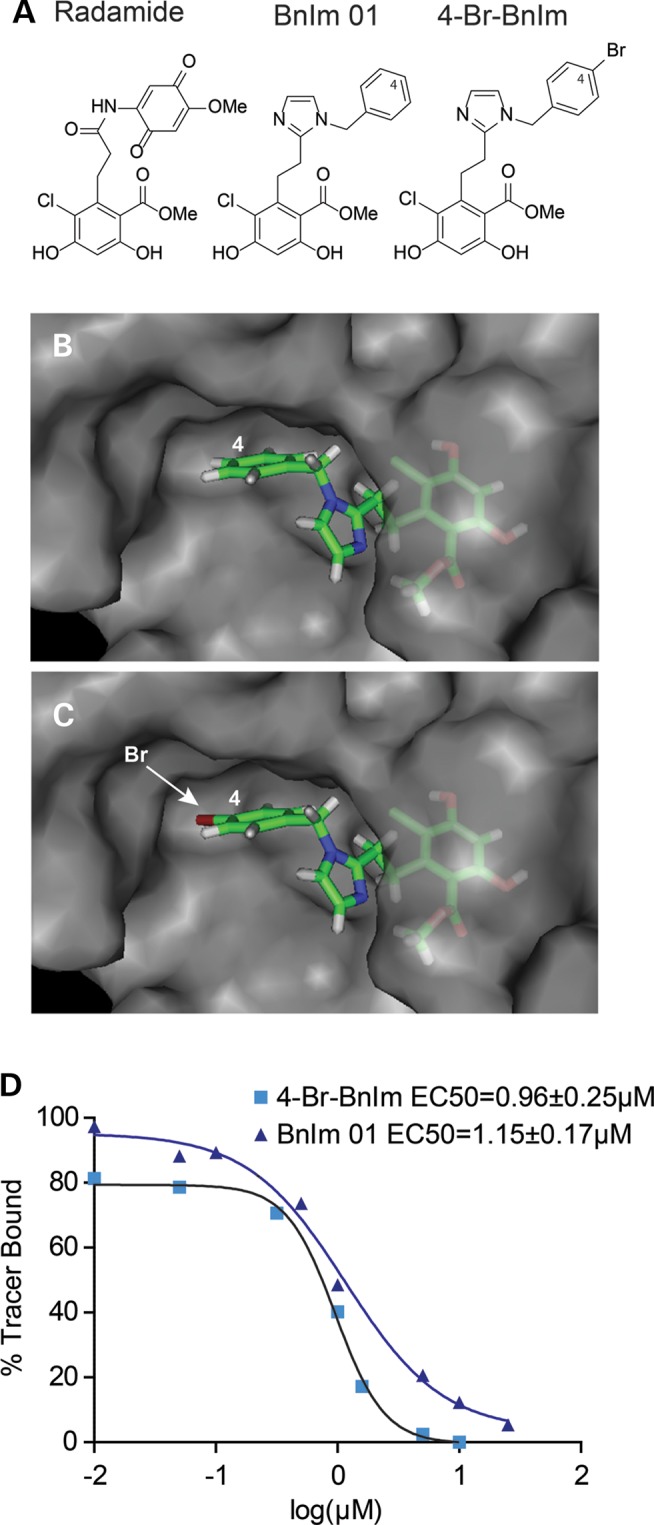
Structure, modeling and activity of Grp94 inhibitors. (A) Structures of radamide, BnIm 01 and 4-Br-BnIm. (B) BnIm 01 docked in a hydrophobic pocket within the ATP binding site of Grp94 (based on radamide-bound structure, PDB: 2GFD). (C) 4-Br-BnIm docked in Grp94 shows how the hydrophobic pocket can be occupied with the hydrophobic substitution at C4 (indicated by arrow), which likely improves specificity. (D) Fluorescence polarization (FP) assay of recombinant Grp94 pre-incubated with FITC-labeled geldanamycin and then increasing concentrations of each compound. Plates were incubated for 24 h and FP measured at 485 ex and 528 em. FP values were normalized to DMSO (100% tracer bound).
To test this hypothesis, we synthesized 4-Br-BnIm (Fig. 2A); modeling predicts this molecule fits more tightly into the hydrophobic pocket of Grp94 than the parent BnIm 01 (Fig. 2C). While in vitro fluorescence polarization binding assays indicate its potency against Grp94 is only slightly increased (1.5 µm with the BnIm 01 compound compared with 1 µm with the 4-Br-BnIm compound) (Fig. 2D), 4-Br-BnIm had an ∼10-fold better effect on I477N mutant myocilin in HEK cells stably over-expressing tetracycline-regulatable FLAG-tagged myocilin (iHEK) compared with BnIm 01 (Fig. 3A and B). This improved potency in cells was likely due to better selectivity for Grp94 over other Hsp90 proteins in cells (24). We observed no changes in Grp78, suggesting that ER stress was not activated by either inhibitor (26), nor did we observe an increase in heat shock proteins 27 or 70, indicating that our compound was selectively targeting Grp94 over the cytosolic Hsp90 isoforms (27). We also did not see any change in Grp94 expression levels, suggesting that the inhibitors are reversibly binding to Grp94. In addition, similar to BnIm 01, 4-Br-BnIm did not cause degradation of other Hsp90-dependent clients Akt and Ras (Fig. 3C) (24). We then evaluated whether 4-Br-BnIm showed improved potency over BnIm 01 against other mutant myocilin species. Indeed, 4-Br-BnIm had greater efficacy against total myocilin levels of each mutant species tested compared with BnIm 01 (Fig. 3D).
Figure 3.

