

Abstract
Objective:
To report neurologic phenotypes and their etiologies determined among 68 patients with either (1) celiac disease (CD) or (2) no CD, but gliadin antibody positivity (2002–2012).
Methods:
Neurologic patients included both those with the CD-prerequisite major histocompatibility complex class II human leukocyte antigen (HLA)-DQ2/DQ8 haplotype, and those without. The 3 groups were as follows: group 1 (n = 44), CD or transglutaminase (Tg)-2/deamidated gliadin immunoglobulin (Ig)A/IgG detected; group 2 (n = 15), HLA-DQ2/DQ8 noncarriers, and gliadin IgA/IgG detected; and group 3 (n = 9), HLA-DQ2/DQ8 carriers, and gliadin IgA/IgG detected. Neurologic patients and 21 nonneurologic CD patients were evaluated for neural and Tg6 antibodies.
Results:
In group 1, 42 of 44 patients had CD. Neurologic phenotypes (cerebellar ataxia, 13; neuropathy, 11; dementia, 8; myeloneuropathy, 5; other, 7) and causes (autoimmune, 9; deficiencies of vitamin E, folate, or copper, 6; genetic, 6; toxic or metabolic, 4; unknown, 19) were diverse. In groups 2 and 3, 21 of 24 patients had cerebellar ataxia; none had CD. Causes of neurologic disorders in groups 2 and 3 were diverse (autoimmune, 4; degenerative, 4; toxic, 3; nutritional deficiency, 1; other, 2; unknown, 10). One or more neural-reactive autoantibodies were detected in 10 of 68 patients, all with autoimmune neurologic diagnoses (glutamic acid decarboxylase 65 IgG, 4; voltage-gated potassium channel complex IgG, 3; others, 5). Tg6-IgA/IgG was detected in 7 of 68 patients (cerebellar ataxia, 3; myelopathy, 2; ataxia and parkinsonism, 1; neuropathy, 1); the 2 patients with myelopathy had neurologic disorders explained by malabsorption of copper, vitamin E, and folate rather than by neurologic autoimmunity.
Conclusions:
Our data support causes alternative to gluten exposure for neurologic dysfunction among most gliadin antibody–positive patients without CD. Nutritional deficiency and coexisting autoimmunity may cause neurologic dysfunction in CD.
Celiac disease (CD) is a chronic immune-mediated enteropathy precipitated by exposure to dietary gluten within wheat, rye, and barley.1 CD has one of the strongest human leukocyte antigen (HLA) associations. Family members of patients with CD who do not have HLA-DQ2 or -DQ8 have low risk of developing CD.2,3 Coexisting autoimmune diseases are common in CD, and include diabetes mellitus and thyroid disease.4 Early neurologic reports included sensory ataxia (due to myeloneuropathy) usually without cerebellar ataxia.5 That phenotype is usually attributable to enteropathy-induced malabsorption of copper or vitamin E.6–8 Rare autopsy cases of inflammatory neurologic disorders arising in patients with CD have also been reported.5,9 A causal link between gluten exposure and nervous system inflammation has remained controversial.10
After their introduction in the 1970s, gliadin antibodies served as the serologic test for CD.11 Low specificity led to their abandonment for the diagnosis of CD.12 International consensus concluded that immunoglobulin (Ig)A antibodies with endomysial, transglutaminase-2 (Tg2), and deamidated gliadin specificities have superior sensitivity and specificity.1 In the mid-1990s, first-generation gliadin antibodies were reported to be more common in patients with idiopathic neurologic disorders than in patients with neurologic disorders of known cause.13 This spawned reports of neurologic disorders triggered by gluten, unified by gliadin antibody positivity.14–18 Those patients may have both CD and the HLA-DQ2/DQ8 haplotype, one of those, or neither.18 Furthermore, Tg6 was reported as a pertinent nervous system–specific antigen.19–21
Herein, we evaluate the significance of positive CD serologies in neurologic patients evaluated at the Mayo Clinic, Rochester, MN (2002–2013).
METHODS
Standard protocol approvals, registrations, and patient consents.
This study was approved by the Mayo Clinic institutional review board (06-09331). The medical record index system (1997–2012) was interrogated for patients who had received ICD-9 billing codes for both CD and a neurologic diagnosis (not necessarily simultaneously). Patient medical records (1,007 total) were reviewed for patients in whom a diagnosis of gluten sensitivity or CD-related neurologic disorder was being considered. Of 111 patients identified, 68 with duodenal biopsy results documented and serum available for additional testing were included.
We reviewed medical records of the 68 patients. We sought to establish the causes of neurologic dysfunction in patients, among both those with CD and those without CD. To accomplish this, we divided patients into 3 groups according to CD-prerequisite HLA haplotype and CD serologic findings. Group 1 patients had the HLA-DQ2 or -DQ8 haplotype and had second-generation CD serologic testing positivity (Tg2-IgA or -IgG, or deamidated gliadin IgA or IgG) during neurologic evaluation, or had a duodenal biopsy-proven diagnosis of CD before that evaluation. Group 2 patients did not have HLA-DQ2 or -DQ8 haplotype, but nonetheless had first-generation CD serologic testing positivity (gliadin IgA or IgG). Group 3 patients had the HLA-DQ2 or -DQ8 haplotype, and had gliadin IgA or IgG testing positivity.
Patients with CD without neurologic disorders.
Twenty-one patients known to be Tg2-IgA seropositive and had CD, but had no neurologic symptoms known, were also tested for Tg6-IgA and -IgG by ELISA.
