

Abstract
The Mo- and V-nitrogenases are two homologous enzymes with distinct structural and catalytic features. Previously, we demonstrated that the V-nitrogenase was nearly 700 times more active than its Mo-counterpart in reducing CO to hydrocarbons. Here, we report a similar discrepancy between the two nitrogenases in the reaction of CO2 reduction, with the V-nitrogenase capable of reducing CO2 to CO, CD4, C2D4 and C2D6, and its Mo-counterpart only capable of reducing CO2 to CO. Further, we show that V-nitrogenase may route the formation of CD4 in part via CO2-derived CO, but it does not catalyze the formation of C2D4 and C2D6 via this route. The exciting observation of C-C coupling by V-nitrogenase from CO2 adds another interesting reaction to the catalytic repertoire of this unique enzyme system; whereas the differential activities of V- and Mo-nitrogenases in CO2 reduction provide an important framework for systematic investigations of this reaction in the future.
Keywords: nitrogenase, carbon dioxide, carbon monoxide, C-C coupling, hydrocarbon
Nitrogenases are a family of complex metalloenzymes that catalyze a key step in global nitrogen cycle: the reduction of atmospheric nitrogen (N2) to a bio-accessible form, ammonia (NH3).[1–4] Apart from N2, nitrogenases are also capable of reducing alternative substrates, such as acetylene (C2H2) and carbon monoxide (CO), thereby displaying a unique versatility in processing small, carbon-containing molecules.[1,5] The molybdenum (Mo)- and vanadium (V)-nitrogenases are two homologous members of this enzyme family, sharing a good degree of homology in primary sequence and cluster composition.[5,6]. Both enzymes are homologous binary systems, which consist of (i) a reductase component (nifH- or vnfH-encoded Fe protein), which contains one subunit-bridging [Fe4S4] cluster and one ATP-binding site per subunit; and (ii) a catalytic component (nifDK-encoded MoFe or vnfDGK-encoded VFe protein), which contains a P-cluster at the α/β-subunit interface and a cofactor (FeMoco or FeVco) within each α-subunit (Fig. 1A). Moreover, both enzymes use the same mode of action during catalysis, which involves the formation of a functional complex between the two component proteins,[7,8], the ATP-dependent transfer of electrons from the [Fe4S4] cluster of the reductase component, via the P-cluster, to the cofactor of the catalytic component, and the eventual reduction of substrates at the cofactor site upon accumulation of a sufficient amount of electrons (Fig. 1A).
Figure 1.
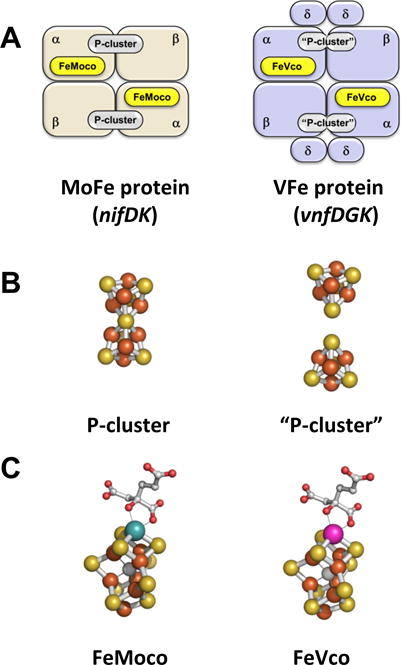
Comparison between the Mo- and V-nitrogenases. Schematic presentations of the catalytic components (A) and structural models of the P-clusters (B) and cofactors (C) in Mo- (left) and V- (right) nitrogenases. Atoms are colored as follows: Fe, orange; S, yellow; Mo, cyan; V, magenta; O, red; C, gray.
