

Abstract
Traditional approaches for translating observations of molecular events into the context of a living organism have suffered from the requirements for either sacrificing animals at multiple time points prior to labor-intensive analyses of multiple tissues, or have relied on subjective observations or measurements of the animals over time. Recently an explosion of dedicated animal imaging modalities and the release of modified clinical imaging devices dedicated for animal imaging have allowed for the design of quantitative real time experiments incorporating fewer animals and providing whole animal analyses. Of these modalities, optical imaging (bioluminescence and fluorescence) has emerged as a powerful research tool, allowing investigators with limited whole animal imaging expertise to rapidly and inexpensively translate models produced in cellular assays into the context of a living animal. Here we will outline the steps necessary for translation of models established in culture systems into rodents.
Keywords: Bioluminescence, fluorescence, whole animal, molecular imaging, non-invasive, reporter
INTRODUCTION
Luciferase has been used as a reporter gene in a variety of cellular assays for many years, while fluorescent proteins are routinely used as a means to examine molecular events microscopically and the development of large panels of distinguishable fluorophores covering a wide spectrum of wavelengths has allowed for greater understanding of molecular events through flow cytometry. The transfer of some of these reporters and assays into whole animal models has greatly accelerated our understanding of molecular events in vivo.
The field of whole animal imaging has rapidly developed over the last decade, with the introduction of multiple modalities, each with individual strengths and weaknesses 1. Many of these modalities represent the conversion of clinical imaging instruments into dedicated animal imaging devices. Several of these modalities provide purely structural data (such as Computed Tomography or X-ray imaging), while others are strongly translation focused (including Molecular Resonance Imaging, MRI and Nuclear Medicine aproaches including Positron Emission Tomography, PET and Single Photon Emission Computed Tomography, SPECT), primarily due to the cost and complexity of their use. Ultrasound imaging has developed some novel applications in the context of small animal imaging due to the ability to image through the entire animal at this scale and the development of high-resolution transducers 2. Although primarily a structural imaging modality, ultrasound has the ability to image in real time (allowing for analayses of blood flow and perfusion), and the application of contrast enhanced ultrasound can provide molecular data.
Here we will focus on optical modalities (bioluminescence and fluorescence). Although not directly translational to the clinic, these modalities have become the primary resource for imaging of small animals in the research environment, due to their ease of use, flexibility, inexpensive nature and the capability for rapid image collection, as well as the ability to directly transfer some models and assays developed in tissue culture into small animals. However, this modality has some limitations in reporter use, depth of imaging capabilities, resolution and background that need to be considered, and that will be covered in this unit.
The depth penetration issues encountered with optical imaging means that these modalities are currently limited to small animal or superficial imaging. Fluorescence and bioluminescence imaging of fish and insect models is routinely used, but primarily imaging is at a microscopic level, and so will not be covered in this unit. The principal animal model used for optical whole animal molecular imaging is the mouse, however rat imaging is feasible and superficial imaging of some larger animals (such as rabbit or non-human primate) have been achieved. Larger animals typically rely on clinical imaging modalities where depth penetration is not an issue.
Here we will cover basic protocols for imaging cells, pathogens and other biological materials (such as peptides or antibodies) in mouse models.
Basic Protocol 1. BIOLUMINESCENCE IMAGING OF ADOPTIVELY TRANSFERRED EUKARYOTIC CELLS;
Non-invasive whole animal imaging provides several advantages over traditional animal models. The ability to repeatedly image the same animals means that they become their own internal controls, and also dramatically reduces the number of animals required to run experiments. In addition, unexpected and expected signals detected during whole animal imaging can be used to define time points and tissues selected for ex vivo analysis, and so better define a disease model, timings of therapeutic responses, and any unexpected gene expression profiles.
Materials
Cells of interest (e.g. mouse tumor cell line)
D-luciferin, 30 mg/ml in PBS, filter sterilized.
Coelenterazine 5mg/ml in Methanol (Millipore Chemicon).
Mice, typically obtained form commercial vendors (e.g. Jackson laboratories or Charles River)
Whole animal bioluminescence imaging system (such as IVIS, Caliper Life Sciences) with heated (37oC) stage and appropriate image analysis software (such as Living Image, Caliper Life Sciences.).