Serum and CSF testing.
Tissue immunofluorescence, immunoprecipitation, and cell-binding assays.
Patient and control serums were assayed with indirect immunofluorescence for IgG and IgA antibodies with neural antigen specificity using the following 2 cryosectioned tissue composites: (1) mouse brain (hippocampus, cerebral cortex, cerebellum, basal ganglia, and thalamus), kidney, and stomach; and (2) monkey brain (cerebellum and cerebrum) and mouse stomach (Inova Diagnostics, San Diego, CA), as previously described.22 Patient serums were assayed for endomysial-IgA using monkey esophagus (EUROIMMUN, Lübeck, Germany). CSF specimens were available in 14 patients, and were also tested by indirect immunofluorescence for neural-reactive autoantibodies.
Radioimmunoprecipitation assays were used to detect serum antibodies with the following specificities: neuronal calcium channels (P/Q-type and N-type), voltage-gated potassium channel (VGKC) complexes, muscle (α1) and neuronal ganglionic (α3) acetylcholine receptors (AChRs), and glutamic acid decarboxylase 65 isoform (GAD65), as previously described.23 ELISA was used to detect striational antibody.23
All serums yielding positive VGKC-complex-IgG results were analyzed by an in-house–validated HEK-293 cell–based immunofluorescence kit assay for IgG reactive with leucine-rich, glioma inactivated 1 or contactin-associated protein 2 (EUROIMMUN). A cell-based assay was also used to detect aquaporin-4 (AQP4)-IgG (EUROIMMUN).
ELISA: Detection of autoantibodies against human recombinant Tg6.
Tg6-IgA and -IgG ELISA testing was performed using an immobilized, purified preparation of human recombinant Tg6 (ZediXplore Tg6-ELISA IgA [catalog no. E003] and Tg6-ELISA IgG [catalog no. E004]; Zedira, Darmstadt, Germany). The absorbance was read at 450 nm (GENios Pro plate reader; Tecan, Research Park Triangle, NC). Specific optical density (OD) for each serum was calculated by averaging the OD of the duplicate Tg6-coated wells and subtracting the average of the duplicate control wells. Quantitative evaluation was performed using a standard curve fitting the kit calibrators provided. The units of Tg6 antibodies (IgA or IgG) per mL of serum were calculated from the standard curve using the specific OD. Specimens with units of Tg6 antibodies per mL of serum 1.5 times greater than the average of normal donor controls (same 4 were included in each assay) were positive (>5.5 U/mL).
Statistics.
Serologic results for neurologic and nonneurologic patients were compared using Fisher exact test.
RESULTS
Summary of findings.
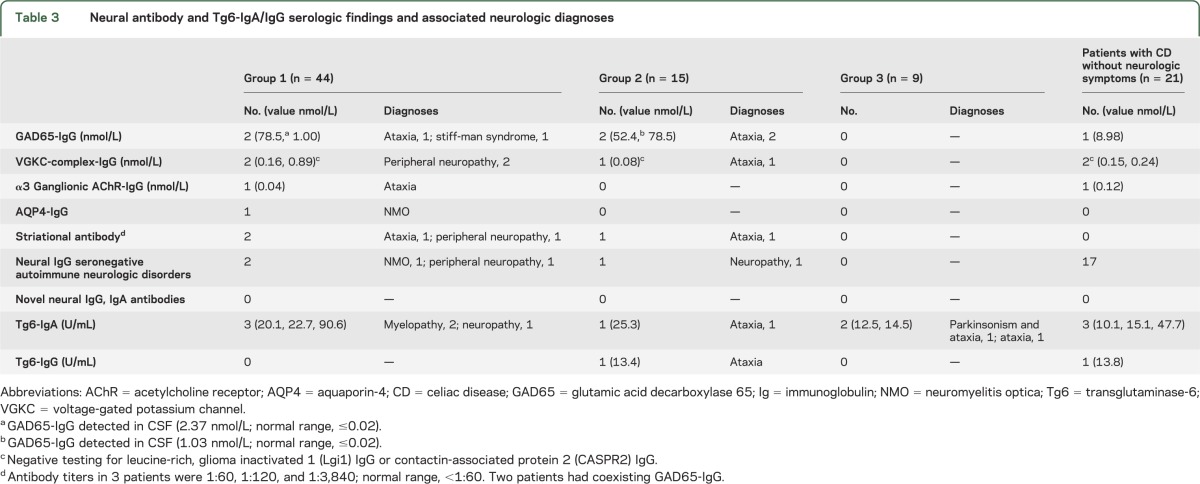
CD-related clinical and serologic data are recorded in table 1. Neurologic diagnoses are recorded in table 2. Neural autoantibody data and Tg6-IgA/IgG data are recorded in table 3.
Table 1.