Despite their homology in structure and function, the two nitrogenases are clearly distinct from each other with regard to their associated metalloclusters. The P-cluster of the Mo-nitrogenase assumes a ‘standard’, [Fe8S7] structure; whereas the P-cluster of the V-nitrogenase consists of a pair of [Fe4S4]-like clusters (Fig. 1B).[5,7–9]. Likewise, despite a striking homology in structure, the cofactors of the Mo- and V-nitrogenases are distinguishable not only by heterometals, but also by electronic properties (Fig. 1C).[10] The differences between the metal clusters in the Mo- and V-nitrogenases underline the differences in the catalytic behavior of these homologous enzymes. It has been documented that the V-nitrogenase is less efficient than its Mo-counterpart in terms of N2 reduction; yet, this nitrogenase can reduce C2H2 to ethane (C2H6), a catalytic activity not observed in the case of Mo-nitrogenase.[6,8]. Perhaps the biggest discrepancy between the catalytic properties of the two nitrogenases is their abilities to reduce CO to hydrocarbons, with the V-nitrogenase showing an overall activity that is nearly 700 times higher than its Mo-counterpart.[11,12] This observation has prompted us to conduct a comparative study between the Mo- and V-nitrogenases to address the questions of (i) whether the two nitrogenases can also reduce CO2 to hydrocarbons and (ii) if they have the same discrepancy in their activities to generate hydrocarbons from this substrate.
Consistent with an earlier report,[13] the Mo-nitrogenase can reduce CO2 to CO (Fig. 2A, triangles). Like its Mo-counterpart, the V-nitrogenase can also catalyze the reduction of CO2 to CO (Fig. 2A, circles) in an ATP-dependent reaction (Fig. S1) that contains 20 mM dithionite at a pH of 8.5. The two nitrogenases display comparable efficiencies in H2O-based reactions, forming approximately the same amount of CO from CO2 over a time period of 180 min (Fig. 2A). Moreover, both nitrogenases exhibit roughly the same increase of activity in the formation of CO from CO2 upon substitution of D2O for H2O, reaching a maximum increase of activity at 120 min (Fig. 2A). Apart from CO, CH4—a further reduced C1 product—can be detected in reactions catalyzed by both Mo- and V-nitrogenases when CO2 is supplied as a substrate (Fig. 2B). However, when H2O is replaced by D2O, the activity of CH4 formation by V-nitrogenase increases from 0 to a maximum of 22.2 nmol/μmol protein (Fig. 2B, solid vs. open circles); whereas the activity of CH4 formation by Mo-nitrogenase decreases from a maximum of 7.3 nmol/μmol protein to 0 (Fig. 2B, solid vs. open triangles). Such a disparate D2O effect implies a difference in the routes to CH4 formation taken by the two nitrogenases.
Figure 2.
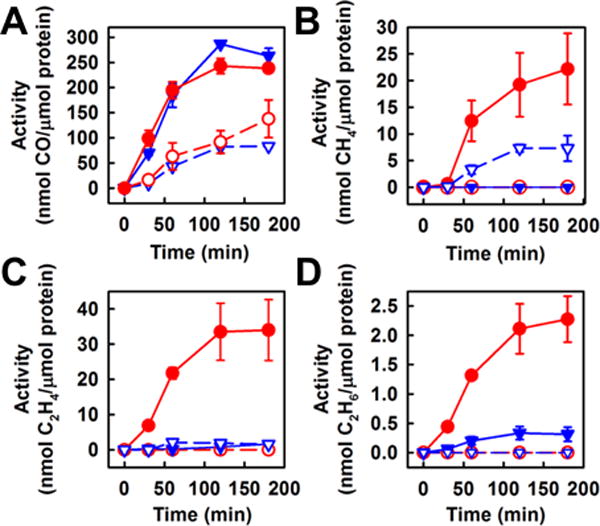
Product formation by Mo- and V-nitrogenases in the presence of CO2. Time-dependent formation of CO (A), CH4 (B), C2H4 (C) and C2H6 (D) by Mo-nitrogenase in H2O (blue open triangle, dashed line) or D2O (blue solid triangle, solid line) and by V-nitrogenase in H2O (red open circle, dashed line) or D2O (red solid circle, solid line). Data are presented as mean ± SD (N=3) after background correction.
