

Abstract
Osteosarcoma is a malignancy of the bone that primarily affects adolescents. Current treatments retain mortality rates, which are higher than average cancer mortality rates for the adolescent age group. We designed a micellar delivery system with the aim to increase drug accumulation in the tumor and potentially reduce side effects associated with chemotherapy. The design features are the use of the hydrophilic d-aspartic acid octapeptide as both the effective targeting agent as well as the hydrophilic micelle corona. Micelle stabilization was accomplished by binding of model drug (doxorubicin) via an acid-sensitive hydrazone bond and incorporating one to four 11-aminoundecanoic acid (AUA) moieties to manipulate the hydrophobic/hydrophilic ratio. Four micelle-forming unimers have been synthesized and their self-assembly into micelles was evaluated. Size of the micelles could be modified by changing the architecture of the unimers from linear to branched. The stability of the micelles increased with increasing content of AUA moieties. Adsorption of all micelles to hydroxyapatite occurred rapidly. Doxorubicin release occurred at pH 5.5, whereas no release was detected at pH 7.4. Cytotoxicity toward human osteosarcoma Saos-2 cells correlated with drug release data.
Introduction
Osteosarcoma is a cancer of the bone that primarily affects adolescents. While improvements in treatment have increased the 5-year survival rate to 65%, it still lags behind overall cancer survival rates for that age group.1 Furthermore, the metastatic or recurring disease 5-year survival rate is still at a meager 20%.2 Current osteosarcoma therapies include surgical resection followed by chemotherapy regimens of doxorubicin (DOX), high-dose methotrexate with leucovorin rescue, cisplatin, and ifosfimide.3 However, therapeutic indexes of these drugs are limited by severe toxicities; DOX, for instance, has well-documented cardiotoxicity.4
Several groups have attempted to reduce this cardiotoxicity and enhance pharmacokinetics via large-molecule conjugation or nanoparticle entrapment. For example, Susa et al. increased intracellular drug accumulation by loading lipid-modified dextran nanoparticles with DOX, effectively overcoming multidrug resistance in vitro.5 Other groups targeted drugs with molecules such as bisphosphonates and acidic oligopeptides, which have a strong affinity to bone.6 Utilizing this targeting methodology, Salerno et al. demonstrated reduction in bone metastases in mouse metastatic breast cancer models.7 Hrubý et al. used bisphosphonate targeting ligands on linear HPMA copolymers containing DOX bound via pH-sensitive hydrazone bonds.8 They demonstrated in vitro HAp binding as well as pH-dependent DOX release, due to the reduced pH associated with the interstitial space in some tumors.
In contrast to their small molecule counterparts, these delivery systems have enhanced anti-neoplastic properties by improving pharmacokinetics and reducing unwanted side effects. Improvements, however, can be made in these complex systems as entrapped drugs may have the potential for premature release, and those with covalently bound drug yield drug-to-polymer weight percentages below 10%. Fundamentally, these are valid systems that exhibit the important elements of having a bone-targeting ligand, drug, degradable linker, and large molecules with favorable pharmacokinetics.
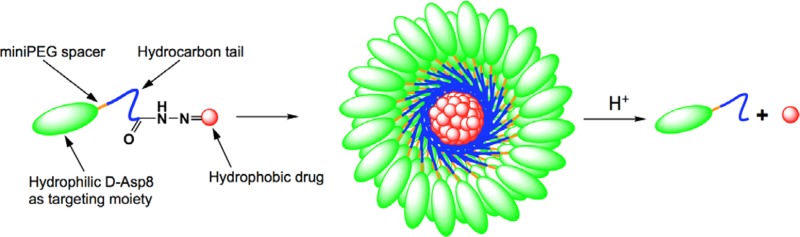
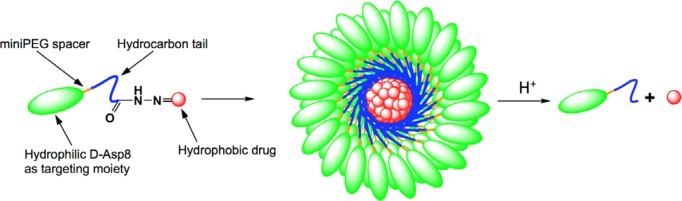
Conjugating a targeting ligand modeled after bone sialoprotein, such as an aspartic acid oligopeptide to DOX via a hydrazone bond, would yield a practical, simple drug that might improve drug accumulation in bone.6 However, it would lack the pharmacokinetics that are associated with larger molecules.9,10 A modest design modification can change this. DOX is a hydrophobic drug, but more importantly it has the tendency toward π–π stacking.11 Furthermore, the aspartic acid oligopeptide-targeting moiety is very hydrophilic. By inserting aliphatic hydrocarbon chains and a flexible miniPEG spacer between DOX and the aspartic acid octapeptide a novel micelle-forming unimer could be assembled (Scheme 1). This design exhibits high drug loading while retaining covalent bonds between the targeting ligand and the drug. The micellar self-assembly increases the size of the targeted delivery system, extending circulation and exposure to the tumor by reducing glomerular filtration. Additionally, the sequestration of DOX to the center of the micelle is designed to reduce metabolism by the myocardium and thus reduce cardiotoxicity.