70% Ethanol
Anesthetic (ideally inhaled, such as isoflurane, but may be injected, such as avertin)
Black walled, clear bottom 96 well plates.
Luciferase assay system (Promega).
Prepare the Cells
- Cells will need to be engineered to stably express luciferase. This is frequently achieved through lentiviral transfection and selection (see support protocol 1). Alternatively, many commonly used cell lines are commercially available with luciferase expression. Finally, several commercially available transgenic mouse strains are available that express luciferase constitutively from all celltypes. These can be used as donors for adoptive transfer of pre-labeled immune cell populations (so negating the need to use viral labeling of immune cells), or even for transplant of intact organs or tissues.In addition to eukaryotic cells, bioluminescence imaging has frequently been used to image infections with a variety of different pathogens. Many viruses can be engineered to express luciferase from infected cells, so allowing the real-time imaging of the infection. Bacteria will typically be engineered to express the Lux-operon following transfection with a suitable plasmid. This operon, from Vibrio fischeri, expresses gene products required for production of both the bacterial luciferase, as well as for production of the luciferin substrate (so negating the need to exogenously add luciferin to infected mice).
The choice of luciferase enzyme incorporated for labeling is important. In order to achieve maximum light output from within a living organism, a significant part of the spectrum of the light produced from any luciferase should be greater than 600nm, as light below this wavelength will be adsorbed, primarily by hemoglobin. The most commonly used luciferase is that of the firefly (Promega), and this produces light with a spectral peak of 560nm, however at 37°C this shifts to 590nm making it suitable for in vivo use 3. Other insect luciferases, such as those of the click beetle or railroad worm produce a variety of spectra and although green luciferases are often brighter in vitro, the increased adsorption at these wavelengths in vivo makes them less useful. Several luciferases have also been cloned from marine organisms, such as Renilla and Aequorea, but these produce blue-green light (peak of 475nm). However, one advantage of Renilla luciferase is that is utilizes a different substrate (coelenterazine) to the insect luciferases (luciferin) and so dual reporter bioluminescence imaging is possible. Also, marine luciferases do not require ATP, and so may also be able to function extracellularly. However, an additional handicap of the marine luciferase is that the coelenterazine substrate is unstable meaning that a higher background signal is typically produced, and the substrate only remains saturating within an animal for a short period of time. This can be used as an advantage when dual imaging of both firefly and Renilla luciferases in a single animal is attempted, as it is possible to image the Renilla luciferase first following addition of coelenterazine, and then to image the firefly luciferase (as soon as 1 hr later, once the coelenterazine has degraded) following addition of luciferin. Both substrates are non-toxic, are rapidly distributed throughout the body, and can cross most membranes, including cellular membranes and the placenta and the blood-brain barrier. A further common strategy is to combine luciferase and GFP co-transfection, and a variety of constructs, including fusion proteins, dual promoter and genes separated with insertion of ribosomal slippage sites have been reported 4. This allows whole animal bioluminescence imaging to be used to identify time points and tissues that are of interest for post mortem analyses, while flow cytometry or fluorescence microscopy can then be used to distinguish host and adoptively transferred or implanted cells.
The choice of promoter will depend on the model being examined. Studies examining cell proliferation, persistence, biodistribution and trafficking will require a strong constitutive promoter (such as that of the retroviral LTR, beta-actin or ubiquitin). However, other studies may depend on a specific promoter or promoter element to examine processes such as gene expression profiles or metabolic activity. The strength of promoter is an important consideration, as weak promoters may not produce sufficient luciferase. This can be overcome with the incorporation of two-step gene expression systems, such that a weak promoter can drive expression of a trancription factor that would induce gene expression from a stronger promoter, driving luciferase production. Other model systems, such as conditional expression of the Cre-recombinase protein and a fLOX-ed stop codon between a promoter and luciferase expression can allow for study of transient gene expression, or monitoring of trafficking or survival of cells after a gene has been transiently expressed.
Prepare the mice
Consideration should also be given to the animal model to be used. Because of the adsorption and diffraction limitations of optical imaging, tissues greater than 1cm deep cannot be imaged. This means that the ideal species to use will be the mouse. Choice of mouse strain may be limited by the availability of cell lines, but in general white mice are preferable (as the pigmentation in the skin of black or brown mice further adsorbs emitted light). Some normally dark mice (e.g. C57B/6) have been bred and are commercially available as albinos. Animal fur also diffracts light, and so shaving or use of depilatory cream may enhance weak signal. Alternatively, athymic nu-/nu-mice may be used if appropriate. Pilot experiments should always be run in vivo to first characterize the model.