CD-related clinical and serologic characteristics
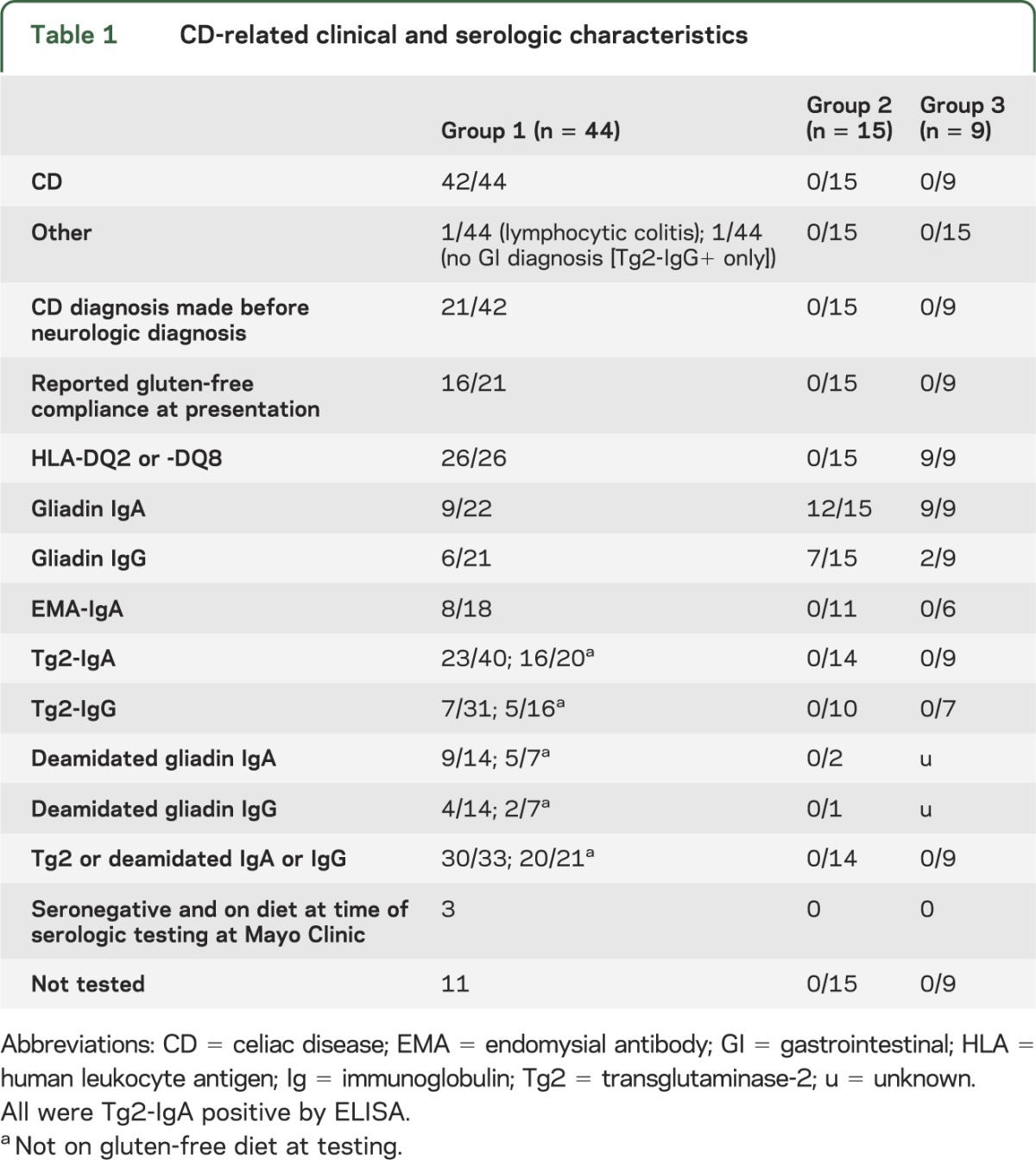
Table 2.
Neurologic disorders and their causes
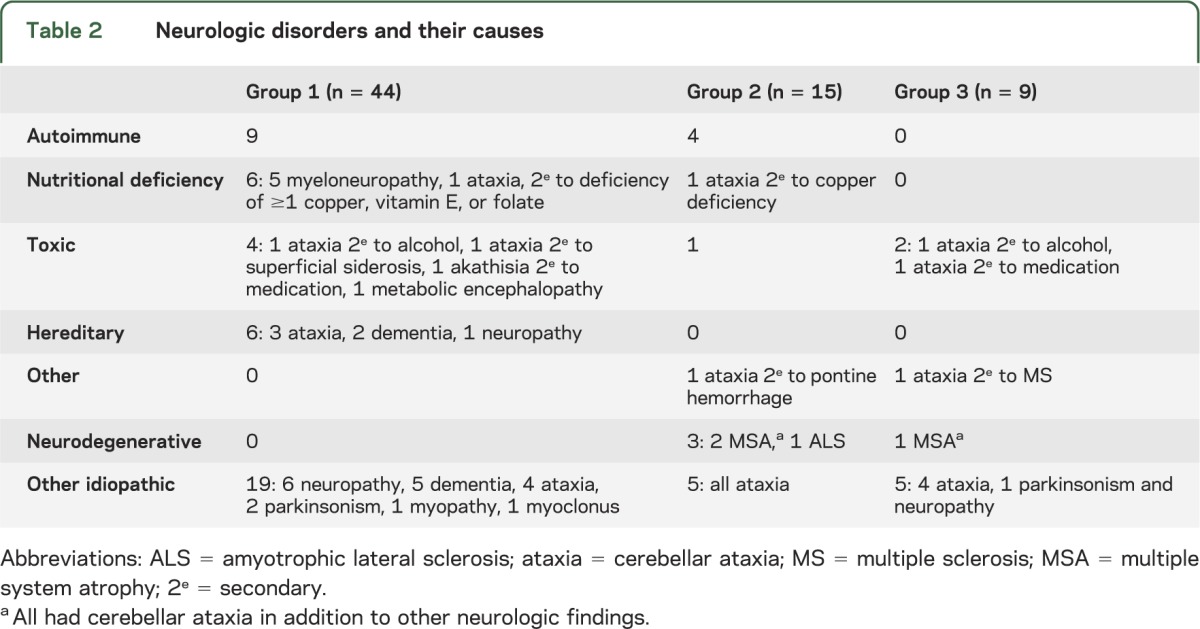
Table 3.