Effects of specific Grp94 inhibitors on levels of intracellular mutant myocilin. (A) Western blot analysis of HEK cells stably over-expressing tetracycline-regulatable FLAG-tagged I477N mutant myocilin lysates and treated with indicated doses of BnIm 01 or 4-Br-BnIm for 24 h. Western blots were probed for myocilin levels, as well as levels of other heat-shock proteins (Hsp27, Hsp70) and ER chaperones (Grp78, Grp94). (B) Quantification of (A) shows the half maximal effective concentration (EC50) of BnIm 01 and 4-Br-BnIm for reducing mutant myocilin. (C) Western blot analysis of HEK cells stably over-expressing tetracycline-regulatable FLAG-tagged I477N mutant myocilin lysates and treated with indicated doses of 4-Br-BnIm for 24 h. Akt and Ras levels were unaffected by treatment. (D) Western blot of lysates from HEK cells transiently overexpressing five forms of mutant myocilin and treated with 30 µm BnIm 01, 4-Br-BnIm or equivalent volume of vehicle (Veh/DMSO) for 24 h.
Grp94 inhibition reduces mutant myocilin levels and toxicity in primary human TM cells
We next sought to evaluate the efficacy of the selective and potent 4-Br-BnIm in primary human TM cells because mutant myocilin is not known to be toxic to cells other than TM cells. Lentiviral vectors were generated to express RFP-tagged WT and Y437H mutant myocilin, along with RFP only as a control. TM cells were transduced with each viral vector for 7 days and subsequently treated with 30 µm BnIm 01, 4-Br-BnIm or an equivalent volume of DMSO (vehicle) for 24 h. Cells were counterstained with DAPI and imaged using confocal microscopy (Fig. 4A). Quantification of RFP density revealed that BnIm 01 and 4-Br-BnIm significantly reduced Y437H mutant myocilin while WT myocilin and the RFP-only vector were largely unaffected by Grp94 inhibition (Fig. 4B). As was the case with the HEK cell system, 4-Br-BnIm resulted in better clearance of Y437H-mutant myocilin than did BnIm 01. We then evaluated whether Grp94 inhibition could be cytoprotective in TM cells ectopically expressing mutant myocilin using an MTS assay. Y437H, but not WT myocilin, significantly reduced TM cell viability (Fig. 4C). Impressively, both BnIm 01 and 4-Br-BnIm rescued this toxicity (Fig. 4C).
Figure 4.

Grp94 inhibitors reduce mutant myocilin levels and toxicity in HTM cells. (A) Immunofluorescence imaging of human trabecular meshwork (HTM) cells transduced with RFP-tagged lentivirus expressing RFP control, RFP-tagged WT myocilin or RFP-tagged Y437H myocilin and treated with 30 µm BnIm 01, 4-Br-BnIm or equivalent volume of DMSO vehicle. Myocilin is shown in red and the nuclear marker DAPI shown in blue (×40 magnification). (B) Quantification of immunofluorescence images detecting RFP density ± standard deviation (n = 3). Significance was measured using Student's t-test; *P < 0.05, **P < 0.01. (C) MTS assay of HTM cells overexpressing WT or Y437H myocilin following treatment with 30 µm BnIm 01, 4-Br-BnIm or vehicle equivalent (DMSO) for 6 h. Cell viability is shown prior to treatment (Pre-Tx) and following treatment (Post-Tx) ± standard deviation (n = 3). Significance was measured using the Student's t-test; *P < 0.05, **P < 0.01.
Grp94 recognizes on-pathway aggregates of myocilin
To better understand the mechanism of interaction between Grp94 and myocilin at the molecular level, we examined the effects of Grp94 on myocilin aggregation using an in vitro assay. Myoc-OLF variants fibrillize under neutral pH and physiological temperatures, producing amyloidogenic fibrils over the course of a few hours (13,20). WT myoc-OLF is stable, but at slightly elevated temperatures or at physiological temperatures with gentle rocking, amyloid formation ensues over several days (13,20).
Our previous work indicated a stable interaction between Grp94 and mutant myocilin (23), but the molecular species recognized by Grp94, namely folded mutant protein or an aggregated state, was not established. First, using our maltose binding protein- myoc-OLF (MBP-OLF) fusion protein (1) in either its well-defined monomeric or characterized protofibrillar state isolated from cell lysates (20), we performed binding studies with recombinant Grp94 using the affinity of MBP for amylose resin. Neither species co-eluted with Grp94 (Supplementary Material, Fig. S1), suggesting that either the binding epitope is obscured by the presence of MBP, or Grp94 recognizes neither species. Next, we evaluated the effects of Grp94 on isolated myoc-OLF aggregation kinetics. Grp94 accelerated myoc-OLF aggregation and led to an aggregate with higher amyloid content as measured by thioflavin T (ThT) fluorescence (Fig. 5A). SDS–PAGE analysis of end-point aggregates clearly showed the presence of both species, with little of either protein remaining in the supernatant (Fig. 5B). This accelerated aggregation was blocked by 4-Br-BnIm pre-incubation (Fig. 5A). Initial rates of aggregation for inhibitor-treated samples appeared to be similar to the case without 4-Br-BnIm, but ThT-sensitive aggregates leveled off after ∼30 h, near those of myoc-OLF in the absence of Grp94. Interestingly, at the conclusion of the experiment at 96 h, abundant Grp94 remained in the supernatant, and the aggregate was predominantly composed of myoc-OLF. These data reveal that 4-Br-BnIm largely abolishes the effect of Grp94-accelerated myocilin aggregation, but there is a complex interplay between the non-covalent interaction between Grp94, 4-Br-BnIm and myoc-OLF aggregation kinetics. Our hypothesis is that at early time points, any Grp94 not bound to 4-Br-BnIm present in solution co-aggregates with myoc-OLF; once this Grp94 species is depleted from the solution, the population of 4-Br-BnIm-inhibited Grp94 no longer recognizes the on-pathway aggregate present, enabling myoc-OLF to continue to aggregate as it does in the absence of Grp94. Moreover, the effect of reduced binding of aggregated myocilin with Grp94 inhibition could be replicated in cells (Fig. 5C). In sum, these data favor a mechanism of action by which Grp94 recognizes a ligand present on an on-pathway multimeric aggregate myocilin structure, rather than on either the misfolded mutant protein or an off-pathway aggregate. Future improvements in Grp94 inhibitors will focus on the ability of the inhibitor to disrupt the interaction between Grp94 and the particular offending myocilin aggregate species.