An initial pilot experiment may be used for example to verify tumor growth patterns and take rates for different injected doses of cancer cell lines, or to establish limits of detection for different labeled cell types. Furthermore, sequences of images should be taken at different times post addition of luciferin substrate in order to establish the optimal timing of imaging. Anesthetic to be used should also be tested to ensure the minimum required dose is used, and if possible, to verify that the anesthetic used does not interfere with the study. Most injected anesthetics are metabolized by the subject and so they may affect metabolic pathways, stress responses or oxygen utilization. Mice also take longer to recover from injected anesthetics and so fewer imaging time points can be used. Inhaled anesthetics (such as isoflurane at 2%) are therefore typically preferable, however there is some evidence that these may produce a low level of background luminescence, and so may not be suitable for detection of very weak signal, especially from the lungs. Protocols for all planned experiments should be approved by institutional review boards.
Deliver D-luciferin substrate to mice. Imaging of insect luciferase will typically require injection of 200 μl of 30mg/ml luciferin intraperitoneally 10–20 minutes prior to imaging, coelenterazine for Renilla luciferase will typically be injected intravenously (tail vein or retino-orbital injection), with 100μl of 5mg/ml injected no longer than 2 minutes prior to imaging.
Anesthetize mice. The exact order of delivery of substrate and anesthesia will depend on the system being used.
Image the mice
- Arrange mice in the imaging chamber so that the expected source of signal is closest to the top of the animal (e.g. ventral view for imaging major organs, dorsal for imaging spine, left side for imaging spleen); Imaging stage should be wiped with 70% ethanol prior to use, and a piece of black paper placed on the stage to protect it. Dividers may be used between the animals (if multiple animals are to be imaged at once), to prevent strong signal from one animal reflecting off its neighbor. All non-essential materials should be removed from the imaging chamber. The stage should be heated to 37°C, and animals should be checked for level of anesthesia for 30 seconds prior to closing the chamber door.Many plastics and other materials (including animal bedding, chow and dander) will produce phosphorescence, and so careful selection of any item used in the imaging chamber is important. In particular, anesthesia nose cones, paper used to protect the stage, plastic dividers and animal shields should always be carefully tested, and all bedding, dander and chow should be cleaned from the stage before imaging.
- Take image. Several variables may be adjusted to improve the quality of the image, including height of stage; focus; length of exposure; binning and aperture setting (depending on the imaging system being used)(Fig 1).The height of the stage should be adjusted so that the animals to be imaged best fit the field of view with minimal empty space. It is recommended to initially try a 60 second image with medium binning if the strength of the signal is not known. If this saturates the camera then quantification is not possible, and so shorter exposure times should be tried and smaller binning used. If at 1 second, a minimum binning image still saturates the Charge-Coupled Device (CCD) camera, then the aperture may be reduced. If the original image produces weak or no signal, then increased exposure times (up to 5 minutes) and increased binning may be attempted. Images longer than 5 minutes will likely not improve the signal, as the background will also increase.
- In order to help locate the source of a signal of unknown origin, it is recommended that the mice are repositioned and re-imaged from different angles.Recent advances in software available for image analysis may include 3D reconstructions, whereby animals are imaged consecutively at different wavelengths, and the relative levels of blue and red light emitted can be used to determine the depth of the signal.
Recover mice. Once imaging is complete, mice should be recovered on a heated stage, until capable of self-righting.
Fig 1.

Different stage heights provide different fields of view, such that multiple animals may be imaged at one time (high throughput) or a single region of one animal may be imaged (high resolution).
Analysis of Data
- Image analysis. A variety of software packages exist for image analysis, including quantification, such as Living Image (Caliper Life Sciences). Bioluminescence signal produced from an implanted or transferred cell population may be quantified within different, user-defined regions of interest and compared to the same regions in differentially treated animals, or in the same animals over time.Many software packages, including Living Image, provide data quantifications in units (photons per second per unit area) that will be adjusted for changes made to image collection parameters (such as length of exposure, stage height, binning or aperture setting). This means that if the imaging settings change during the time course of an experiment (for example as a tumor grows shorter exposure times may be required), directly comparable quantitative data can still be produced.