Scheme 1. Illustration of Micelle Formation from Amphiphilic Unimer Consisting of d-Aspartic Acid Octapeptide (d-Asp8), miniPEG Spacer, Hydrophobic Tail Based on 11-Aminoundecanoic Acid, and Doxorubicin Bound via an Acid-Sensitive Hydrazone Bond.
In order to test the viability of the proposed micellar delivery system as well as the relationship between structure and properties, four novel DOX-containing unimers with varying hydrophobicity as well as architecture have been synthesized. Each unimer has been analyzed regarding its ability to form micelles, its size, and its adsorption to hydroxyapatite. In addition, drug release and in vitro osteosarcoma toxicity have been analyzed.
Results and Discussion
Drug carriers are often employed to increase solubility of hydrophobic drugs as well as boost their pharmacokinetics. In designing a new carrier effective against osteosarcoma it is important to produce a molecule that has a defined structure, produces micelles with reproducible polydispersity, and is stable. Rather than focusing on increasing solubility, we decided to utilize the hydrophobic nature of chemotherapeutics to stabilize a micellar delivery system. The targeting moiety, d-aspartic acid octapeptide (d-Asp8), was selected for its optimized targeting potential,12 its stability toward protease degradation associated with d-peptides, as well as its hydrophilic nature.13,14 The addition of 11-aminoundecanoic acid (AUA) resulted in increased hydrophobicity. 8-Amino-3,6-dioxaoctanoic acid (miniPEG) was placed between the AUA and the d-Asp8 for additional flexibility of the backbone. An acid-sensitive hydrazone bond was incorporated at the unimer’s C-terminus to bind DOX, a model drug selected for its hydrophobic nature as well as an ability to π–π stack with itself.15,16 As such, the prototype unimer DOX-A1-D8 was formed (Scheme 2).
Scheme 2. Synthesis of Amphiphilic Unimers with Linear and Branched Architecture.

Thermodynamic stability of micelles increases as the hydrophilic/lipophilic balance (HLB) is lowered, e.g., by increasing the weight percent of the hydrophobic moiety.17 In order to verify this theory regarding stability in our micelles, an additional AUA moiety was added to DOX-A1-D8 to form DOX-A2-D8. Continually adding hydrophobic AUA to the micelle has its potential drawbacks; if the hydrophobic portions far exceed the hydrophilic portions in a linear unimer the conical shape of a single unimer may be lost and the risk of an inverse micelle or other undesirable structures increases.18 To counteract this effect, yet retain the ability to increase stability by adding hydrophobic moieties, a modification was needed. Branched head groups will in theory increase the lateral area of the headgroup, thereby retaining the conical structure of the micelle. DOX-A2-K-D4 demonstrates a simple addition of this branched headgroup (when compared to DOX-A2-D8), while DOX-A4-K-D4 doubles the number of hydrophobic AUAs (Scheme 2).
Assessment of Micelle Formation and Hydrodynamic Diameter via Dynamic Light Scattering
Verification of unimer self-assembly was determined by DLS. Scattered light count rates exponentially increase as nucleation and assembly of micelles occur. Therefore, plotting the normalized light-scattering count rate (counts per second) vs concentration (LOG-LOG) graph (Figure 1) made the point of nucleation of the micelles readily apparent. Micelle thermodynamic stability increased as expected when the number of AUA moieties was increased from two to four: DOX-A2-K-D4 0.0027 mg/mL to DOX-A4-K-D4 0.00036 mg/mL. By contrast very little difference in thermodynamic stability was observed between branched and nonbranched micelles DOX-A2-D8, 0.0035 mg/mL vs DOX-A2-K-D4, 0.0027 mg/mL. Of note, by comparing documented blood volume (<100 mL/kg of mouse) with standard in vivo DOX dosages (3 mg/kg/dose) even the least stable micelle DOX-A1-D8 at 0.0053 mg/mL is expected to remain above its critical micelle concentration after dilution during dosing.19−21
Figure 1.

Determination of the critical micellar concentration (CMC) of DOX-containing unimers. A logarithmic plot of the intensity ratio in counts per second (CPS) vs the logarithm of the unimer concentration. The figure demonstrates the increased thermodynamic stability of more hydrophobic micelles.
The architecture, branched vs linear, had a profound effect on the size of the micelles. The higher degree of conicality created as head groups are branched should reduce the diameter of the micelle.18 Indeed, the diameters of both branched unimers, DOX-A2-K-D4 and DOX-A4-K-D4, were 28.4 and 28.0 nm, respectively, smaller than their linear counterparts, DOX-A1-D8, DOX-A2-D8, which were 53.3 and 50.4 nm. All four micelles had polydispersities below 0.1 as determined by DLS (Figure 2).
Figure 2.

Size distribution of micelles as determined by dynamic light scattering in 0.01 M HEPES (pH 7.4; 1 mg·mL–1). A clear division between linear and branched unimers was observed: linear unimers form larger micelles than do branched unimers.