Figure 5.

Grp94 recognition of aggregated quaternary OLF structure and acceleration of myocilin aggregation can be blocked by Grp94 inhibition. (A) Myoc-OLF aggregation kinetics analysis performed by monitoring the increase in ThT fluorescence of samples over a 96 h period. (B) SDS–PAGE analysis characterization of end-point aggregation assay samples from (A). S: supernatant, W: supernatant after wash of aggregate pellet, P: final resolubilized pellet. Molecular mass markers listed as kDa. (C) Co-IP of myocilin from HEK cells transiently overexpressing Y437H myocilin and treated with BnIm, 4-Br-BnIm or an equivalent volume of DMSO vehicle, performed 48 h after transfection and 24 h after drug treatment. Western blot analysis indicates levels of Grp94 association after inhibitor treatment.
Post-translational modifications render wild-type myocilin a client of Grp94 via misfolding
In vitro studies have described several ways in which WT myocilin can aggregate in the absence of a mutation, raising the possibility that misfolding of WT myocilin could be occurring in sporadic cases of POAG, for example by induction of misfolding in relation to post-translational modifications. Impaired glycosylation can prevent protein secretion, but it is also known to cause protein misfolding. For myocilin, we predict this misfolding could lead to production of early aggregates recognized by Grp94. Therefore, we tested the effects of tunicamycin, a reagent known to inhibit N-linked glycosylation (28), on WT myocilin and its subsequent ability to associate with Grp94 in cells. Indeed, transiently over-expressed WT myocilin, which is not fully secreted (23), did become insoluble in cells treated with tunicamycin (Fig. 6A), consistent with our cell-free data. Then, using the iHEK cell model of WT myocilin overexpression, in which all WT myocilin is secreted, we found that Grp94 associated with WT myocilin only in cells treated with tunicamycin, further corroborating our in vitro observations (Fig. 6B). Conversely, WT myocilin did not interact with Grp94 in these cells when treated with brefeldin A (BFA), a chemical that blocks ER/Golgi transport and secretion without affecting myocilin folding, further suggesting that the effects of tunicamycin on the myocilin/Grp94 interaction are due to myocilin misfolding, not impaired secretion (Fig. 6C). Then, using the same iHEK cell model of WT myocilin overexpression, we found that tunicamycin blocked myocilin secretion, but this myocilin was sensitized to Grp94 inhibition (Fig. 6D and E), further suggesting that misfolded myocilin becomes a Grp94 client.
Figure 6.

WT myocilin becomes a client of Grp94 when myocilin is misfolded. (A) Western blot analysis of Triton soluble/insoluble fractions from iHEK WT myocilin cell lysates treated with 6 µm tunicamycin for 24 h. (B) Co-IP of myocilin and subsequent western blot analysis of iHEK WT myocilin cell lysates treated with 6 µm concentration of tunicamycin; inputs indicate western blot of lysates without co-IP. (C) Co-IP of myocilin and subsequent western blot analysis of iHEK WT myocilin cell lysates treated with 3 µm concentration of Brefeldin A; inputs indicate western blot of lysates without co-IP. (D) Western blot analysis of iHEK WT myocilin cells treated with 6 µm concentration of tunicamycin followed by a dose–response of BnIm 01, 4-Br-BnIm or equivalent volume of DMSO vehicle. (E) Quantification of Western blot analysis on dose–response from (D) showing myocilin levels as a percent of vehicle following normalization to loading control (Gapdh).
DISCUSSION
Here, we have demonstrated the effectiveness of a second-generation Grp94-selective inhibitor in clearing a number of POAG-relevant mutant myocilins in several cell lines, including, importantly, primary human TM cells. Mechanistically, in vitro studies indicate that Grp94 recognizes on-pathway aggregation intermediates, suggesting that aberrant quaternary myocilin structure drives Grp94 client recognition rather than exposed peptide motifs of individual misfolded or mutant proteins. Grp94 accelerates myocilin aggregation and co-precipitates with the aggregates, but this can largely be reduced by Grp94 inhibition by pre-incubation with 4-Br-BnIm. When forced to misfold, WT myocilin becomes an intracellular client of Grp94 that is then sensitized to Grp94 inhibition. Taken together, our work demonstrates that reducing mutant or aggregated WT myocilin via inhibiting its interaction with Grp94 represents a viable therapeutic strategy for myocilin-associated forms of glaucoma (Fig. 7).
Figure 7.

Grp94 accelerates myocilin aggregation, a process that can be prevented by Grp94 inhibition. Myocilin misfolding causes it to form small aggregates that bind Grp94. Grp94 facilitates amyloid formation, preventing clearance of myocilin and causing toxicity. Grp94 inhibitors prevent Grp94 from interacting with myocilin, allowing a secondary autophagic pathway to facilitate clearance.