The difference between the V- and Mo-nitrogenases in CO2 reduction is further illustrated by the difference in their abilities to form C-C bonds from CO2. In the presence of H2O, little or no C2 product can be detected in the reactions of CO2 reduction by either Mo- or V-nitrogenase (Fig. 2C and D, open triangles and open circles). In the presence of D2O, however, C2D4 (Fig. 2C, solid circles) and C2D6 (Fig. 2D, solid circles) can be detected as products of CO2 reduction by V-nitrogenase; whereas these C2 products are hardly detectable in the same reaction catalyzed by Mo-nitrogenase (Fig. 2C and D, solid triangles). Thus, as is observed in the case of CH4 formation, there is a clear increase in the activities of C2D4 and C2D6 formation by V-nitrogenase upon substitution of D2O for H2O; whereas these activities remain marginal in the reaction catalyzed by Mo-nitrogenase following such a substitution. Moreover, like the formation of CH4, the formation of C2 products by V-nitrogenase is ATP-dependent, as no C2D4 and C2D6 can be detected in the absence of ATP (Fig. S1).
GC-MS analysis supplies further evidence for the difference between Mo- and V-nitrogenases in hydrocarbon formation from CO2. When 12CO2 is replaced by 13CO2, 13CD4 can be detected in the V-nitrogenase-catalyzed reaction in D2O (Fig. 3B); yet, 13CH4 is absent from the Mo-nitrogenase-catalyzed reaction in H2O (Fig. 3A). This observation confirms CO2 as a carbon source for the CD4 generated by V-nitrogenase while suggesting a different carbon source for the same C1 product generated by Mo-nitrogenase. Aside from CD4, CO2 also gives rise to the C2 products in the reaction catalyzed by V-nitrogenase, as 13C2D4 (Fig. 3C) and 13C2D6 (Fig. 3D) can be detected in the presence of D2O upon substitution of 13CO2 for 12CO2. Together, the GC-MS and activity data highlight the difference between the reactions of CO2 reduction by V- and Mo-nitrogenases, showing the ability of V-nitrogenase to form C1 and C2 hydrocarbons along with CO and the inability of its Mo-counterpart to generate products other than CO under these experimental conditions. Given the previous observation that V-nitrogenase can reduce CO to hydrocarbons,[11,12] the co-production of CO and hydrocarbons by this enzyme as products of CO2 reduction raises a relevant question of whether it is the CO2-derived CO that gives rise to the hydrocarbon products.
Figure 3.
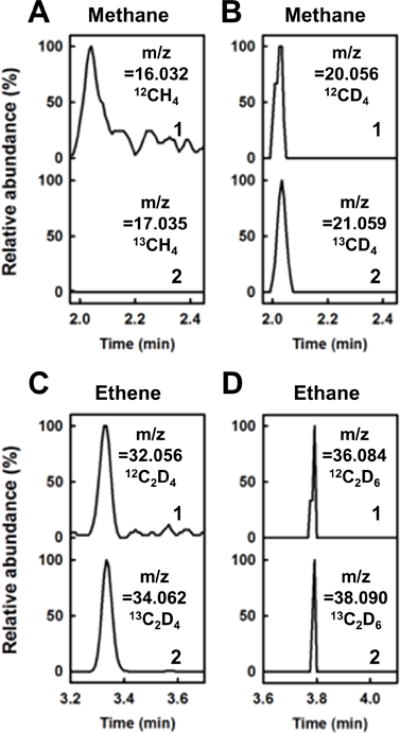
GC-MS analyses of hydrocarbon products formed by Mo- and V-nitrogenases. The products were generated by Mo-nitrogenase in H2O (A) or by V-nitrogenase in D2O (B–D) when 12CO2 (1) or 13CO2 (2) was supplied. The mass-to-charge (m/z) ratios at which the products were traced are indicated.