Neural antibody and Tg6-IgA/IgG serologic findings and associated neurologic diagnoses
For the 68 patients, median neurologic symptom-onset age was 53 years (range, 11–82); 40 patients were women. Median follow-up duration after neurologic diagnosis was 12 months (range, 1–156).
A cause for neurologic dysfunction was discernible in 39 patients (57%), with coexisting neurologic autoimmunity being the most common (13 patients [19%]). One or more neural-reactive IgG autoantibodies were detected in 10 of those 13 patients (GAD65-IgG, 4; VGKC-complex-IgG, 3; striational antibody, 3; ganglionic AChR-IgG, 1; AQP4-IgG, 1) and 4 of 21 patients with CD without neurologic symptoms (p < 0.001). Two of 4 patients seropositive for GAD65-IgG also had this antibody detected in CSF. None had novel neural IgA or IgG antibodies detected by tissue immunofluorescence of mouse or monkey brain.
CD was thought the likely cause of neurologic dysfunction in 9 patients (13%), all of whom were from group 1, either because of a gastrointestinal malabsorption syndrome or because of improvement that occurred with a gluten-free diet. A malabsorption-induced deficiency of neurologically critical vitamin E, folate, or copper was documented in 6 of those 9 patients. The remaining 3 patients had physician-reported neurologic improvements with gluten-free diet despite no cause identified on testing.
Tg6-IgA or -IgG was detected by ELISA in 7 of 68 neurologic patients and in 4 of 21 patients with CD without neurologic symptoms (p = 0.28). Among the 7 seropositive patients, neurologic diagnoses previously recorded included the following: idiopathic cerebellar ataxia, 3; myelopathy secondary to malabsorption of copper and vitamin E or folate, 2; autoimmune neuropathy, 1; and idiopathic cerebellar ataxia and parkinsonism, 1. Tg6-IgA median U/mL values were 21.4 for the cases (range, 12.5–92.6) and 15.1 for the patients with CD without neurologic symptoms (range, 10.1–47.7). Tg6-IgG was detected in only one neurologic patient (13.4 U/mL) and one control (13.8 U/mL). Three Tg6-IgA–seropositive patients and one Tg6-IgG–positive patient received a gluten-free diet. These comprised 1 myeloneuropathy and 1 neuropathy in group 1; 1 cerebellar ataxia in group 2; and 1 cerebellar ataxia in group 3. The Tg6-IgA–seropositive patient from group 1 with CD and myeloneuropathy secondary to malabsorption of copper had neurologic improvements.
Group 1: 44 patients seropositive for Tg2 or deamidated gliadin antibodies, or with CD diagnosis established.
CD was diagnosed in 42 of 44 patients. CD had been confirmed histologically before neurologic evaluation in 21 patients; median diagnosis duration was 5 years (range, 0.5–37). Of these 21 patients, 16 reported compliance with gluten-free diet at the time of neurologic symptom onset and presentation to Mayo Clinic (9 were Tg2 or deamidated gliadin antibody negative at neurologic presentation; 5 remained antibody positive, and 2 were not retested) and 5 reported poor dietary compliance (all were Tg2 or deamidated gliadin antibody positive at neurologic presentation).
The CD diagnosis was established histologically during neurologic evaluation at Mayo Clinic in 21 patients, 7 of whom had no gastrointestinal symptoms at that time. All but one was seropositive for Tg2 or deamidated gliadin antibody, and the diagnosis of CD was established by histology alone. The 2 remaining patients did not have CD; one had lymphocytic colitis that improved with gluten-free diet and one had no intestinal disease detected (but was Tg2-IgG positive only). Of 4 patients who were Tg2-IgA or deamidated gliadin IgA negative at presentation, none had IgA deficiency.
The timing of gastrointestinal symptom onset in relation to neurologic symptom onset was available in 28 patients. Gastrointestinal symptoms preceded neurologic symptom onset in 17 patients (median duration, 13 years; range, 1–40), had simultaneous or near simultaneous onset in 6, and started after neurologic symptom onset in 5 (median duration, 7 years; range, 1–50). Gastrointestinal symptoms reported were as follows: diarrhea, 20; weight loss, 9; bloating and/or flatulence, 5; constipation, 3; abdominal pain, 3; dysphagia, 1; and nausea, 1. Other presenting disorders that precipitated investigation for CD were dermatitis herpetiformis and iron deficiency anemia.
Neurologic phenotypes included the following: cerebellar ataxia, 13; peripheral neuropathy, 11; cognitive disorders, 8; myeloneuropathy, 5; parkinsonism, 2; neuromyelitis optica, 2; stiff-man syndrome, 1; dyskinesias, 1; and myoclonus, 1.
Autoimmune neurologic disorders, diagnosed in 9 patients, included (tables 2 and 3, figure): peripheral neuropathy, 4 (2 were VGKC-IgG positive, 1 was striational antibody seropositive and had 9 CSF-exclusive oligoclonal bands, and 1 was Sjögren syndrome serology [SS]-A and -B positive); cerebellar ataxia, 2 (1 was α3 ganglionic AChR antibody positive, and the other was GAD65-IgG and striational antibody positive); neuromyelitis optica, 2 (1 was AQP4-IgG positive); and stiff-man syndrome, 1 (GAD65-IgG positive).