Our findings also help explain the paradox of the failure of ERAD to triage misfolded myocilin. Previously, we showed that mutant, but not wild-type, myocilin, was recognized by Grp94, and that the degradation rate of mutant myocilin was increased when Grp94 was depleted or inhibited (23). This result was surprising because it suggested that Grp94 was interfering with mutant myocilin clearance, rather than facilitating its removal through the default ERAD pathway. Grp94 attempts to triage mutant myocilin through ERAD but is unsuccessful, leading to its accumulation and mis-sorting, which results in ER stress (2,23). Our current work shows that Grp94 cannot triage mutant myocilin through ERAD because it recognizes an already-aggregated state of myocilin, and this association further accelerates aggregation into an amyloid structure known to be resistant to resolubilization and refolding. This is not the first time that an Hsp90 protein was shown to accelerate the aggregation of an amyloid-prone protein: Hsp90α was previously shown to stimulate the aggregation of tau (29). The evolutionary reason for accelerated aggregation by a chaperone protein remains unclear, but one possibility is that amyloid fibrils are typically less toxic than amorphous protein aggregates (30,31). Thus, perhaps chaperones can stimulate amyloid structure to avoid the production of amorphous structures that are more prone to non-specific interactions with other proteins in the cell. However, in the case of myocilin amyloid, this innate function of chaperones triggers an ER stress response, that culminates in TM cell death. Thus, in this case, pharmacological inhibition of Grp94 reduces the interaction with aggregated myocilin, and, as we showed previously, engages autophagic mechanisms that are better equipped for handling protein aggregates while avoiding ER stress activation.
The BnIm scaffold holds considerable promise for selective Grp94 inhibition without significant cellular side effects. New analogs that further exploit the hydrophobic pocket occupied by the C4 position are currently under development. Efforts are also currently underway to better understand the complex interaction between Grp94 and myoc-OLF. Although the N-terminal ATP-binding domain of Grp94 contains the site of 4-Br-BnIm inhibition, other domains might provide the client protein recognition site; inhibition may simply lock Grp94 in a configuration that occludes binding. Such structural information could be valuable in developing new inhibitors remote from the N-terminal ATP binding site. Finally, the cellular mechanisms that lead to clearance of myocilin after Grp94 inhibition are still not clear, but once defined, these too could be important targets for therapeutic development against myocilin and allow for a better understanding of how aggregated proteins are removed from the ER.
MATERIALS AND METHODS
Cell culture
HEK 293T cells were grown and maintained in Dulbecco's modified Eagle's Medium supplemented with 10% FBS (Invitrogen), penicillin (100 units/ml), streptomycin (100 µg/ml) and 1% GlutaMAX (Invitrogen) at 37°C under 5% CO2.
Stably transfected iHEK cells expressing myocilin were grown and maintained in Dulbecco's Modified Eagle's Medium supplemented with 10% FBS (Invitrogen), penicillin (100 units/ml), streptomycin (100 µg/ml) and 1% GlutaMAX (Invitrogen) at 37°C under 5% CO2. For cell selection, the cells were supplemented with hygromycin B (200 µg/ml) (InvivoGen) and G418 (100 µg/ml) (Gibco). To induce myocilin expression in these cells, they were treated with 5 µg/µl tetracycline 24 h prior to transfection, or 48 h prior to treatment.
Human trabecular meshwork cells (ScienCell) were plated on poly-l-lysine coated plates at a ratio of 1:10 poly-l-lysine to ddH2O. Cells were kept and maintained in Fibroblast Medium (Sciencell) supplemented with 2% fetal bovine serum (ScienCell cat. 0010), 1% fibroblast growth supplement (ScienCell cat. 2352) and 1% penicillin/streptomycin solution (ScienCell cat. 0503). HTM cells were maintained at 37°C under 5% CO2.
cDNA and siRNA transfection
All myocilin cDNA constructs were a generous gift from Dr Vincent Raymond [Laval University Hospital (CHUL) Research Hospital]. All siRNA was purchased from Qiagen (Valencia, CA, USA) (23).
Plasmid transfections were carried out in serum-free opti-mem (Invitrogen) medium. cDNA was mixed with Lipofectamine 2000 (Invitrogen) transfection reagent at a ratio of 1 µg cDNA: 2.5 µl Lipofectamine 2000. cDNA was left on the cells for 48 h before harvest. siRNA transfections were carried out in serum-free opti-mem (Invitrogen) medium. siRNA was mixed with siLentFect (BioRad) lipid reagent for RNAi transfection. Next, 40 nm of siRNA was added to cells, so that the siRNA to siLentFect ratio was 1 µl siRNA to 2 µl siLentFect. siRNA was kept on the cells for 24 h prior to cDNA transfection or drug treatment.
Antibodies
Myocilin monoclonal antibody was purchased from R&D Systems (Minneapolis, MN, USA). Grp94, Grp78, Hsp70, Hsp27 and DYKDDDK (Flag) monoclonal antibodies were purchased from Cell Signaling Technologies. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) antibody was purchased from Meridian Life Science (Saco, ME, USA). Actin antibody was purchased from Sigma. Secondary antibodies were all HRP-linked and purchased from Southern Biotechnolofies (Birmingham, AL, USA). All antibodies were added to blots in a 1:1000 dilution in 7% milk.
Pharmacological treatment
Grp94 inhibitors were solubilized in DMSO (Sigma), and added to cells at varying concentrations. All drug treatments were carried out for 24 h prior to cell harvest, and DMSO never exceeded 1% of total volume of cell medium.
Cell harvest
For all cell harvesting, culture media were aspirated and cells were washed 2× in ice cold phosphate-buffered solution (pH 7.4) (PBS). Mammalian Protein Extraction Reagent (M-PER) buffer (Pierce) containing protease inhibitor mixture (Calbiochem), 100 mm phenylmethylsulfonyl fluoride (PMSF) and phosphatase inhibitor II and III mixtures (Sigma) were added to the cells at a 1:100 dilution and cells were scraped. Cells in lysis buffer were collected and incubated on ice for 10 min to allow lysis to occur. Cell lysates were spun down at 16 000g to remove nuclear supernatant, and a bicinchoninic acid assay (BCA) reaction was carried out to determine protein concentration.
Western blotting and co-immunoprecipitation
Western blotting and Co-Immunoprecipitation were performed as previously described (7). Cell lysates were prepared with 2× Laemmli sample buffer (Bio-Rad) and denatured by boiling for 5 min at 100°C. Prepared lysates were then loaded onto a 10-well 10% Tris–glycine gel (Invitrogen) or 18-well, 10% criterion gel (Bio-Rad). After running, gels were transferred onto PVDF membranes (Millipore) and blocked for 1 h at room temperature with 7% milk.