Cryo-Electron Microscopy (CryoEM)
In addition to DLS, the size of the micelles was confirmed via CryoEM. The shape was regular and spherical (Figure 3). One anomaly that was observed in both DLS and CryoEM, however, was the surprisingly large size of the micelles. Each unimer length is far shorter than the radius of an individual micelle. Due to the discrepancy in expected size vs what was experimentally observed, the self-assembly into a liposomal structure rather than a micelle was hypothesized. Further analysis of the cryoEM images did not reveal an evident bilayer; thus, we believe that the structures are still micelles. Also of note, the overall size of the micelles is above the renal threshold, but the size of the unimers is under the renal threshold. Although blood vessel shear forces and protein–micelle interactions drastically complicate predictions, it is feasible that under ideal conditions micelles could circulate above their critical micelle concentration; their size, above the renal threshold, could extend circulation time.22,23 Over time, the micelles could extravasate from the bloodstream as they bind to the bone at the tumor site and eventually the blood concentration would dip below the critical micelle concentration; micelles then destabilize into unimers, which are readily cleared as is the covalently bound DOX.
Figure 3.

TEM image of micelles in 0.01 M HEPES (1 mg·mL–1) at room temperature. The images confirm the sizes observed in the DLS studies as well as demonstrate the spherical nature of the micelles.
HAp Binding
Bone is a complex weave of organic fibers and inorganic mineral giving it rigidity as well as some elasticity. The inorganic portion, hydroxyapatite HAp, composed of Ca10(PO4)6(OH)2, increases in crystallinity over time. It is this higher crystalline state that aspartic acid oligopeptides (AO) preferentially bind to.24,25 In osteosarcoma patients, highly active osteoclasts produce extensive resorption surfaces by exposing highly crystalline HAp surfaces which AO is able to target.26,27 This specificity of AO for highly crystalline HAp additionally may reduce nonspecific binding to the majority of the noncancerous bone.
In an attempt to simulate bone binding, a HAp binding assay was carried out. A low ratio of HAp to unimers was selected where saturation of the HAp occurred and an excess of all four unimers was present in measurable amounts, demonstrating the difference in binding between the four micelles. Slight increases in HAp concentration yielded complete binding nearly immediately, reducing the discrepancy between each of the micelles (data not shown). Still, each micelle reached their respective binding maximum (Bmax) nearly immediately, as previously noted.8,28 Differences were observed in the amount of unimer needed to saturate the given HAp although the rate of binding was nearly identical. A general trend of lower saturation levels was observed for larger molecules. The largest unimer, DOX-A4-K-D4, reached Bmax at 55 ± 3.0%, whereas the lowest MW unimer, DOX-A1-D8, reached Bmax at 91 ± 3.1% (Figure 4) and as others have noted may be due to steric hindrance.29 An alternative explanation is that due to the high surface energy of HAp, more hydrophobic molecules have reduced binding.30 This hypothesis is unlikely as it would predict that slight variations in HAp concentration would have little effect on Bmax. There was very little difference between branched and linear unimers of close molecular weights, DOX-A2-D8 and DOX-A2-K-D4, reaching Bmax at 78 ± 4.6% and 83 ± 6.4%, respectively.
Figure 4.

In vitro adsorption of DOX-containing micelles (52.5 μM) to HAp at subsaturation levels of HAp (7.5 mg·mL–1).
Release Kinetics
Osteosarcoma provides a unique microenvironment, which can be exploited by a pH-sensitive hydrazone bond. The hydrazone bond is stable at pH 7.4 but labile at acidic pHs. Therefore, it would be cleaved in endosomal compartments (assuming cell internalization).9 Supposing strong adsorption of micelles with HAp occurs, an osteoclast-assisted release mechanism in the interstitial space is hypothesized. Osteoclasts, as stated previously, are highly active in osteosarcoma. The micelles that preferentially bind to resorption surfaces will be covered by osteoclasts. The osteoclasts then produce a sealing zone or strong bond to the bone. A resorption lacuna will be formed underneath the osteoclasts as these osteoclasts release cathepsin K, degrading the collagen fiber network, as well as protons, reducing the pH and degrading the HAp.31 The reduced pH could cleave hydrazone bond and release the DOX from the unimers. Incidentally, other bone metastases also recruit osteoclasts. In turn, the osteoclasts, during resorption, release essential growth factors trapped in bone.32 These micelles utilize osteoclast-assisted release, and therefore might have the potential to treat bone metastases.
Though lacunae have a pH of around 4–4.5, other groups measure hydrazone release kinetics at pH of 5.5 mimicking conditions in an endosome.33,34 By using a pH of 5.5 for our studies we are able to compare to what others have observed while gaining insight into what we might expect with lacunae release. Trends in release seem to mirror the micellar thermodynamic stability established in the CMC experiments. As stability of micelles increases, the rate of DOX release decreases (Figure 5).
Figure 5.

In vitro release profiles of DOX from DOX-containing micelles (at pH 5.5 and pH 7.4 at 37 °C) measured by HPLC. Of note, all of the pH 7.4 data are on the baseline due to lack of detectable DOX release.