Figure. EMA-IgA, GAD65-IgG, and VGKC-complex-IgG detected by indirect immunofluorescence.

EMA-IgA, which has Tg2 specificity, is non–organ-specific, staining interstitial connective tissue in endomysium (A, monkey esophagus) and blood vessels (B, monkey cerebellum). GAD65-IgG (C) and VGKC-complex-IgG (D) stain neural tissue (mouse cerebellum) but not kidney (arrows) or gut (not shown). EMA = endomysial antibody; GAD65 = glutamic acid decarboxylase 65; Ig = immunoglobulin; Tg2 = transglutaminase 2; VGKC = voltage-gated potassium channel.
Neurologic disorders were attributable to malabsorption-induced nutritional deficiency in 6 patients. These disorders included myeloneuropathies (with sensory ataxia) secondary to deficiency of ≥1 of copper, vitamin E, and folate, 5; and myelopathy secondary to copper deficiency, 1 (the patient also had cerebellar ataxia). Other etiologies included genetic disorders, 6; chronic alcoholism-induced cerebellar ataxia, 1; metabolic encephalopathy, 1; tardive akathisia, 1; and ataxia caused by superficial CNS siderosis, 1. The remaining 19 patients had neurologic disorders of unknown cause (neuropathy, 6; dementia, 5; cerebellar ataxia, 4; parkinsonism, 2; multifocal myoclonus, 1; and myopathy, 1). Zinc deficiency, of unclear neurologic significance, was detected in 6 of those patients (neuropathy, 3; myelopathy, 2; and cognitive impairment, 1).
Group 2: 15 patients seropositive for gliadin IgA or IgG only, without HLA-DQ2 or -DQ8 haplotype.
All 15 patients had gliadin IgA/IgG detected in the course of evaluation for cerebellar ataxia (13) and peripheral nervous system disease (2). Six patients had gastrointestinal symptoms (diarrhea, 4; appetite loss, 1; constipation, 1). None had CD. Final neurologic diagnoses for the 13 patients with cerebellar ataxia were as follows: cause unknown, 5; autoimmune, 3 (2 had GAD65-IgG, 1 had VGKC-complex-IgG, and 1 had striational antibody); multiple system atrophy, 2; copper deficiency, 1; chronic alcohol abuse, 1; and pontine hemorrhage, 1. Diagnoses in the 2 remaining patients were autoimmune neuropathy and amyotrophic lateral sclerosis. The patient with autoimmune neuropathy had SS-A and SS-B antibodies and rheumatoid factor detected in serum, and an elevated protein and IgG synthesis rate detected in CSF.
Group 3: 9 patients seropositive for gliadin IgA or IgG, with HLA-DQ2 or -DQ8 haplotype.
All 9 patients had gliadin IgG or IgA detected in the course of evaluation for ataxia (8) and parkinsonism (1). Three patients had gastrointestinal symptoms, one each of diarrhea, constipation, and heartburn. None had CD. Final neurologic diagnoses for the 8 patients with ataxia were as follows: cause unknown, 4; chronic alcohol abuse, 1; medication-induced (reversible), 1; multiple sclerosis, 1; and multiple system atrophy, 1. The remaining patient had idiopathic parkinsonism and neuropathy.
Treatment.
Gluten-free diet.
Group 1.
Improvements reported in 6 of 33 patients were major in 4 and minor in 2. Three had a diagnosis of CD made during evaluation for neurologic symptoms and 3 had preexisting CD for 2 years or less. All 6 had gastrointestinal symptoms before neurologic symptoms.
Three patients had myeloneuropathy and malabsorption-related deficiencies of copper or vitamin E, which improved with treatment. The mechanism of neurologic improvement in the 3 other patients was unknown. A 37-year-old woman had fatigue and impaired frontal-subcortical function. CD was diagnosed but she did not have an apparent vitamin or trace element deficiency. She initiated and maintained a strict gluten-free diet. Neuropsychometric reassessment after 3 years revealed normal cognition, and she resumed work as a languages teacher. Improvements in sensory symptoms were reported in 2 patients with peripheral neuropathy after initiating a gluten-free diet. Neurologic progression was reported in 16 patients and no change in 11 patients.
Of 18 patients who presented with neurologic symptoms in the context of an established CD diagnosis, 8 had persisting positive Tg2 or deamidated gliadin serologies, indicative of ongoing contamination of diet with gluten. Of those, 3 patients had improvements in neurologic as well as gastrointestinal symptoms with improved dietary compliance.
Groups 2 and 3.
Of 4 patients treated with gluten-free diet, all continued to deteriorate neurologically at last follow-up.
Other treatments and outcomes.
Other treatments were undertaken in 10 patients. In group 1, autoimmune neurologic disorders stabilized in 4 other patients with immunotherapy (neuropathy, 2; neuromyelitis optica [NMO], 1; ataxia, 1), and tardive akathisia stabilized in one patient after withdrawal of neuroleptic medication. Hepatic encephalopathy improved in one patient with portosystemic shunt placement. In group 2, GAD65-IgG–associated ataxia stabilized in 2 patients after treatment with immunotherapy. In group 3, ataxia resolved in one patient after discontinuing rabeprazole.