Co-Imunoprecipitation cell harvest was achieved in M-PER buffer supplemented with protease inhibitor mixture, PMSF and phosphatase inhibitor II and III mixtures. A BCA reaction was performed to determine protein concentration, and the lysates were incubated with myocilin antibody by rocking for 4 h to overnight at 4°C. Next, 50 µl of Protein G Dynabeads® (Novex by Life Technologies) were added to the samples and incubated by rocking overnight at 4°C. Samples were washed with ice-cold PBS and loaded onto gels for western blot analysis.
Triton solubility
HEK 293T cells were transfected with either Grp94 siRNA or Control siRNA 24 h prior to mutant myocilin transfection. After a 48 h mutant myocilin transfection, cells were washed twice with ice-cold PBS and lysed in buffer containing 100 mm Tris–HCl (pH 7.4), 3 mm EGTA, 5 mm MgCl2, 0.5% Triton X-100, protease inhibitor cocktail Calbiochem) and 1 mm PMSF and incubated for 2 min on ice. Cells were spun down in a tabletop centrifuge at 16 000g for 10 min. Supernatant was removed and kept as the Triton soluble fraction. The pellet was washed with ice-cold PBS, and then re-suspended in 2× Laemmli sample buffer with 9 m urea. The pellet was sonicated and denatured. Soluble fractions were prepared with 2× Laemmli sample buffer, denatured and run on western blot for analysis.
Tetrazolium assay (MTS)
Assay is performed using CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay (Promega) kit. Cells are plated in a 96-well plate, and transfection/drug treatment is carried out as previously reported. Manufacturer's protocol was followed to complete assay. Assay is read at 490 nm on a standard plate reader. The tetrazolium in the MTS solution will react with live cells to form formazan, which is detectable at 490 nm. The more formazan detected, the more viable the cells.
Lentiviral production and transduction
Lentiviral vectors were created by sub-cloning RFP-tagged cDNA from a mammalian expression vector to the pLEX lentiviral vector. Second generation lentiviral production required both a packaging plasmid (pPax2) and an envelope plasmid (VSV-G). Lentiviral vector, packaging plasmid and envelope plasmid were transfected into HEK 293T cells using Lipofectamine 2000 transfection reagent (Invitrogen) in serum-free media. Cells with transfection reagent were placed in a 37°C incubator with 5% CO2 for 4 h. After 4 h, viral supernatant was removed and fresh serum-free media was added to the cells. After 48–72 h the media containing viral particles was collected. The virus-containing media was then centrifuged to remove cell debris, and filtered with a 0.45 µm syringe filter. Virus-containing media was aliquotted and frozen at −80°C for long-term storage.
For lentiviral transduction, virus-containing media were added to cells in a 1:1 dilution with complete media. Polybrene (Santa Cruz) was added to the lentiviral mixture at 8 µg/ml. HTM cell transduction was achieved after 7 days.
Immunofluorescence and imaging
HTM Cells were plated on poly-l-lysine coated coverslips, and transduced with RFP-tagged lentivirus, treated as described earlier. Cells were then fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100 in PBS, and glycine-fixed in 0.1% glycine in PBS. DAPI was used for a nuclear stain at 1:20 000 dilutions in PBS and incubated for 5–10 min. Coverslips were then glued to glass tissue slides using ProLong® Gold Antifade Reagent (Invitrogen). Images of cells were taken using the Olympus FV1000 MPE Multiphoton Laser Scanning Microscope. Image analysis of intensity was carried out using ImageJ64 software (NIH).
Synthesis
4-Br-BnIm 4-bromo-benzylamine (32 μl, 0.26 mmol) was added dropwise to a stirred solution of readily available aldehyde (24) (125 mg, 0.26 mmol) in MeOH (2 ml) followed by addition of ammonium bicarbonate (20 mg, 0.26 mmol) and dropwise addition of 40% w/v glyoxal (29 μl, 0.26 mmol). The reaction was stirred at 25°C for 12 h. Upon consumption of the starting aldehyde, as seen by thin-layer chromatography, tetrabutylammonium fluoride (0.52 ml, 1.0 m solution in THF) was added and the reaction was allowed to stir for an additional 30 min. The reaction was then quenched with saturated ammonium chloride (5 ml) and then extracted with EtOAc (10 ml × 3). The organic layers were combined, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue was subjected to flash chromatography (95:5 CH2Cl2/MeOH) to provide 4-Br-BnIm as an off white solid (45 mg, 38% yield). 1H NMR (400 MHz, CDCl3/MeOD) 7.45–7.41 (m, 2 H), 7.00 (d 1.39, 1 H), 6.88 (d 8.37, 2 H), 6.82 (d 1.38, 1 H), 6.45 (s, 1 H), 5.00 (s, 2 H), 3.86 (s, 3 H), 3.50-3.44 (m, 2 H), 2.88-2.82 (m, 2 H). 13C NMR (500 MHz, CDCl3/MeOD) 171.9, 163.5, 159.3, 148.8, 142.6, 136.1, 133.5 (2 C), 129.6 (2 C), 127.6, 123.5, 121.4, 116.0, 107.3, 104.07, 54.0, 50.4, 32.1, 27.2 HRMS (ESI) calculated for C20H19BrClN2O4 (M + H)+ 465.0217, found 465.0237.
Computational modeling based on co-crystal structure
Molecular docking studies were done using Surflex-Dock module in Sybyl v8.0 from Tripos International (St Louis, MO, USA). Co-crystal structures of radamide bound to Grp94 and yeast Hsp82 were used for docking the designed molecules (25). Visual interpretation and rendering of the images were carried out using the PyMOL Molecular Graphics System v1.5.0.4. from Schrödinger, LLC.