Verification of imaging results. It is important to verify patterns of gene expression and the source of any signals ex vivo in a subset of animals. Bioluminescence signal from firefly luciferase will remain strong for up to 45 minutes post mortem, and so individual organs can be imaged to verify the tissue of origin of any signal (although the signal no longer remains quantifiable). Luciferase expression can be verified ex vivo using luciferase assay systems (on frozen, ground tissues, and normalized to protein level), and luciferase quantified by Polymerase Chain Reaction (PCR). The limited availability of reliable luciferase targeting antibodies means techniques such as Western Blot or immunohistochemistry are not recommended.
SUPPORT PROTOCOL 1: LENTIVIRAL LABELING OF CELL LINES
The most common method for labeling cells or cell lines in order to express luciferase for subsequent in vivo imaging is through lentiviral labeling, typically using third generation, four plasmid systems to ensure safety. Commercially available kits can be cloned to express latest generation luciferase enzymes (such as from the pGL4 plasmid, Promega).
Materials
293T cell lines
Lentiviral expression kits (third generation, four plasmid packaging systems are recommended, can be cloned to express luciferase)
DMEM, 10% FCS, 1% Penicillin-Streptomycin, 1% Glutamine
Lipofectamine 2000(Invitrogen)
OPTI-MEM media
Polybrene
Puromycin (or other selection antibiotic as appropriate)
Generation of Lentiviral vectors
18–24 hours prior to transfection, plate 293T cells at 2–3 million cells per 10 cm plate in DMEM+ 10% FBS. Gently shake forward and backward, then side to side, to spread cells evenly about the plate. Allow to attach to plate for 10 to 24h.
- Verify cells are at 60 to 80% confluence prior to proceeding.At this subconfluent stage cells are most transfectable and will survive the transfection best, giving the highest titer virus possible. Never let cells reach confluence, this will reduce transfection efficiency in the short term.
Transfection
In one tube mix 1500ul OPTI-MEM + 60ul Lipofectamine
- In a second tube mix 1500ul OPTI-MEM + 24ug of insert DNA plasmid + 4ug packaging DNA plasmids.Ideally plasmid DNA should be from an Endo-free midi or maxi-prep, different ratios and quantities of insert and packaging DNA should be tried to optimize protocol. Alternative transfection reagents and procedures are also available.
Mix the two tubes together and wait 15–45 minutes
In the meantime aspirate media from plate of 293T cells and very carefully (down the side of the plate) add 15ml OPTI-MEM (do not add serum or antibiotics to OPTI-MEM media).
After the 15–45 minutes, slowly drip the plasmid/lipofectamine mix onto the cells, with each drip onto a different area of the plate.
- Return plate to incubator overnight.Virus is more stable if incubation is carried out at 32° Celsius, although 37 is acceptable.
- After 24h pipette supernatant from transfected 293T cells into 15 mL tubes and centrifuge at 300 ×g for 5 minutes to pellet cell debris.Filtering through 0.45 um filter may be used as an alternative approach to remove cells debris. Supernatant can be frozen at −80 °C for later infection, although titer drops by one-half for each freeze-thaw cycle, so using fresh is recommended. Fresh media may then be added to the 293T cells and a second round of viral production collected after another 24h. Titering of virus is recommended. 293T cells may also be imaged for bioluminescence as a means to verify initial transfection efficiency.
Prepare Target Cells
Split cells to be labeled at approximately 200,000 cells per well in 6-well plates in DMEM, 10% FCS (1% Penicillin-Streptomycin, 1 % Glutamine). If using suspension cells, they should be growing in log phase at time of infection if possible. Incubate overnight.
Remove 50% of media from each target cell well and replace with equal volume of 293T supernatant.
Add 3 μL polybrene (polybrene is 1000X at 5 mg/ml) to each target cell plate; with gentle and thorough shaking.
- Spin infection (improves efficiency of infection): spin cells (ideally in sealed 6-well plates) at 240 ×g for up to 1 hour at 30°C. Fresh virus can be applied and the cells spun again for 45 minutes.Some cells, especially B-cells and T-cells are sensitive to polybrene and so may need lower levels.