DISCUSSION
Among our patients seropositive for first-generation gliadin antibodies, but not Tg2 or deamidated gliadin antibodies (groups 2 and 3), and without histologic evidence of CD, causes of neurologic dysfunction alternative to a gluten-triggered etiology were demonstrated in the majority. For group 2, two-thirds had an alternative cause, and for group 3, 4 of 9 had an alternative cause. Furthermore, the limited numbers of patients treated with gluten-free diet from those 2 groups continued to deteriorate neurologically. Cerebellar dysfunction was the presenting disorder in almost all patients in groups 2 and 3. The predominance of patients with cerebellar ataxia (as opposed to sensory ataxia) in those groups is likely consistent with test-ordering bias in light of the reported gliadin antibody–cerebellar ataxia association.14
Consistent with previous reports, in patients without CD, first-generation gliadin antibodies lacked specificity for neurologic disorders purported to be related to gluten exposure.10,24,25 Using tissue immunofluorescence, we did not detect any novel IgA or IgG neural antibodies that might serve as biomarkers of gluten-triggered neurologic disorders.20,26 For specificity, our contemporary tissue immunofluorescence protocol includes gastric and renal tissue controls (side by side with brain tissue) and preabsorption of serums to remove non–organ-specific antibodies. Tissue fixation is an evolving field, and future studies could reevaluate those patients in whom an autoimmune neurologic cause was suspected but who were seronegative for currently characterized antibodies.
In contrast to groups 2 and 3, the neurologic phenotypes encountered in group 1 patients were diverse, and in one-quarter of instances were attributable to neurologic autoimmunity coexisting with CD. All but 2 in this group also had CD, consistent with the high specificity of Tg2 and deamidated gliadin IgAs for that disease.12 Most patients with autoimmune neurologic diagnoses had well-characterized neural-reactive IgG (but not IgA) biomarkers detected in serum.23,27–29 These data are consistent with previous reports of coexisting neurologic autoimmunity in CD, and the reported high frequency of nonneurologic organ-specific autoimmune disorders occurring in CD, particularly type 1 diabetes and thyroid disease.30–32
The next most common diagnosis in group 1 was vitamin or trace element deficiency secondary to CD-induced malabsorption from small bowel. Consistent with previous reports, the most common malabsorption-related neurologic phenotype was myeloneuropathy in the setting of copper, vitamin E, or folic acid deficiency, and improvements in symptoms could be attributed to dietary supplementation with these nutrients and gluten-free diet.5–8 For a minority of patients with CD, the mechanism of neurologic improvement with gluten-free diet was unknown. The authors speculate that gluten-free diet could reverse malabsorption of a trace element not usually recognized as a cause of neurologic dysfunction. Zinc deficiency was detected in 6 patients, including 3 with peripheral neuropathy, but was of uncertain neurologic significance. Zinc deficiency, known primarily as a cause of cutaneous disorders, is detected in 65% of patients newly diagnosed with CD.33 However, peripheral neuropathy has been described in animal models of zinc deficiency.34,35 Among patients with CD in whom we did not find a cause for neurologic dysfunction, a gluten-triggered inflammatory etiology was possible,36,37 but there is an absence of data to confirm the existence of this, and specific biomarkers for this are lacking.
The clinical utility of Tg6-IgA and -IgG as markers of gluten-triggered neurologic disorders is controversial.21,25 We detected these antibodies among patients with CD (with or without neurologic disorders) and patients without CD, including those without the CD-requisite haplotype. Others have also reported Tg6-IgA and -IgG among 5% to 15% of non-CD controls.21,25 Because there is a high level of conservation between Tg isoforms, crossreactivity of Tg2 antibody with Tg6 could occur.20 Expression of the Tg6 isoform has been reported in skin as well as the nervous system.27 In addition to nervous system tissues, GAD65-IgG is reactive with pancreatic islet cells,38 and is detected in 8% of the general population.39 Despite this, GAD65-IgG is a clinically useful neurologic disease biomarker. As demonstrated in our neurologic patients, GAD65 autoantibody values (>20 nmol/L in 3 of 4 cases) usually exceed those encountered in type 1 diabetics without neurologic symptoms (usually 0.03–2.00 nmol/L).29
For neurologic patients with positive CD serologies, CD should be confirmed histologically before attributing neurologic disorders to gluten exposure. Alternative etiologies for neurologic symptoms need to be considered, both in patients with CD and those with only positive CD serologies. For patients with confirmed CD and new-onset neurologic symptoms, coexisting IgG-related autoimmune neurologic disorders (e.g., GAD65 or VGKC-complex autoimmunity) and nutritional deficiency rank highest on the list of potentially treatable disorders. Some patients with CD may have improved neurologic symptoms attributable to gluten-free diet. Reversal of nutritional deficiencies explains neurologic improvements in some patients with CD but not all.
ACKNOWLEDGMENT
The authors thank Vickie Mewhorter, Amy Moses, and Carol Van Dyke for excellent technical support.
GLOSSARY
- AChR
acetylcholine receptor
- AQP4
aquaporin-4
- CD
celiac disease
- GAD65
glutamic acid decarboxylase 65
- HLA
human leukocyte antigen
- ICD-9
International Classification of Diseases, ninth revision
- Ig
immunoglobulin
- NMO
neuromyelitis optica
- OD
optical density
- Tg
transglutaminase
- VGKC
voltage-gated potassium channel
AUTHOR CONTRIBUTIONS
A.M.: study concept and design, literature search, data collection, data analysis, data interpretation, drafting and critical revision of manuscript, study supervision. V.A.L.: provision of laboratory testing, data collection, data interpretation, study supervision. S.J.P.: data interpretation, critical revision of manuscript. T.J.K.: data collection, data analysis. J.A.M.: study concept and design, data collection, data analysis, data interpretation, critical revision of manuscript.