This question can be addressed by directly supplying CO to the V-nitrogenase in a concentration simulating the maximum concentration of CO achieved in the ‘equilibrated state’ of CO2 reduction by this enzyme (see Fig. 2A) and monitoring the formation of C1 and C2 hydrocarbons in D2O over a time period of 180 minutes. Interestingly, the CO-based formation of CD4 by V-nitrogenase (Fig. 4A, dotted circles) displays an activity increase of 12.6 nmol/μmol protein between 0 and 30 minutes; whereas the CO2-based formation of CH4 exhibits a nearly identical activity increase of 11.8 nmol/μmol protein between 30 and 60 minutes after an initial ‘lag’ phase between 0 and 30 minutes (Fig. 4A, solid circles). This observation suggests the possibility for V-nitrogenase to route the formation of C1 hydrocarbon via CO, as the 30-minute delay could be correlated with a need for the enzyme to accumulate a sufficient amount of CO2-derived CO to initiate further reduction of CO to CD4. On the other hand, the time courses of CD4 formation from CO (Fig. 4A, dotted circles) and CO2 (Fig. 4A, solid circles) diverge beyond 60 minutes, with a gradual decrease of activity in the case of the former and a gradual increase of activity in the case of the latter. The difference between the two time courses (Fig. S2A) could represent the portion of CD4 that is generated independently from CO2-derived CO. Consistent with this hypothesis, there is a notable difference between the CO- and CO2-based reactions in the percentage activity of CH4 formation in H2O relative to that in D2O (Fig. S3A), with the CO-based reaction favoring the formation of CH4 in H2O over that in D2O (42%) considerably more than the CO2-based reaction (0%). It is possible, therefore, that the V-nitrogenase generates CH4 both from CO2-derived CO and from CO2 and/or other CO2-derived intermediate(s).
Figure 4.
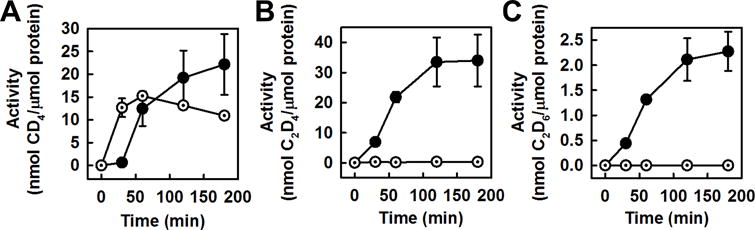
Formation of hydrocarbon products by V-nitrogenase. Time-dependent formation of CD4 (A), C2D4 (B) and C2D6 (C) from CO2 (solid circle) or CO (dotted circle) by V-nitrogenase in D2O. CO was added in a concentration of 110 pm in assays involving the direct formation of products from CO, which was equivalent to the maximum concentration of CO that could be generated from CO2 reduction by V-nitrogenase (also see Fig. 2). Data are presented as mean ± SD (N=3) after background correction.
Contrary to CD4, both C2D4 and C2D6 seem to be produced by V-nitrogenase via a CO-independent route, as no C2 products can be detected (Fig. 4B and C, dotted circles) upon direct addition of CO in the same amount produced by V-nitrogenase from CO2 reduction in the ‘equilibrated state’ (see Fig. 2A). This observation suggests that, instead of CO, CO2 and/or other CO2-derived intermediates are responsible for the formation of C2 hydrocarbon products by V-nitrogenase. Indeed, as is observed in the case of CH4 formation, there is a significant difference between the CO- and CO2-based reactions in the percentage activity of C2H4 (Fig. S3B) or C2H6 (Fig. S3C) formation in H2O relative to that in D2O, with the CO-based reaction favoring the formation of C2 products in H2O over that in D2O (C2H4, 92%; C2H6, 65%) considerably more than the CO2-based reaction (C2H4, 0.7%; C2H6, 0%). Such a disparate deuterium effect on the CO- and CO2-based reactions further implies that V-nitrogenase routes the formation of C2 hydrocarbons via CO2 or other CO2-derived intermediate(s). The lack of contribution of CO to the formation of C2 hydrocarbons in this case could be explained by an insufficient CO concentration achieved by the reduction of CO2, which does not allow the formation of C-C bonds. More excitingly, it defines the ability of V-nitrogenase to directly use CO2 as a substrate for the initial C-C coupling and the subsequent carbon chain extension.