We see a sigmoidal curve that may indicate a certain degree of destabilization is necessary in order to achieve rapid release. Ultimately, pH 5.5 release kinetics falls in the normal range of what is observed in the literature.8,9,35,36 However, at pH 7.4 we observed no detectable release, which appears to be slightly lower than what others have reported.8,9,35,36
A lower than expected release of DOX from DOX-A4-K-D4 micelles was observed. This is most probably connected to increased thermodynamic stability of the micelles. We hypothesize that more thermodynamically stable secondary packing is responsible for the low release rate of DOX from DOX-A4-K-D4 micelles. Before self-assembly into micelles, the unimers were eluted at higher elution time (12.5 min) indicating hydrophobic character. However, after extended incubation (48 h, 37 °C) the peak moved to 3.6 min, indicating a very hydrophilic molecule such as an intact micelle. Release kinetics of the DOX-A4-K-D4 following incubation at 37 °C was consistent with the HPLC observations.
Cytotoxicity
IC50 values of DOX-containing micelles were determined and correlated with the drug release data. Free DOX had a lower IC50 value of 0.077 ± 0.009 μM than the micelles (p < 0.01) (Figure 6). Three of the micelles (DOX-A1-D8 0.39 ± 0.012 μM, DOX-A2-D8 0.39 ± 0.021 μM, and DOX-A4-K-D4 0.45 ± 0.026 μM) did not differ significantly from each other (p > 0.05). However, DOX-A2-K-D4 consistently produced slightly higher mean IC50 values (0.61 ± 0.086 μM) than the linear micelles (p < 0.05), it was not statistically different from DOX-A4-K-D4 (p > 0.05), its most similar compound. Both DOX-A2-K-D4 and DOX-A4-K-D4 demonstrated slower release kinetics that may have reduced the IC50 slightly for the test. Most importantly, each unimer delivered its payload and demonstrated that it is pharmacologically active and has potential for future in vivo studies. DOX-A4-K-D4 ended up being the most interesting because it forms the most stable micelle, yet retains nearly all the activity of the others. In addition, the cytotoxicity of A1-D8 (control; unimer without DOX) was minimal (94 ± 5.2% cell viability at 1000 μM).
Figure 6.

Cytotoxicity of free DOX and micelles with variable structure toward human osteosarcoma Saos-2 cells following 72 h incubation.
Conclusions
The premise of this study was to design, characterize, and optimize unimers for the treatment of osteosarcoma. Each unimer component has one or more purposes for being included. Most importantly, the targeting ligand provided both hydrophilicity to the amphiphile as well as targeting to HAp. The drug, being covalently bound to the unimers via hydrazone bond, provided hydrophobic stability in addition to its therapeutic properties. DOX-A1-D8 is the simplest unimer containing drug, degradable hydrazone bond, a single AUA, miniPEG, and a d-Asp8 targeting ligand. By adding an additional AUA to DOX-A1-D8 we increased hydrophobicity (DOX-A2-D8) as well as the thermodynamic stability of the micelles. Branched unimers incorporating longer hydrophobic chains were synthesized; DOX conjugates of branched unimers (DOX-A2-K-D4 and DOX-A4-K-D4) possessed higher stability and smaller size of the micelles when compared to linear architectures. Each unimer maintained its affinity to hydroxyapatite adsorption when in a micellar assembly. In addition, at reduced pH (5.5) the hydrazone bond hydrolyzed and released unaltered DOX. The rate of DOX release can be extended by increasing the stability of the micelles. Ultimately, each micelle was able to release unmodified drug in vitro.
Future studies may benefit from several of the micelle’s characteristics. The sizes of the assembled micelles are above the renal threshold and we expect to see increased circulation and improved pharmacokinetics as compared to a targeted small molecule. Other key aspects of this delivery system include its potential for diverse applications. For example, this construct might be used to target a variety of bone maladies with other hydrophobic drugs that contain ketone or aldehyde groups in their structures. Additional imaging moieties may be introduced and coassembled into micelles providing location and biodistribution information about the drug delivery system.
Experimental Procedures
Materials
Solvents, dimethylformamide (DMF), dichloromethane (DCM), methanol (MeOH), dimethyl sulfoxide (DMSO), ethyl acetate, ether, and acetonitrile (ACN) were purchased from VWR, Fisher Scientific or Sigma-Aldrich and were reagent grade or better. Piperidine, diisopropyl ethylamine (DIPEA), trifluoroacetic acid (TFA), triisoproyl silane (TIS), 11-aminoundecanoic acid (AUA), hydrazine, sodium carbonate (Na2CO3), and Dulbecco’s Modified Eagle Medium F-12 (DMEM) were purchased from Sigma-Aldrich. Magnesium sulfate and sodium sulfate were purchased from Fisher Scientific and Macron Chemicals, respectively. 1-(9-Fluorenyl)methylchloroformate (Fmoc-Cl), Fmoc N-hydroxysuccinimide ester (Fmoc-OSu), and 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) were purchased from AKsci. 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), chloro-trityl resin, hydroxybenzotriazole (HOBt), and N-9-fuorenylmethoxycarbonyl-d-aspartic acid (Fmoc-d-Asp-OH) were purchased from P3Biosystems. Diisopropylcarbodiimide (DIC) was purchased from Research Chemicals. N-2-N-6-Bis(9-fluorenylmethyloxycarbonyl)-l-lysine (Fmoc-Lys(Fmoc)-OH) was purchased from Aapptec and 9-fluorenylmethoxycarbonyl-8-amino-3,6-dioxaoctanoic acid (Fmoc-miniPEG) was purchased from BioBlocks. CAPTAL S hydroxyapatite (HAp) was purchased from Plasma Biotal Ltd. Holey carbon grids were purchased from Electron Microscopy Sciences. Sephadex LH20 beads were purchased from Amersham Pharmacia Biotech AM. Doxorubicin (DOX) was a generous gift from Meiji Seika Kaisha, Tokyo, Japan. 2-(2-Methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt (CCK-8) was purchased from Dojindo. Human osteosarcoma Saos-2 cells were purchased from ATTC. Fetal bovine serum (FBS) was purchased from Hyclone.