STUDY FUNDING
Supported in part by grant DK 057892 from the NIH (J.A.M.).
DISCLOSURE
A. McKeon receives research support from the Guthy-Jackson Charitable Foundation and MedImmune Inc. V. Lennon receives royalties for technology relating to AQP4 antibodies for diagnosis of NMO, is a named inventor on filed patents that relate to functional AQP4/NMO-IgG assays and NMO-IgG as a cancer marker and receives research support from the NIH (NS065829). S. Pittock is a named inventor on filed patents that relate to functional AQP4/NMO-IgG assays and NMO-IgG as a cancer marker and receives research support from the NIH (NS065829), the Guthy-Jackson Charitable Foundation, and Alexion Pharmaceuticals Inc. T. Kryzer receives royalties for technology relating to AQP4 antibodies for diagnosis of NMO, and is a named inventor on filed patents that relate to functional AQP4/NMO-IgG assays. J. Murray receives grant support from Alba Therapeutics (>US$50,000); Dr. Murray has served on advisory boards for Alvine Pharmaceuticals (<US$10,000) and Nexpep (<US$10,000). Dr. Murray has served as a consultant for (none >US$10,000) Ironwood, Flamentera, Actogenix, Ferring Research Institute, Bayer Healthcare Pharmaceuticals, Vysera Biomedical, 2G Pharma, ImmunosanT, and Shire US. Dr. Murray receives research support from the NIH (DK057892). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Ludvigsson JF, Leffler DA, Bai JC, et al. The Oslo definitions for coeliac disease and related terms. Gut 2013;62:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sollid LM, Markussen G, Ek J, Gjerde H, Vartdal F, Thorsby E. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J Exp Med 1989;169:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tighe MR, Hall MA, Barbado M, Cardi E, Welsh KI, Ciclitira PJ. HLA class II alleles associated with celiac disease susceptibility in a southern European population. Tissue Antigens 1992;40:90–97. [DOI] [PubMed] [Google Scholar]
- 4.Ventura A, Magazzu G, Greco L. Duration of exposure to gluten and risk for autoimmune disorders in patients with celiac disease. SIGEP Study Group for Autoimmune Disorders in Celiac Disease. Gastroenterology 1999;117:297–303. [DOI] [PubMed] [Google Scholar]
- 5.Cooke WT, Smith WT. Neurological disorders associated with adult coeliac disease. Brain 1966;89:683–722. [DOI] [PubMed] [Google Scholar]
- 6.Halfdanarson TR, Kumar N, Hogan WJ, Murray JA. Copper deficiency in celiac disease. J Clin Gastroenterol 2009;43:162–164. [DOI] [PubMed] [Google Scholar]
- 7.Harding AE, Muller DP, Thomas PK, Willison HJ. Spinocerebellar degeneration secondary to chronic intestinal malabsorption: a vitamin E deficiency syndrome. Ann Neurol 1982;12:419–424. [DOI] [PubMed] [Google Scholar]
- 8.Mauro A, Orsi L, Mortara P, Costa P, Schiffer D. Cerebellar syndrome in adult celiac disease with vitamin E deficiency. Acta Neurol Scand 1991;84:167–170. [DOI] [PubMed] [Google Scholar]
- 9.Dimberg EL, Crowe SE, Trugman JM, et al. Fatal encephalitis in a patient with refractory celiac disease presenting with myorhythmia and carpal spasm. Mov Disord 2007;22:407–411. [DOI] [PubMed] [Google Scholar]
- 10.Lock RJ, Pengiran Tengah DS, Unsworth DJ, Ward JJ, Wills AJ. Ataxia, peripheral neuropathy, and anti-gliadin antibody: guilt by association? J Neurol Neurosurg Psychiatry 2005;76:1601–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Signer E, Burgin-Wolff A, Berger R, Birbaumer A, Just M. Antibodies to gliadin as a screening test for coeliac disease: a prospective study. Helv Paediatr Acta 1979;34:41–52. [PubMed] [Google Scholar]
- 12.Rostom A, Murray JA, Kagnoff MF. American Gastroenterological Association (AGA) Institute technical review on the diagnosis and management of celiac disease. Gastroenterology 2006;131:1981–2002. [DOI] [PubMed] [Google Scholar]
- 13.Hadjivassiliou M, Gibson A, Davies-Jones GA, Lobo AJ, Stephenson TJ, Milford-Ward A. Does cryptic gluten sensitivity play a part in neurological illness? Lancet 1996;347:369–371. [DOI] [PubMed] [Google Scholar]
- 14.Hadjivassiliou M, Grunewald RA, Chattopadhyay AK, et al. Clinical, radiological, neurophysiological, and neuropathological characteristics of gluten ataxia. Lancet 1998;352:1582–1585. [DOI] [PubMed] [Google Scholar]
- 15.Hadjivassiliou M, Grunewald RA, Lawden M, Davies-Jones GA, Powell T, Smith CM. Headache and CNS white matter abnormalities associated with gluten sensitivity. Neurology 2001;56:385–388. [DOI] [PubMed] [Google Scholar]
- 16.Hadjivassiliou M, Rao DG, Wharton SB, Sanders DS, Grunewald RA, Davies-Jones AG. Sensory ganglionopathy due to gluten sensitivity. Neurology 2010;75:1003–1008. [DOI] [PubMed] [Google Scholar]