The ability of certain variants of Mo-nitrogenase to reduce CO2 to CH4 was reported recently.[14] To our surprise, contrary to what has been reported for these variants of Mo-nitrogenase, the wild-type Mo-nitrogenase cannot reduce CO2 to CH4; rather, it uses an unknown carbon source to generate CH4 in the presence of CO2 and H2O. Considering the presence of an interstitial carbide[15–18] and a homocitrate moiety in the FeMoco,[15,17] it can be postulated that CO2 or its derivative in H2O somehow promotes the release of the central carbide ligand or the carbon-containing groups of homocitrate in the form of CH4. Alternatively, the side chain groups of certain amino acids at the active site of Mo-nitrogenase may also serve as a carbon source for the production of CH4 in the presence of CO2. Remarkably, despite the unclear nature of the carbon source, the formation of CH4 by Mo-nitrogenase is ATP-dependent and requires the presence of both component proteins; moreover, it only occurs in the presence of CO2 and H2O (Fig. S4). This observation points to a redox-dependent characteristic of this reaction, as the requirement for ATP and both components is specifically associated with the transfer of electrons through the enzyme system, which may permit the initial binding and processing of CO2 or its derivative in H2O and the subsequent interaction between CO2 or CO2-derived intermediate(s) and the carbon species that eventually gives rise to CH4. Given the overall homology between the Mo- and V-nitrogenases, one would expect the V-nitrogenase to catalyze the same, unspecific formation of CH4 as its Mo-counterpart from a different carbon source than CO2. While this possibility cannot be ruled out, our current data (see Fig. 3A and B) clearly demonstrate that the CH4 formed by V-nitrogenase is derived, at least in part, from CO2. Further investigation of the origin of the different routes taken by the two nitrogenases to CH4 formation could be informative, particularly with regard to the initial binding and processing of CO2 by this enzyme system.
Based on the hydrocarbon products identified so far in the gas phase, the V-nitrogenase generates carbon-containing compounds at a slow rate from CO2 reduction, forming 0.3 mol CO, 0.02 mol CH4, 0.04 mol C2H4 and 0.002 mol C2H6 per mol protein. Nevertheless, the ability of V-nitrogenase to form hydrocarbons, particularly the C2 products, from CO2, is a most exciting finding of the current study, because it adds another exciting reaction to the catalytic repertoire of this unique enzyme system. As was observed in the case of CO reduction,[12] the V-nitrogenase is superior to its wild-type Mo-counterpart in generating hydrocarbons from CO2. The disparate CO-reducing activities of V- and Mo-nitrogenases were compared with the differential capacities of synthetic V- and Mo-compounds to reductively couple two CO moieties into functionalized acetylene ligands;[19] whereas an alteration of CO-reducing activities was reported for MoFe protein variants containing modified residues at the active site.[20] By analogy, the disparate CO2-reducing activities of the two nitrogenases could also stem from the structural/redox differences between FeVco and FeMoco, as well as the protein environments surrounding the two cofactors (see Fig. 1). Additionally, the different structural/redox properties of the P-clusters in the two nitrogenases could further contribute to the differences between their abilities to reduce CO2 (see Fig. 1). In fact, the ability of nitrogenase to generate hydrocarbons from CO2 was first described in the case of a cofactor-deficient variant of MoFe protein[21] and attributed to its unique ‘P-cluster’ that contains a [Fe4S4]-like cluster pair instead of the ‘normal’ [Fe8S7] P-cluster.[22] Interestingly, the P-cluster of V-nitrogenase also consists of a pair of [Fe4S4]-like clusters[5,8,9] and could, in principle, serve as a site for CO2 reduction on its own; only in the case of the holo V-nitrogenase, the presence of the cofactor ‘downstream’ of the P-cluster along the electron transfer pathway (see Fig. 1) may effectively ‘funnel’ the electrons toward the cofactor site and only allow a small, ‘leaky’ activity of CO2 reduction at the P-cluster site. The possibility of two reaction sites (i.e., P-cluster and cofactor) and different reaction routes (i.e., via CO or other CO2-derived intermediates) for CO2 reduction makes it a challenging task to elucidate the mechanistic details of this reaction. Nevertheless, the work reported herein provides an important framework for systematic investigations of this unique reaction in the future, which will hopefully lead to development of nitrogenase-based strategies to recycle the greenhouse gas into the useful carbon fuel.
Supplementary Material
Footnotes
This work was supported by NIH grant GM-67626 (M.W.R.)
Supporting information for this article including experimental procedures and Figures S1–S3 is given via a link at the end of the document.