Fluorescence polarization assays
Increasing concentrations of compound were incubated in 96-well plate (black well, black bottom) format with recombinant cGrp94 (Enzo Life Sciences, 10 nm) and FITC-labeled geldanamycin (Enzo Life Sciences, 6 nm) in assay buffer (20 mm HEPES, pH 7.3, 50 mm KCl, 5 mm MgCl2, 2 mm DTT, and 20 mm Na2MoO4, 0.01% NP-40, and 0.1 mg/ml BGG) at a final volume of 100 μl per well. Plates were incubated for 24 h at 4°C. Fluorescence polarization values were measured using excitation and emission filters of 485 and 528 nm, respectively. Polarization values were then normalized to DMSO (100% tracer bound) and apparent Kd values determined using GraphPad Prism software.
Recombinant protein expression and purification
The myocilin OLF domain was expressed as a tobacco etch virus (TEV) protease-cleavable maltose binding protein (MBP) fusion, as described (13). TEV protease was prepared in-house as previously published (32), using the pRK793 plasmid (Addgene). Grp94 (Canis lupus familiaris, full-length with C-terminal hexahistidine tag, cloned in Novagen pET21a vector) was expressed in E. coli BL21(DE3) cells. Cells were grown in Luria-Bertani broth (BD) supplemented with 100 mg/l ampicillin (Amresco) at 37°C with shaking at 225 rpm. Once cells reached an OD600 = 0.6–0.8, they were induced with 0.5 mm isopropyl-β-d-1-thiogalactopyranoside (Calbiochem) and allowed to shake an additional 3 h prior to harvesting by centrifugation. Cells were solubilized in Tris buffer (50 mm Tris, 0.5 m NaCl at pH 8.0) containing protease inhibitor cocktail (complete Tablets EDTA-free, Roche), lysed via passage through a French Press, and the resulting lysate was clarified with ultracentrifugation. The Grp94 protein was affinity purified using a HisTrap HP 5 ml column (GE Healthcare Life Sciences) with an imidazole gradient (50 mm Tris, 0.5 M NaCl at pH 8.0 with 0–0.5 m imidazole) followed by size-exclusion (HiPrep 16/60 Sephacryl S-300 h, GE Healthcare) with phosphate buffer (PBS, 10 mm sodium phosphate dibasic, 10 mm potassium phosphate monobasic, 0.2 M NaCl, pH 7.2).
Myocilin-OLF aggregation assay
The aggregation assay was a slight modification from that previously described (20). Thioflavin T (ThT, Sigma Aldrich) was prepared as a 1 mg/ml stock solution in nanopure water, from which a working stock of 200 μm was made via dilution into PBS. A 100 mm stock of 4-Br-BnIm was prepared in DMSO and kept at −20°C prior to use. All samples contained 10 μm ThT, and brought to 150 μl final volume in PBS buffer. Proteins (myoc-OLF or Grp94) were at 30 μm each. The final concentration of DMSO in DMSO- or 4-Br-BnIm-containing samples was 0.5% (v/v); at concentrations of DMSO exceeding 0.5%, the interaction of myoc-OLF with Grp94 was either altered or myoc-OLF aggregation was enhanced. Assay sample solutions were prepared in 1.5 ml centrifuge tubes. All sample contents, with the exclusion of myoc-OLF, when 4-Br-BnIm inhibition was tested, samples were incubated with 250 μM inhibitor in the absence of myoc-OLF; the final concentration of inhibitor was less than the starting amount in all cases, as some undissolved material was always present and had to be eliminated to prevent scattering effects during data collection and myoc-OLF protein destabilization. Samples were mixed and incubated in the centrifuge tubes for 1 h in a 42°C water bath. Samples were centrifuged at room temperature for 10 min at 18 000g to remove any particulate, undissolved inhibitor, and then filtered with 0.22 μm Millex-GV syringe-driven filter units (Millipore). Purified myoc-OLF, at room temperature, was added next to appropriate samples. Fully prepared samples were transferred (final volumes 150 μl) to a 96-well (black well, black bottom) microplate (Grenier) that was subsequently sealed with clear MicroAmp PCR film (Applied Biosystems). ThT fluorescence measurements were performed at 10 min intervals for a total of 96 h using a Biotek Synergy H2 microplate reader (excitation filter, λex = 440 nm, emission filter λem = 485 nm). Each sample had a corresponding blank containing all components besides myoc-OLF, which was subtracted to give net signal; samples and blanks were prepared in at least triplicate.
Characterization of aggregation assay products by gel electrophoresis
Aggregation assay samples were harvested for denaturing gel electrophoresis (SDS–PAGE) (33) analysis of aggregate and supernatant contents. Replicates for Grp94 only, myoc-OLF only, Grp94 + myoc-OLF and Grp94 + 4-Br-BnIm + myoc-OLF were pooled in microcentrifuge tubes, recovering ∼450 μl/sample. Insoluble material was isolated by centrifugation for 10 min at 18 000 g; samples with Grp94 only had no insoluble materials. Pellets were washed four times with 1 ml PBS; the last wash was saved to establish fair representation of pellets via gel electrophoresis. Finally, pellets were resuspended in 450 μl PBS with agitation. Samples of original supernatants, final washes and pellets were mixed 1:1 with gel loading buffer for SDS–PAGE analysis. After boiling for 3 min, samples were loaded onto 12% SDS–PAGE gel. Gels were stained with Coomassie blue (33).