Fmoc-Hydrazine (9-Fluorenylmethyl carbazate)
Fmoc-hydrazine was synthesized as previously described37 with modifications. Briefly, 1 g Fmoc-Cl was dissolved in 20 mL of precooled ether, and this solution was added dropwise to 5× excess hydrazine suspension in ether over 1 h, immediately producing a white precipitate. The reaction was stirred overnight and then added to 200 mL ethyl acetate under strong stirring. Water was slowly added and the organic solution was washed twice. The organic layer was transferred to a flask, and dried with anhydrous Na2SO4. Removal of ethyl acetate was performed by evaporation under vacuum to get a white flurry product. Yield: 75%, mp 169.2–170.8 °C.
1H NMR (DMSO-d6) δ 8.36 (s, 1H, CONH), 7.31–7.90 (m, 8H, Ar), 4.28 (m, 2H, CHCH2), 4.23 (m, 1H, CHCH2), 4.12 (br, s, 2H, NH2).
Fmoc-11-aminoundecanoic Acid
(11-(9-Fluorenylmethoxycarbonylamino)undecanoic acid)) (Fmoc-AUA) was synthesized as previously described38 with slight modification. 11-Aminoundecanoic acid (1 g, 5 mmol) was suspended in 50 mL of H2O–dioxane (v/v 4:1). The pH of the suspension was adjusted to 9 using 10% Na2CO3. After the mixture became a clear solution under reflux, Fmoc N-hydroxysuccinimide ester (Fmoc-OSu; 1.68 g, 5 mmol) in dioxane (50 mL) was added dropwise over 20 min. The reflux was kept overnight. Then, the cloudy mixture was diluted with H2O and acidified with 1 N HCl to pH 3. Dichloromethane (2 × 200 mL) was added to extract the product. The organic phase was washed with brine twice, dried with anhydrous magnesium sulfate, and then concentrated by rotary evaporation under reduced pressure. A white powder (1.83 g) was obtained with yield 86%; mp 127.8–128.5 °C.
1H NMR (DMSO-d6) δ 7.30–7.90 (m, 8H, Ar), 7.25 (t, 1H, CONH), 4.28 (m, 2H, CHCH2), 4.18 (m, 1H, CHCH2), 2.95 (m, 2H, NHCH2), 2.17 (m, 2H, CH2CO), 1.47 (2H, NHCH2CH2). 1.38 (2H, CH2CH2CO), 1.24 (12H, (CH2)6).
Solid Phase Peptide Synthesis
Synthesis of unimers was performed by standard Fmoc solid phase peptide synthesis. 2-Chlorotrityl chloride resin (1.11 mmol/g) was loaded with 0.4 mmol/g Fmoc-hydrazine in DCM (a small amount of DMF was added due to solubility) overnight. A 4-fold excess of DIPEA was added as a base. The resin was then capped with mixture of DCM:MeOH:DIPEA 17:2:1 (20 mL × 4) followed by washing three times with DCM and DMF, consecutively. After removal of Fmoc-group with 20% (v/v) piperidine in DMF, a 2.5-fold excess of Fmoc-AUA was added using DIC/HOBt as coupling agents. The completion of each coupling step was verified by Kaiser test. To prepare branched unimers, Fmoc-Lys(Fmoc)-OH was added following AUA. The branched unimers have 4 aspartic acids on each branch with a total of eight instead of eight consecutive aspartic acids on the linear unimers (Scheme 2). Upon completion the resin was washed with DMF, DCM, and MeOH and dried in a desiccator. Unimers were then cleaved using 95:2.5:2.5 TFA:TIS:H2O and precipitated in diethyl ether. Precipitated product was dried and purified on a preparative HPLC column (Agilent Zorbax 300SB-C18) using water with 0.1% trifluoroacetic acid (TFA) as the aqueous phase and ACN with 0.1% TFA as the organic phase. Purified fractions had their volume reduced under low-pressure rotoevaporation followed by freeze–drying. Purity was confirmed using HPLC (see Supporting Information). Molecular weight for each unimer was confirmed using positive mode MALDI-ToF mass spectrometry (see Supporting Information). Linear unimers are abbreviated A1-D8 and A2-D8 indicating that they have eight consecutive aspartic acids (D8) and delineating whether they have one (A1) or two (A2) AUA moieties, respectively. The branched unimers are labeled A2-K-D4 and A4-K-D4 indicating their lysine branch (K) and the four consecutive aspartic acids (D4), as well as two (A2) or four (A4) AUAs, respectively. Both linear and branched unimers contain 8-amino-3,6-dioxaoctanoic acid (miniPEG) between the aspartic acids and the rest of the sequence.