Place plate at 37 Celsius (with gentle shaking) and incubate for 4–8 hours, spin cells, and wash away virus supernatant.
At 24 hours post-infection, change media on target cells to fresh DMEM, 10% FCS and re-place at 37 Celsius.
- At 48 hours post-infection cells can be initially assayed for luciferase expression.If you are using an antibiotic marker such as puromycin resistance (recommended) you can add puromycin to media and wait a further 72h and pick colonies) (N.B. the concentration of puromycin used will depend on the target cells, you will need to run an initial assay to determine the dose required to just kill off the target cells). Remove the selection after 7–10 days to determine stability of insert (you can passage in marker again every 2 months to maintain selection).You may want to pick a single colony following selection and test this, or else you can mix together all labeled cells.You should verify properties of cells after labeling (for example, verify that rate of growth and morphology are unchanged, plus any particular attributes that may be critical to the model).
BASIC PROTOCOL 2: FLUORESENCE IMAGING
Although bioluminescence imaging has traditionally been the most widely used modality, due to its sensitivity and large dynamic range, it suffers from several limitations, including the inability to reliably image more than one reporter within a single animal. Fluorescence imaging has historically suffered with issues related to autofluorescence and limited depth penetration in the past, but novel fluorophores and advances in imaging technology have meant that fluorescence imaging may soon become the more routinely used approach.
Considerations for fluorescence imaging
Choice of reagents to be imaged. Depending on the model to be examined, a variety of biological agents may be labeled (including peptide, antibody, DNA, sugars, small molecules and cells). Fluorophore binding to these agents may require some complex chemistry, or may be possible through simple one step procedures with commercially available kits (such as N-hydroxysuccinimide (NHS) esterification of Cy dyes to cysteine residues). Consideration should be given to any possible changes to the properties of the reagents as a result of fluorophore tagging (such as increased size or changes in hydrophobicity). Cells can also be labeled in a variety of different ways, including loading with fluorescently labeled quantum dots, membrane labeling, peptide labeling, or expression of fluorescent protein. Issues such as the reduction in fluorophore concentration after cell division; processing of fluorophore, or uptake into other cells such as macrophages after cell death; effects of cellular manipulation on cellular function; or length of time required for imaging of cell tracking after transfer should be considered when choosing a labeling protocol. In addition, although multiple fluorescent proteins are available only a small number have the required spectral properties that make them suitable for whole animal imaging (see below)5. However, cells labeled with appropriate fluorescent proteins can be treated in a similar way to luciferase labeled cells (with the exceptions that (i) there is no need for exogenous addition of substrate; (ii) sensitivity of detection is likely to be reduced relative to bioluminescence; (iii) ATP is not required for fluorescence; and (iv) fluorescent proteins tend to be more stable than luciferase, and so are less useful as a means to follow gene expression profiles).
Choice of fluorophore(s). One of the major advantages of fluorescence imaging is that multiple (up to four) channels can be imaged in the same animal. Different detection devices offer different fluorophore channels but, as for bioluminescence imaging, the key consideration is the wavelength of light needed to pass through tissue. However, this applies both for excitation and emission, with wavelengths between 600nm and 1000nm providing greatly enhanced tissue penetration properties. This means that fluorophores in the far red and near infrared range are most suited to whole animal imaging. The high level of autofluorescence at lower wavelengths also complicates imaging in this range. Indeed it is recommended that mice be fed alfalfa-free imaging diets for both bioluminescence and fluorescence imaging to reduce the levels of background produced by chlorophyllin the stomach and intestinal tract. This wavelength recommendation does reduce the number of fluorescent proteins that can be readily imaged at any depth in living animals, however several recently described proteins do have adsorption and emission spectra in appropriate wavelength ranges.
Choice of Imaging Apparatus. Novel imaging devices are being introduced to the market at a rapid rate, while other investigators have chosen to build their own custom models. This means a large range of technical approaches to image collection have been incorporated. The most effective devices typically use transillumination of the subjects and will utilize laser excitation light. The imaging device incorporated will therefore further determine the limits of sensitivity and quantification available to investigators.