In vitro MBP-OLF/Grp94 complexation
After equilibration with PBS buffer, amylose resin (250 μl, New England Biolabs) was transferred to microcentrifuge tubes, and mixed with MBP-OLF fusion protein samples (either in soluble aggregate (1) or monomer form, isolated from cell lysates); 1 ml of 6.7 μm protein was added in total. The resin–protein mixture was incubated at room temperature for 30 min, then centrifuged for 10 min at 18 000g. The supernatant, containing unbound protein, was set aside for SDS–PAGE analysis. The resin was next washed three times with 1 ml volumes of PBS. Grp94 was mixed with uncharged or MBP-OLF-charged amylose resin, and then samples were allowed to incubate at room temperature for 30 min. The samples were again pelleted via centrifugation, setting aside unbound protein for SDS–PAGE analysis. The protein-bound resin was subsequently washed 3 × 1 ml PBS. Bound proteins were eluted with 1 ml of PBS supplemented with 10 mm maltose (Sigma Aldrich) for analysis by SDS–PAGE. All protein samples were evaluated without further manipulation, mixing 1:1 with gel loading buffer. Gel samples were run on a 12% SDS–PAGE gel and stained as earlier.
Quantification and statistical analysis
Quantification of western blots was carried out using ImageJ64 software. Graphs are plotted based on Intensity Density/Area values. Statistical analysis was performed as indicated in the figure legends.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from the NIH (C.A.D.; NS073899: R.L.L.; EY021205: B.B.; CA109265) and Bright Focus Foundation.
Supplementary Material
Acknowledgements
The authors would like to thank Dr Stanislav Tomerav for providing the tetracycline-inducible HEK cell models (iHEK) and Dr Shannon Hill for helpful discussions.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Burns J.N., Orwig S.D., Harris J.L., Watkins J.D., Vollrath D., Lieberman R.L. Rescue of glaucoma-causing mutant myocilin thermal stability by chemical chaperones. ACS Chem. Biol. 2010;5:477–487. doi: 10.1021/cb900282e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zode G.S., Kuehn M.H., Nishimura D.Y., Searby C.C., Mohan K., Grozdanic S.D., Bugge K., Anderson M.G., Clark A.F., Stone E.M., et al. Reduction of er stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J. Clin. Invest. 2011;121:3542–3553. doi: 10.1172/JCI58183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Groef L., Van Hove I., Dekeyster E., Stalmans I., Moons L. Mmps in the trabecular meshwork: Promising targets for future glaucoma therapies? Invest. Ophthalmol. Vis. Sci. 2013;54:7756–7763. doi: 10.1167/iovs.13-13088. [DOI] [PubMed] [Google Scholar]
- 4.Gupta N., Yucel Y.H. Glaucoma as a neurodegenerative disease. Curr. Opin. Ophthalmol. 2007;18:110–114. doi: 10.1097/ICU.0b013e3280895aea. [DOI] [PubMed] [Google Scholar]
- 5.Shepard A.R., Jacobson N., Millar J.C., Pang I.H., Steely H.T., Searby C.C., Sheffield V.C., Stone E.M., Clark A.F. Glaucoma-causing myocilin mutants require the peroxisomal targeting signal-1 receptor (pts1r) to elevate intraocular pressure. Hum. Mol. Genet. 2007;16:609–617. doi: 10.1093/hmg/ddm001. [DOI] [PubMed] [Google Scholar]
- 6.Bruttini M., Longo I., Frezzotti P., Ciappetta R., Randazzo A., Orzalesi N., Fumagalli E., Caporossi A., Frezzotti R., Renieri A. Mutations in the myocilin gene in families with primary open-angle glaucoma and juvenile open-angle glaucoma. Arch. Ophthalmol. 2003;121:1034–1038. doi: 10.1001/archopht.121.7.1034. [DOI] [PubMed] [Google Scholar]
- 7.Jacobson N., Andrews M., Shepard A.R., Nishimura D., Searby C., Fingert J.H., Hageman G., Mullins R., Davidson B.L., Kwon Y.H., et al. Non-secretion of mutant proteins of the glaucoma gene myocilin in cultured trabecular meshwork cells and in aqueous humor. Hum. Mol. Genet. 2001;10:117–125. doi: 10.1093/hmg/10.2.117. [DOI] [PubMed] [Google Scholar]
- 8.Burns J.N., Turnage K.C., Walker C.A., Lieberman R.L. The stability of myocilin olfactomedin domain variants provides new insight into glaucoma as a protein misfolding disorder. Biochemistry. 2011;50:5824–5833. doi: 10.1021/bi200231x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDowell C.M., Luan T., Zhang Z., Putliwala T., Wordinger R.J., Millar J.C., John S.W., Pang I.H., Clark A.F. Mutant human myocilin induces strain specific differences in ocular hypertension and optic nerve damage in mice. Exp. Eye. Res. 2012;100:65–72. doi: 10.1016/j.exer.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zode G.S., Sharma A.B., Lin X., Searby C.C., Bugge K., Kim G.H., Clark A.F., Sheffield V.C. Ocular-specific er stress reduction rescues glaucoma in murine glucocorticoid-induced glaucoma. J. Clin. Invest. 2014 doi: 10.1172/JCI69774. 124, 1956–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aroca-Aguilar J.D., Sanchez-Sanchez F., Ghosh S., Coca-Prados M., Escribano J. Myocilin mutations causing glaucoma inhibit the intracellular endoproteolytic cleavage of myocilin between amino acids arg(226) and ile(227) J. Biol. Chem. 2005;280:21043–21051. doi: 10.1074/jbc.M501340200. [DOI] [PubMed] [Google Scholar]
- 12.Fingert J.H., Heon E., Liebmann J.M., Yamamoto T., Craig J.E., Rait J., Kawase K., Hoh S.T., Buys Y.M., Dickinson J., et al. Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum. Mol. Genet. 1999;8:899–905. doi: 10.1093/hmg/8.5.899. [DOI] [PubMed] [Google Scholar]