Conjugation to DOX
Purified unimers were conjugated to DOX via hydrazone bond using a 1:2 unimer to DOX molar ratio. Dry DMSO was added to the dry ingredients until the mixture fully dissolved and mixed freely (approximately 10 μL DMSO to 1 mg DOX/unimer). The solution was mixed in the dark at room temperature for 3 days and the progression of the reaction was tracked by HPLC (data not shown). Following the reaction, free DOX was removed using an LH20 column. The DMSO reaction solution was diluted 5-fold in methanol and added to an LH20 column. Methanol was used as an eluent and collected fractions were dried under nitrogen yielding DOX-A1-D8, DOX-A2-D8, DOX-A1-K-D4, and DOX-A4-K-D4. The final DOX content in micelles was determined using UV–vis spectrophotometry by measuring the methanol solution absorbance at 495 nm. Purity was confirmed using HPLC (see Supporting Information).
Dynamic Light Scattering (DLS)
Size, polydispersity, and critical micelle concentration (CMC) measurements were all taken using a Wyatt DynaPro Plate Reader II and analyzed using Dynamics 7 software. Each DOX-bound unimer was dissolved in 0.01 M HEPES, pH 7.4. For CMC measurement, dilutions were made in triplicate from 5 mg/mL down to 0.01 μg/mL on 96-well plates (NUNC optical bottom black polystyrene plates), final volumes per well being 200 μL. Plates were then sealed and refrigerated overnight prior to running DLS at 20 °C. For size/polydispersity measurements, samples with concentration of 1 mg/mL were selected to coincide with cryoEM measurements (see below).
Cryo-Electron Microscopy (CryoEM)
CryoEM (FEI Tecnai 12) was used to confirm the size of the micelles as well as observe their shape. Samples were prepared at room temperature at 1 mg/mL DOX-bound unimers in 0.01 M HEPES. The samples were then manually added to holey-carbon-coated copper grids. A FEI Vitrobot then blotted and plunged the samples into liquid ethane. The samples were transferred to liquid nitrogen until imaged.
Hydroxyapatite Adsorption Assay
Hydroxyapatite (HAp) adsorption was assessed for each DOX-bound unimer. HAp was added to microcentrifuge tubes (3 mg/50 μL HEPES vehicle) followed by 350 μL of 60 μM DOX-bound unimer solution. The centrifuge tube was then vortexed for the allotted time and centrifuged at 7000 rpm for 1 min. The supernatant was measured at 480 nm on a Cary 400Bio UV-Spectrophotometer. Data points measured in triplicates included times 0, 15 s, 30 s, 1 min, and 2 min.
DOX Release Kinetics
DOX release kinetics was measured using an Agilent 1100 HPLC with heated autosampler plate. Samples were prepared in triplicate at 0.1 mg/mL DOX-bound unimer in pH 5.5 and 7.4 in 1 M HEPES solutions. Samples were placed in the HPLC autosampler, which retained a temperature of 37 °C over the course of the experiment. Samples were run at 0, 1, 2, 3, 5, 10, and 24 h. pH dependent DOX release was measured using HPLC UV absorption and calculated using the following equation:
Cytotoxicity
Cytotoxicity of DOX bound unimers and free DOX toward human osteosarcoma Saos-2 cells was assessed using the CCK-8 bioassay. Saos-2 cells were maintained in DMEM/F12 medium supplemented with 10% fetal bovine serum. Plates (CELLSTAR TC 96-well) were seeded at a cell density of 4000 cells/well and were administered dilutions of each unimer in triplicates. Concentrations administered reflected hydrazone bound DOX content in each micelle and was confirmed using spectrophotometer measurements in MeOH. Following a 72 h treatment with drug, cytotoxicity was measured using CCK-8 per manufacturers instructions. Each assay was repeated 3 times, IC50 values were expressed as the mean ± SEM of three experiments. The data were analyzed using one-way analysis of variance to compare more than two groups, with p-values <0.05 considered to be significant.
Acknowledgments
We thank Dr. David Belnap for his help with cryogenic electron microscopy. We are grateful to Dr. Huaizhong Pan for his help advising the preliminary research. The research was supported in part by NIH grant RO1 GM69847 (to J.K.).