- 17.Hadjivassiliou M, Sanders DS, Grunewald RA. Multiple sclerosis and occult gluten sensitivity. Neurology 2005;64:933–934. [DOI] [PubMed] [Google Scholar]
- 18.Hadjivassiliou M, Sanders DS, Grunewald RA, Woodroofe N, Boscolo S, Aeschlimann D. Gluten sensitivity: from gut to brain. Lancet Neurol 2010;9:318–330. [DOI] [PubMed] [Google Scholar]
- 19.Hadjivassiliou M, Aeschlimann P, Strigun A, Sanders DS, Woodroofe N, Aeschlimann D. Autoantibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann Neurol 2008;64:332–343. [DOI] [PubMed] [Google Scholar]
- 20.Thomas H, Beck K, Adamczyk M, et al. Transglutaminase 6: a protein associated with central nervous system development and motor function. Amino Acids 2013;44:161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hadjivassiliou M, Aeschlimann P, Sanders DS, et al. Transglutaminase 6 antibodies in the diagnosis of gluten ataxia. Neurology 2013;80:1740–1745. [DOI] [PubMed] [Google Scholar]
- 22.O'Toole O, Lennon VA, Ahlskog JE, et al. Autoimmune chorea in adults. Neurology 2013;80:1133–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKeon A, Lennon VA, Lachance DH, Fealey RD, Pittock SJ. Ganglionic acetylcholine receptor autoantibody: oncological, neurological, and serological accompaniments. Arch Neurol 2009;66:735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bushara KO, Goebel SU, Shill H, Goldfarb LG, Hallett M. Gluten sensitivity in sporadic and hereditary cerebellar ataxia. Ann Neurol 2001;49:540–543. [PubMed] [Google Scholar]
- 25.Lindfors K, Koskinen O, Laurila K, et al. IgA-class autoantibodies against neuronal transglutaminase, TG6 in celiac disease: no evidence for gluten dependency. Clin Chim Acta 2011;412:1187–1190. [DOI] [PubMed] [Google Scholar]
- 26.Fukui M, Kuramoto K, Yamasaki R, et al. Identification of a highly reactive substrate peptide for transglutaminase 6 and its use in detecting transglutaminase activity in the skin epidermis. FEBS J 2013;280:1420–1429. [DOI] [PubMed] [Google Scholar]
- 27.Klein C, Lennon VA, Aston PA, et al. Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping. JAMA Neurol 2013;70:229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solimena M, Folli F, Aparisi R, Pozza G, De Camilli P. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in stiff-man syndrome. N Engl J Med 1990;322:1555–1560. [DOI] [PubMed] [Google Scholar]
- 29.Pittock SJ, Yoshikawa H, Ahlskog JE, et al. Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction. Mayo Clin Proc 2006;81:1207–1214. [DOI] [PubMed] [Google Scholar]
- 30.Briani C, Doria A, Ruggero S, et al. Antibodies to muscle and ganglionic acetylcholine receptors (AchR) in celiac disease. Autoimmunity 2008;41:100–104. [DOI] [PubMed] [Google Scholar]
- 31.Hadjivassiliou M, Aeschlimann D, Grunewald RA, Sanders DS, Sharrack B, Woodroofe N. GAD antibody-associated neurological illness and its relationship to gluten sensitivity. Acta Neurol Scand 2011;123:175–180. [DOI] [PubMed] [Google Scholar]
- 32.Stordal K, Bakken IJ, Suren P, Stene LC. Epidemiology of coeliac disease and comorbidity in Norwegian children. J Pediatr Gastroenterol Nutr 2013;57:467–471. [DOI] [PubMed] [Google Scholar]
- 33.Wierdsma NJ, van Bokhorst-de van der Schueren MA, Berkenpas M, Mulder CJ, van Bodegraven AA. Vitamin and mineral deficiencies are highly prevalent in newly diagnosed celiac disease patients. Nutrients 2013;5:3975–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terril-Robb LA, Clemons DJ, Besch-Williford C, O'Brien DP, O'Dell BL. Morphophysiologic characterization of peripheral neuropathy in zinc-deficient guinea pigs. Proc Soc Exp Biol Med 1996;213:50–58. [DOI] [PubMed] [Google Scholar]
- 35.O'Dell BL, Conley-Harrison J, Browning JD, Besch-Williford C, Hempe JM, Savage JE. Zinc deficiency and peripheral neuropathy in chicks. Proc Soc Exp Biol Med 1990;194:1–4. [DOI] [PubMed] [Google Scholar]
- 36.Boscolo S, Lorenzon A, Sblattero D, et al. Anti transglutaminase antibodies cause ataxia in mice. PLoS One 2010;5:e9698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hadjivassiliou M, Maki M, Sanders DS, et al. Autoantibody targeting of brain and intestinal transglutaminase in gluten ataxia. Neurology 2006;66:373–377. [DOI] [PubMed] [Google Scholar]
- 38.Solimena M, Folli F, Denis-Donini S, et al. Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy, and type I diabetes mellitus. N Engl J Med 1988;318:1012–1020. [DOI] [PubMed] [Google Scholar]
- 39.Walikonis JE, Lennon VA. Radioimmunoassay for glutamic acid decarboxylase (GAD65) autoantibodies as a diagnostic aid for stiff-man syndrome and a correlate of susceptibility to type 1 diabetes mellitus. Mayo Clin Proc 1998;73:1161–1166. [DOI] [PubMed] [Google Scholar]