- 13.Hill S.E., Donegan R.K., Lieberman R.L. The glaucoma-associated olfactomedin domain of myocilin forms polymorphic fibrils that are constrained by partial unfolding and peptide sequence. J. Mol. Biol. 2014;426:921–935. doi: 10.1016/j.jmb.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gobeil S., Rodrigue M.-A., Moisan S., Nguyen T.D., Polansky J.R., Morissette J., Raymond V. Intracellular sequestration of hetero-oligomers formed by wild-type and glaucoma-causing myocilin mutants. Invest. Ophthalmol. Vis. Sci. 2004;45:3560–3567. doi: 10.1167/iovs.04-0300. [DOI] [PubMed] [Google Scholar]
- 15.Joe M.K., Sohn S., Hur W., Moon Y., Choi Y.R., Kee C. Accumulation of mutant myocilins in er leads to er stress and potential cytotoxicity in human trabecular meshwork cells. Biochem. Biophys. Res. Commun. 2003;312:592–600. doi: 10.1016/j.bbrc.2003.10.162. [DOI] [PubMed] [Google Scholar]
- 16.Vollrath D., Liu Y. Temperature sensitive secretion of mutant myocilins. Exp. Eye Res. 2006;82:1030–1036. doi: 10.1016/j.exer.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Z.H., Vollrath D. A cellular assay distinguishes normal and mutant tigr/myocilin protein. Hum. Mol. Genet. 1999;8:2221–2228. doi: 10.1093/hmg/8.12.2221. [DOI] [PubMed] [Google Scholar]
- 18.Kim B.S., Savinova O.V., Reedy M.V., Martin J., Lun Y., Gan L., Smith R.S., Tomarev S.I., John S.W., Johnson R.L. Targeted disruption of the myocilin gene (myoc) suggests that human glaucoma-causing mutations are gain of function. Mol. Cell. Biol. 2001;21:7707–7713. doi: 10.1128/MCB.21.22.7707-7713.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam D.S., Leung Y.F., Chua J.K., Baum L., Fan D.S., Choy K.W., Pang C.P. Truncations in the tigr gene in individuals with and without primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci. 2000;41:1386–1391. [PubMed] [Google Scholar]
- 20.Orwig S.D., Perry C.W., Kim L.Y., Turnage K.C., Zhang R., Vollrath D., Schmidt-Krey I., Lieberman R.L. Amyloid fibril formation by the glaucoma-associated olfactomedin domain of myocilin. J. Mol. Biol. 2012;421:242–255. doi: 10.1016/j.jmb.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lutjen-Drecoll E., May C.A., Polansky J.R., Johnson D.H., Bloemendal H., Nguyen T.D. Localization of the stress proteins alpha b-crystallin and trabecular meshwork inducible glucocorticoid response protein in normal and glaucomatous trabecular meshwork. Invest. Ophthalmol. Vis. Sci. 1998;39:517–525. [PubMed] [Google Scholar]
- 22.Polansky J.R., Fauss D.J., Chen P., Chen H., Lutjen-Drecoll E., Johnson D., Kurtz R.M., Ma Z.D., Bloom E., Nguyen T.D. Cellular pharmacology and molecular biology of the trabecular meshwork inducible glucocorticoid response gene product. Ophthalmologica. 1997;211:126–139. doi: 10.1159/000310780. [DOI] [PubMed] [Google Scholar]
- 23.Suntharalingam A., Abisambra J.F., O'Leary J.C., Koren J., Zhang B., Joe M.K., Blair L.J., Hill S.E., Jinwal U.K., Cockman M., et al. Glucose-regulated protein 94 triage of mutant myocilin through endoplasmic reticulum-associated degradation subverts a more efficient autophagic clearance mechanism. J. Biol. Chem. 2012;287:40661–40669. doi: 10.1074/jbc.M112.384800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duerfeldt A.S., Peterson L.B., Maynard J.C., Ng C.L., Eletto D., Ostrovsky O., Shinogle H.E., Moore D.S., Argon Y., Nicchitta C.V., et al. Development of a grp94 inhibitor. J. Am. Chem. Soc. 2012;134:9796–9804. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Immormino R.M., Metzger L.E.T., Reardon P.N., Dollins D.E., Blagg B.S., Gewirth D.T. Different poses for ligand and chaperone in inhibitor-bound hsp90 and grp94: Implications for paralog-specific drug design. J. Mol. Biol. 2009;388:1033–1042. doi: 10.1016/j.jmb.2009.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee A.S. The er chaperone and signaling regulator grp78/bip as a monitor of endoplasmic reticulum stress. Methods. 2005;35:373–381. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 27.Proctor C.J., Lorimer I.A.J. Modelling the role of the hsp70/hsp90 system in the maintenance of protein homeostasis. Plos One. 2011;6 doi: 10.1371/journal.pone.0022038. e22038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Freitas J.C.M., Silva B.D.D., de Souza W.F., de Araujo W.M., Abdelhay E.S.F.W., Morgado-Diaz J.A. Inhibition of n-linked glycosylation by tunicamycin induces e-cadherin-mediated cell-cell adhesion and inhibits cell proliferation in undifferentiated human colon cancer cells. Cancer Chemother. Pharmacol. 2011;68:227–238. doi: 10.1007/s00280-010-1477-8. [DOI] [PubMed] [Google Scholar]
- 29.Blair L.J., Nordhues B.A., Hill S.E., Scaglione K.M., O'Leary J.C., III, Fontaine S.N., Breydo L., Zhang B., Li P., Wang L., et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Invest. 2013;123:4158–4169. doi: 10.1172/JCI69003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eisenberg D., Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldschmidt L., Teng P.K., Riek R., Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl Acad. Sci. USA. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tropea J.E., Cherry S., Waugh D.S. Expression and purification of soluble his(6)-tagged tev protease. Methods Mol. Biol. 2009;498:297–307. doi: 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook J., Russell D.W. Molecular Cloning: A Laboratory Manual. NY: Cold Spring Harbor Laboratory Press, Cold Spring Harbor; 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.