Contributor Information
Johannes G. Rebelein, Department of Molecular Biology and Biochemistry, University of California, Irvine, Irvine, CA 92697-3900
Yilin Hu, Email: yilinh@uci.edu, Department of Molecular Biology and Biochemistry, University of California, Irvine, Irvine, CA 92697-3900.
Markus W. Ribbe, Email: mribbe@uci.edu, Department of Molecular Biology and Biochemistry, and Chemistry, University of California, Irvine, Irvine, CA 92697-3900
References
- 1.Burgess BK, Lowe DJ. Chem Rev. 1996;96:2983–3012. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 2.Rees DC, Tezcan AF, Haynes CA, Walton MY, Andrade S, Einsle O, Howard JB. Philos Trans A Math Phys Eng Sci. 2005;363:971–984. doi: 10.1098/rsta.2004.1539. [DOI] [PubMed] [Google Scholar]
- 3.Ribbe MW, Hu Y, Hodgson KO, Hedman B. Chem Rev. 2014;114:4063–4080. doi: 10.1021/cr400463x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffman BM, Lukoyanov D, Yang ZY, Dean DR, Seefeldt LC. Chem Rev. 2014;114:4041–4062. doi: 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu Y, Lee CC, Ribbe MW. Dalton Trans. 2012;41:1118–1127. doi: 10.1039/c1dt11535a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eady RR. Chem Rev. 1996;96:3013–3030. doi: 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]
- 7.Schindelin H, Kisker C, Schlessman JL, Howard JB, Rees DC. Nature. 1997;387:370–376. doi: 10.1038/387370a0. [DOI] [PubMed] [Google Scholar]
- 8.Lee CC, Hu Y, Ribbe MW. Proc Natl Acad Sci USA. 2009;106:9209–9214. doi: 10.1073/pnas.0904408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu Y, Corbett MC, Fay AW, Webber JA, Hedman B, Hodgson KO, Ribbe MW. Proc Natl Acad Sci USA. 2005;102:13825–13830. doi: 10.1073/pnas.0506967102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fay AW, Blank MA, Lee CC, Hu Y, Hodgson KO, Hedman B, Ribbe MW. J Am Chem Soc. 2010;132:12612–12618. doi: 10.1021/ja1019657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee CC, Hu Y, Ribbe MW. Science. 2010;329:642. doi: 10.1126/science.1191455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu Y, Lee CC, Ribbe MW. Science. 2011;333:753–755. doi: 10.1126/science.1206883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seefeldt LC, Rasche ME, Ensign SA. Biochemistry. 1995;34:5382–5389. doi: 10.1021/bi00016a009. [DOI] [PubMed] [Google Scholar]
- 14.Yang ZY, Moure VR, Dean DR, Seefeldt LC. Proc Natl Acad Sci USA. 2012;109:19644–19648. doi: 10.1073/pnas.1213159109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Einsle O, Tezcan FA, Andrade SL, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 16.Wiig JA, Hu Y, Lee CC, Ribbe MW. Science. 2012;337:1672–1675. doi: 10.1126/science.1224603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spatzal T, Aksoyoglu M, Zhang L, Andrade SL, Schleicher E, Weber S, Rees DC, Einsle O. Science. 2011;334:940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lancaster KM, Roemelt M, Ettenhuber P, Hu Y, Ribbe MW, Neese F, Bergmann U, DeBeer S. Science. 2011;334:974–977. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carnahan EM, Protasiewicz JD, Lippard SJ. Acc Chem Res. 1993;26:90–97. [Google Scholar]
- 20.Yang ZY, Dean DR, Seefeldt LC. J Biol Chem. 2011;286:19417–19421. doi: 10.1074/jbc.M111.229344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corbett MC, Hu Y, Naderi F, Ribbe MW, Hedman B, Hodgson KO. J Biol Chem. 2004;279:28276–28282. doi: 10.1074/jbc.M403156200. [DOI] [PubMed] [Google Scholar]
- 22.Lee CC, Hu Y, Ribbe MW. Proc Natl Acad Sci USA. 2012;109:6922–6926. doi: 10.1073/pnas.1202429109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.