Glossary
Abbreviations
- ACN
acetonitrile
- AUA
11-aminoundecanoic acid
- CCK-8
2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt
- d-Asp8
d-aspartic acid octapeptide
- DIPEA
diisopropyl ethylamine
- DMEM/F12
Dulbecco’s Modified Eagle Medium F-12
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- DOX
Doxorubicin
- FBS
fetal bovine serum
- Fmoc-Cl
1-(9-fluorenyl)methylchloroformate
- Fmoc-d-Asp-OH
N-9-fuorenylmethoxycarbonyl-d-aspartic acid
- Fmoc-Lys(Fmoc)-OH
N-2-N-6-bis(9-fluorenylmethyloxycarbonyl)-l-lysine
- HAp
hydroxyapatite
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- MeOH
methanol
- miniPEG
8-amino-3,6-dioxaoctanoic acid
- Saos-2
human osteosarcoma
- SEM
standard error of the mean
- TFA
trifluoroacetic acid
- TIS
triisoproyl silane
Supporting Information Available
Physicochemical characterization of compounds, unimers, and micelles (HPLC, MALDI-ToF MS, NMR). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Kufe D. W., Pollock R. E., Weichselbaum R. R., Bast R. C. Jr., Gansler T. S., Holland J. F., and Frei E. III (2003) Holland-Frei Cancer Medicine, 6th ed., BC Decker. [Google Scholar]
- PosthumaDeBoer J.; van Royen B.; Helder M. (2013) Mechanisms of therapy resistance in osteosarcoma: a review. Oncology Discovery 1, 8–15. [Google Scholar]
- Luetke A.; Meyers P. A.; Lewis I.; Juergens H. (2014) Osteosarcoma treatment - where do we stand? A state of the art review. Cancer Treat. Rev. 40, 523–532. [DOI] [PubMed] [Google Scholar]
- Horie T.; Ono K.; Nishi H.; Nagao K.; Kinoshita M.; Watanabe S.; Kuwabara Y.; Nakashima Y.; Takanabe-Mori R.; Nishi E. (2010) Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovasc. Res. 87, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susa M.; Iyer A. K.; Ryu K.; Hornicek F. J.; Mankin H.; Amiji M. M.; Duan Z. (2009) Doxorubicin loaded polymeric nanoparticulate delivery system to overcome drug resistance in osteosarcoma. BMC Cancer 9, 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low S. A.; Kopeček J. (2012) Targeting polymer therapeutics to bone. Adv. Drug Delivery Rev. 64, 1189–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salerno M.; Cenni E.; Fotia C.; Avnet S.; Granchi D.; Castelli F.; Micieli D.; Pignatello R.; Capulli M.; Rucci N.; Angelucci A.; Del Fattore A.; Teti A.; Zini N.; Giunti A.; Baldini N. (2010) Bone-targeted doxorubicin-loaded nanoparticles as a tool for the treatment of skeletal metastases. Curr. Cancer Drug Targets 10, 649–659. [DOI] [PubMed] [Google Scholar]
- Hrubý M.; Etrych T.; Kučka J.; Forsterová M.; Ulbrich K. (2006) Hydroxybisphosphonate-containing polymeric drug-delivery systems designed for targeting into bone tissue. J. Appl. Polym. Sci. 101, 3192–3201. [Google Scholar]
- Prabaharan M.; Grailer J. J.; Pilla S.; Steeber D. A.; Gong S. (2009) Amphiphilic multi-arm-block copolymer conjugated with doxorubicin via pH-sensitive hydrazone bond for tumor-targeted drug delivery. Biomaterials 30, 5757–5766. [DOI] [PubMed] [Google Scholar]
- Hrubý M.; Koňák Č.; Ulbrich K. (2005) Polymeric micellar pH-sensitive drug delivery system for doxorubicin. J. Controlled Release 103, 137–148. [DOI] [PubMed] [Google Scholar]
- Eksborg S. (1978) Extraction of daunorubicin and doxorubicin and their hydroxyl metabolites: Self-association in aqueous solution. J. Pharm. Sci. 67, 782–785. [DOI] [PubMed] [Google Scholar]
- Sekido T.; Sakura N.; Higashi Y.; Miya K.; Nitta Y.; Nomura M.; Sawanishi H.; Morito K.; Masamune Y.; Kasugai S.; Yokogawa K.; Miyamoto K. (2001) Novel drug delivery system to bone using acidic oligopeptide: pharmacokinetic characteristics and pharmacological potential. J. Drug Targeting 9, 111–121. [DOI] [PubMed] [Google Scholar]
- Zhou N.; Luo Z.; Luo J.; Fan X.; Cayabyab M.; Hiraoka M.; Liu D.; Han X.; Pesavento J.; Dong C.-Z.; Wang Y.; An J.; Kaji H.; Sodroski J. G.; Huang Z. (2002) Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with d-peptides derived from chemokines. J. Biol. Chem. 277, 17476–17485. [DOI] [PubMed] [Google Scholar]
- Verdoliva A.; Pannone F.; Rossi M.; Catello S.; Manfredi V. (2002) Affinity purification of polyclonal antibodies using a new all-D synthetic peptide ligand: comparison with protein A and protein G. J. Immunol. Methods. 271, 77–88. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Bromberg L.; Lin S. N.; Alan Hatton T.; Tam K. C. (2007) Complexation and release of doxorubicin from its complexes with Pluronic P85-b-poly(acrylic acid) block copolymers. J. Controlled Release 121, 137–145. [DOI] [PubMed] [Google Scholar]
