

Abstract
Aging induces myriad cellular and, ultimately, physiological changes that cause a decline in an organism's functional capabilities. Although the aging process and pathways that regulate it have been extensively studied, only in the last decade have we begun to appreciate that dynamic histone methylation may contribute to this process. In this review, we discuss recent work implicating histone methylation in aging. Loss of certain histone methyltransferases and demethylases changes lifespan in invertebrates, and alterations in histone methylation in aged organisms regulate lifespan and aging phenotypes, including oxidative stress-induced hormesis in yeast, insulin signaling in Caenorhabiditis elegans and mammals, and the senescence-associated secretory phenotype in mammals. In all cases where histone methylation has been shown to impact aging and aging phenotypes, it does so by regulating transcription, suggesting that this is a major mechanism of its action in this context. Histone methylation additionally regulates or is regulated by other cellular pathways that contribute to or combat aging. Given the numerous processes that regulate aging and histone methylation, and are in turn regulated by them, the role of histone methylation in aging is almost certainly underappreciated.
1. The aging process
1.1. Physiological changes associated with aging
Aging is associated with a number of detrimental physiological effects that impact the health and overall function of an organism. Among humans and other mammals, these include a decline in immune function, increasing susceptibility to diseases, chronic inflammation, reduction of muscle mass (sarcopenia), increased incidence of cancer, and the onset of age-related degenerative disorders such as Alzheimer's and Huntington's diseases [1]. Although these phenotypes are manifest at a systemic or organismal level, they are ultimately caused by changes in cellular functions, and, indeed, molecular pathways that contribute to, or help slow, aging have been identified. Many processes, including autophagy, mitochondrial (oxidative phosphorylation) efficiency, and proteosome function, decline with age, while incidence of DNA damage increases; these have been implicated in various aging phenotypes [2–5]. A decrease in mitochondrial efficiency causes increased production of reactive oxygen species (ROS), which can damage macromolecules, including DNA, and also function as second messengers, thus ectopically activating signaling; both processes are thought to contribute to aging pathologies [6–8]. In addition to a potential increase in damaged molecules in aged cells due to accumulation of ROS, there is also lowered turnover of damaged or insoluble proteins by the proteosome and proteins and other macromolecules by autophagy [5,9]. The accumulation of protein aggregates that results from decreases in proteosome function and autophagy contribute to the pathophysiology of Alzheimer's and Huntington's diseases [9,10]. Increased ROS and damaged macromolecules are significant sources of cellular stress, and it is well known that activating stress response pathways can promote longevity and slow the progression of aging [11].
Improperly repaired DNA damage causes mutations, and the accumulation of mutations within one cell's genome can eventually lead to cancer; old cells are, of course, subject to more cumulative DNA damage and mutations than young ones. Cancer, during which cells undergo dysregulated cell divisions and disrupt the organism's physiology, is sufficiently detrimental that a process termed cellular senescence is thought to have evolved to counter it [12]. During cellular senescence, tumor suppressor genes are activated to irreversibly halt progression of the cell cycle [13]. As an organism ages, the number of senescent cells increases. Indeed, a general decline in stem cell function has been reported with age, which is thought to contribute to tissue degeneration [14–16]. This decline in function results from both reduced numbers of stem cells in older animals, possibly as the result of cellular senescence, and a reduction in their multilineage differentiation capacity [17–22]. The complex age-associated phenotypes are thus the result of alterations to cellular processes that occur during and/or as a result of the aging process.