Commentary
Background Information
The recent and rapid development of the field of small animal imaging has seen a variety of different modalities enter the market, each with their own advantages and limitations. The use of molecular imaging approaches tonon-invasively interrogate events occurring within a living animal have many advantages over more traditional assays performed on small animals. In particular, the fact that groups of animals do not need to be sacrificed at each time point is a significant advantage scientifically (as more accurate results can be obtained by making each animal its own internal control during the course of an experiment; and, because the whole animal is examined, this ensures no unexpected target tissue or organs are missed in the analyses); ethically (as reduced numbers of animals are required to achieve the same results); and financially (as the labor-intensive analyses of multiple tissues for every time point is no longer required). In addition, the quantitative nature of many imaging modalities reduces the level of subjectivity associated with many live-animal assays (such as measures of morbidity or even tumor caliper measurements).
There is a wide range of whole animal molecular imaging modalities available. However, of these, optical imaging approaches are currently the most widely and extensively used. This is due to their ease of use (the need for a dedicated operator is rare), relatively rapid imaging capabilities (with an experienced user capable of imaging up to 50 mice in approximately 2 hours, so allowing multiple imaging time points within a single experiment), flexibility (with a wide range of biological markers and processes being amenable to imaging), sensitivity (with the possibility of imaging down to a single cell subcutaneously), large dynamic range (of up to 12 logs) and the relative inexpensiveness.
However, the fact that light scatters and is absorbed in tissues means that the resolution is often lower with optical imaging than other imaging modalities, and the limited depth penetration means that the modality is currently limited to small rodent models.
Critical parameters
Because optical imaging relies on imaging of reporters (luciferase enzymatic action or the action of fluorophores), there is a requirement for introducing the reporters into the animals. This can be through ex vivo manipulation of cell populations, development of transgenic strains, or through addition of tagged or labeled molecules. Some initial model planning and development of reagents is therefore critical. For example, although bioluminescence imaging of two reporters in one animal is feasible, using expression of both an insect and a marine luciferase (such that a pathogen and the immune response might be imaged simultaneously for example), it will need to be considered that (i) the marine luciferase light has poor tissue penetration, so its sensitivity is greatly reduced; and (ii) that any immune cells to be imaged will need to be labeled ex vivo, or adoptively transferred from a luciferase-expressing transgenic mouse.
Although fluorescence whole animal imaging has progressed rapidly over the last five years, the choice of fluorophores is still limited by the properties of light adsorption in tissues, and so fluorophores with both excitation and emission spectra above 600nm are recommended. This can limit the simple transfer of tissue culture models (where fluorescent proteins such as GFP or dsRed are often incorporated) into effective animal imaging.
Other issues investigators have to consider include the availability and access to imaging equipment, and the requirement to obtain Institutional approval to complete experiments as planned.
Troubleshooting
The most commonly encountered problem with bioluminescence imaging is a lack of reported consistency between animals. Although this can occasionally be due to poor injection of luciferin substrate, the most common cause is a lack of consistency in the timing between injection and imaging. Although relatively consistent signal is usually produced between 10 and 30 minutes after injection of substrate, this may vary between models, and so it is recommended to repeat image animals after a single substrate injection during a pilot experiment to better appreciate the timing of peak signal, and the rate of subsequent signal ‘drop-off’. As such, it may be necessary to increase luciferin dose, carefully co-ordinate timing of imaging, or even in extreme cases, incorporate osmotic pumps to deliver a constant and steady dose of luciferin.
The most commonly encountered problem with fluorescence imaging is poor choice of fluorophores. In particular, many investigators will continue to attempt to image GFP in whole animals despite knowing the poor likelihood of success.
Anticipated results
The expected results are shown in Fig 2. The level of quantification, sensitivity and reliability of data obtained will depend on the model under investigation, the reporters used and the imaging apparatus available. Under some conditions a single cell can be detected in a living animal, in others 100,000 cells or more in a single location will be required for reliable imaging. For bioluminescence imaging this can be partly predicted based on in vitro quantification of cell signal, with a signal of less than 1.0 photon per second per cell considered weak, and signals of up to 1000 Ph/sec per cell possible. Some imaging apparatus will only allow for 2D imaging, while others have full tomographic imaging capabilities, allowing co-registration with other imaging modalities. However, as long as a model is used consistently throughout an experiment, detection of subtle changes in the level and location of a signal over time and under different conditions can be expected. This sensitivity is often greater than traditional approaches (for example the relative consistency of measurements of tumor burden by caliper measurement and bioluminescence imaging will allow for smaller groups when imaging is used in order to achieve significance).