- Kataoka K.; Matsumoto T.; Yokoyama M.; Okano T.; Sakurai Y.; Fukushima S.; Okamoto K.; Kwon G. S. (2000) Doxorubicin-loaded poly(ethylene glycol)–poly(β-benzyl-l-aspartate) copolymer micelles: their pharmaceutical characteristics and biological significance. J. Controlled Release 64, 143–153. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Hernández K.; Chécot F.; Gnanou Y.; Lecommandoux S. (2005) Toward ‘smart’ nano-objects by self-assembly of block copolymers in solution. Prog. Polym. Sci. 30, 691–724. [Google Scholar]
- Israelachvili J. N. (1991) Intermolecular and surface forces, Academic Press, London. [Google Scholar]
- Riches A. C.; Sharp J. G.; Thomas D. B.; Smith S. V. (1973) Blood volume determination in the mouse. J. Physiol. 228, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M. L.; Friedhuber A. M.; Dunstan D. E.; Choong P. F. M.; Dass C. R. (2010) The performance of doxorubicin encapsulated in chitosan–dextran sulphate microparticles in an osteosarcoma model. Biomaterials 31, 541–551. [DOI] [PubMed] [Google Scholar]
- Dass C. R.; Ek E. T.; Contreras K. G.; Choong P. F. (2006) A novel orthotopic murine model provides insights into cellular and molecular characteristics contributing to human osteosarcoma. Clin. Exp. Metastasis 23, 367–380. [DOI] [PubMed] [Google Scholar]
- Moghimi S. M.; Hunter A. C.; Murray J. C. (2001) Long-Circulating and target-specific nanoparticles: Theory to practice. Pharmacol. Rev. 53, 283–318. [PubMed] [Google Scholar]
- Lukyanov A. N.; Torchilin V. P. (2004) Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv. Drug Delivery Rev. 56, 1273–1289. [DOI] [PubMed] [Google Scholar]
- Miller S.; Pan H.; Wang D.; Bowman B.; Kopečková P.; Kopeček J. (2008) Feasibility of using a bone-targeted, macromolecular delivery system coupled with Prostaglandin E1 to promote bone formation in aged, estrogen-deficient rats. Pharm. Res. 25, 2889–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Miller S. C.; Shlyakhtenko L. S.; Portillo A. M.; Liu X.-M.; Papangkorn K.; Kopečková P.; Lyubchenko Y.; Higuchi W. I.; Kopeček J. (2007) Osteotropic peptide that differentiates functional domains of the skeleton. Bioconjugate Chem. 18, 1375–1378. [DOI] [PubMed] [Google Scholar]
- Lamoureux F.; Richard P.; Wittrant Y.; Battaglia S.; Pilet P.; Trichet V.; Blanchard F.; Gouin F.; Pitard B.; Heymann D. (2007) Therapeutic relevance of osteoprotegerin gene therapy in osteosarcoma: blockade of the vicious cycle between tumor cell proliferation and bone resorption. Cancer Res. 67, 7308–7318. [DOI] [PubMed] [Google Scholar]
- Rodan S. B.; Rodan G. A.; Simmons H. A.; Walenga R. W.; Feinstein M. B.; Raisz L. G. (1981) Bone resorptive factor produced by osteosarcoma cells with osteoblastic features is PGE2. Biochem. Biophys. Res. Commun. 102, 1358–1365. [DOI] [PubMed] [Google Scholar]
- Wang D.; Miller S.; Sima M.; Kopečková P.; Kopeček J. (2003) Synthesis and evaluation of water-soluble polymeric bone-targeted drug delivery systems. Bioconjugate Chem. 14, 853–859. [DOI] [PubMed] [Google Scholar]
- Ouyang L.; Huang W.; He G.; Guo L. (2009) Bone targeting prodrugs based on peptide dendrimers, synthesis and hydroxyapatite binding in vitro. Lett. Org. Chem. 6, 272–277. [Google Scholar]
- Aronov D.; Rosen R.; Ron E. Z.; Rosenman G. (2006) Tunable hydroxyapatite wettability: effect on adhesion of biological molecules. Process Biochem. 41, 2367–2372. [Google Scholar]
- Teitelbaum S. L. (2000) Bone resorption by osteoclasts. Science 289, 1504–1508. [DOI] [PubMed] [Google Scholar]
- Roodman G. D. (2004) Mechanisms of bone metastasis. N. Engl. J. Med. 350, 1655–1664. [DOI] [PubMed] [Google Scholar]
- Georges S.; Ruiz Velasco C.; Trichet V.; Fortun Y.; Heymann D.; Padrines M. (2009) Proteases and bone remodelling. Cytokine Growth Factor Rev. 20, 29–41. [DOI] [PubMed] [Google Scholar]
- Seksek O.; Biwersi J.; Verkman A. S. (1996) Evidence against defective trans-Golgi acidification in cystic fibrosis. J. Biol. Chem. 271, 15542–15548. [DOI] [PubMed] [Google Scholar]
- MacKay J. A.; Chen M.; McDaniel J. R.; Liu W.; Simnick A. J.; Chilkoti A. (2009) Self-assembling chimeric polypeptide–doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat. Mater. 8, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo H. S.; Lee E. A.; Park T. G. (2002) Doxorubicin-conjugated biodegradable polymeric micelles having acid-cleavable linkages. J. Controlled Release 82, 17–27. [DOI] [PubMed] [Google Scholar]
- Carpino L. A.; Han G. Y. (1972) 9-Fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 37, 3404–3409. [Google Scholar]
- Galoppini C.; Meini S.; Tancredi M.; Di Fenza A.; Triolo A.; Quartara L.; Maggi C. A.; Formaggio F.; Toniolo C.; Mazzucco S.; Papini A.; Rovero P. (1999) A New class of pseudopeptide antagonists of the Kinin B1 receptor containing alkyl spacers. J. Med. Chem. 42, 409–414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.