Fig 2.
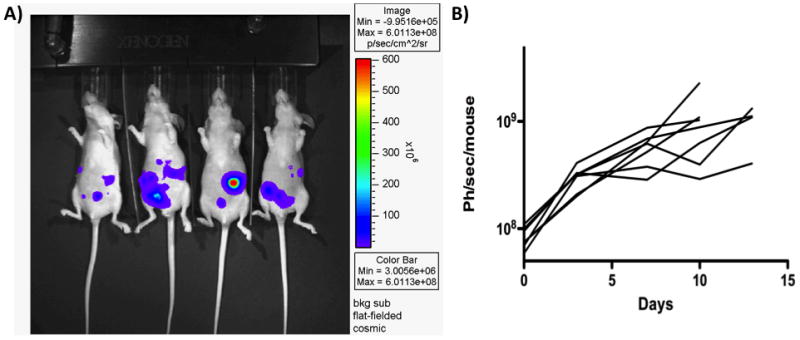
(A) Representative image produced for mice with luciferase labeled ovarian cancer cell line implanted into the peritoneal cavity. Individual tumor nodules can be visualized. (B) The tumor burden from these mice was quantified as a measure of light output and plotted over time (with each graph representing an individual mouse implanted with the same tumor cell line).
Time considerations
One advantage of optical imaging is the relatively quick image collection relative to traditional methods, and also relative to many other imaging modalities. Typically bioluminescence imaging will require prior injection with luciferin and anesthesia, but up to 5 animals can be imaged simultaneously, and images will typically take less than 5 minutes to collect. However, some of the more sensitive fluorescence imaging devices incorporate scanning lasers, and so imaging (of a single animal at a time) may take 20 to 30 minutes. Additional time should be considered for post-collection image analyses.
Acknowledgments
Funding for the Thorne lab is provided by NIH/NCI, The Alliance of Cancer Gene Therapy and The Pittsburgh Foundation.
Suppliers and special equipment appendix
(1)pGL4 Luciferase Reporter Vectors: Promega. Promega Corporation, 2800 Woods Hollow Road Madison, WI 53711, USA. Tel: 608.274.4330, Fax: 608.277.2516 (www.promega.com)
(2)Coelenterazine: Millipore. 290 Concord Road, Billerica, MA 01821, USA. Tel: 781.533.6000 (www.millipore.com)
(3)Lipofectamine2000 transfection reagent: Invitrogen. 5791 Van Allen Way, PO Box 6482, Carlsbad, California 92008, USA. Tel: 760.603.7200 Fax: 760.602.6500 (www.invitrogen.com)
(4)IVIS Imaging System: Caliper Life Sciences. 68 Elm Street, Hopkinton, MA 01748, USA. Tel: 508.435.9500, Fax: 508.435.3439 (www.caliperls.com)
(5)Living Image Software: Caliper Life Sciences. 68 Elm Street, Hopkinton, MA 01748, USA. Tel: 508.435.9500, Fax: 508.435.3439 (www.caliperls.com)
(6)Luciferase assay system: Promega Corporation, 2800 Woods Hollow Road Madison, WI 53711, USA. Tel: 608.274.4330, Fax: 608.277.2516 (www.promega.com)
Literature cited
- 1.Thorne SH, Contag CH. Using in vivo Bioluminescence Imaging to Shed Light on Cancer Biology. Proc IEEE. 2005;93:750–762. [Google Scholar]
- 2.Villanueva FS. Molecular imaging of cardiovascular disease using ultrasound. J Nucl Cardiol. 2008;15:576–586. doi: 10.1016/j.nuclcard.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao H, Doyle TC, Coquoz O, Kalish F, Rice BW, Contag CH. Emission spectra of bioluminescent reporters and interaction with mammalian tissue determine the sensitivity of detection in vivo. J Biomed Opt. 2005;10:41210. doi: 10.1117/1.2032388. [DOI] [PubMed] [Google Scholar]
- 4.Contag CH, Bachmann MH. Advances in in vivo bioluminescence imaging of gene expression. Annu Rev Biomed Eng. 2002;4:235–260. doi: 10.1146/annurev.bioeng.4.111901.093336. [DOI] [PubMed] [Google Scholar]
- 5.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]