1.2. Model organisms and the study of longevity
Some physiological and many cellular aspects of aging are conserved among eukaryotes, and, indeed, much insight into the molecular mechanisms of aging has come from work done in various eukaryotic models, such as the budding yeast Saccharomyces cerevisiae, the nematode worm Caenorhabditis elegans, the fruit fly Drosophila melanogaster, and the house mouse Mus musculus, as well as mammalian tissue culture and progeria disorders, which cause premature aging, in humans. Like mammals, yeast are subject to the phenomenon of replicative senescence, in which individual cells are only able to undergo a limited number of mitoses [23]. Similarly, age-induced sarcopenia occurs in C. elegans and Drosophila [24,25], which may indicate that stem cell exhaustion contributes to aging in Drosophila as well. Although other physiological aspects of aging, including decreased immune function, chronic inflammation, and increased incidence of cancer, have not been shown to occur in invertebrate models, the molecular pathways and dysfunctions associated with aging are remarkably conserved. The age-associated decline in autophagy was first observed in yeast and only later found in C. elegans, Drosophila, and mice [2]. Activation of stress response pathways increases lifespan in yeast, C. elegans, and Drosophila, as well as delays cellular senescence in mammalian cell culture [26]. Similarly, restricting caloric intake (caloric or dietary restriction), increases lifespan from yeast to humans, was first described in rodents, and has been extensively studied in C. elegans [27]. In C. elegans, repression of the insulin and insulin-like signaling (IIS) pathway was shown to promote longevity [28], and correlative studies have subsequently suggested that this mechanism may be conserved in mice and humans [29]. Thus, model organisms, particularly invertebrate models, have been the source of much of our knowledge regarding the molecular pathways that drive aging physiology.
1.3. Chromatin and aging
Another more recently discovered molecular phenotype associated with aging is a dysregulation of transcription, as manifested by changes in gene expression. This phenomenon has been observed in many organisms, though it is not clear whether this dysregulation causes pathophysiologies associated with aging or is itself the result of the aging process [30–35]. Along with alterations in the transcriptome, in many organisms, chromatin structure changes with age [36], and, indeed, some of these changes have been shown to cause age-associated phenotypes in yeast [37]. In general, chromatin is thought to take on a more euchromatic and transcriptionally active (“open”) conformation as an organism ages [38]. In particular, an increase in histone acetylation, mediated by decreased Sirtuin levels, contributes to this phenotype in some organisms [37,39,40], though global decreases in histone levels have also been observed [37,41,42]. Another indication that chromatin state impacts the aging process comes from the field of induced pluripotent stem cells (iPS cells). During the induction of pluripotency, terminally differentiated cells from adults are made to take on characteristics of embryonic stem cells [43]; in essence, to become younger. This reversion to a younger state is accompanied by, and depends on, altered chromatin structure [44]. Thus, changes to chromatin structure are likely to contribute to aging phenotypes.
In this review, we discuss the role of histone methylation in the aging process across eukaryotes. Although increased histone acetylation has been shown to cause age-associated phenotypes [37,39,40], to date, there are only a few studies showing a causal relationship between altered histone methylation and longevity or age-associated pathologies (with the notable exception of cellular senescence in mammals). Additional work has revealed changes in global methylation or methylation patterns in old versus young organisms, which suggests the possibility that misregulation of histone methylation may cause aging pathologies. We also discuss the roles of histone methylation in cellular pathways known to cause age-related phenotypes. Taken together, these data suggest many mechanisms by which changes in histone methylation may contribute to aging pathologies.
2. Global changes in histone methylation patterns in aged organisms and progeria models
Several studies in mammalian systems and Drosophila have identified age-associated changes in histone methylation states. These findings are summarized in Table 1. In human cells cultured from patients with Hutchinson-Gilford progeria syndrome (HGPS), a genetic condition that causes premature aging, there is an upregulation of trimethylated lysine 20 on histone H4 (H4K20me3), and a downregulation of both trimethylated lysine 9 on histone H3 (H3K9me3) and trimethylated lysine 27 on histone H3 (H3K27me3). Along with the decrease in H3K9me3, reduced association of the heterochromatin compaction protein HP1a with pericentrosomal DNA was observed [45]. The loss of H3K27me3 in HGPS was recently shown to be largely from gene poor regions [46], which is consistent with the loss of peripheral heterochromatin seen in cells from HGPS patients and invertebrate models of this disorder [47,48]. Thus, accelerated aging in HGPS is associated with a reduction in heterochromatin, though it should be noted that H4K20me3, which increases in cells derived from HGPS patients, also marks heterochromatin. Interestingly, HP1-associated heterochromatin has been shown to be required to slow aging phenotypes in Drosophila [49]. While H3K9me3 levels are increased in aging flies [50], a reduction in dimethylated H3K9 (H3K9me2), to which HP1 can also bind [51,52], is seen [49]. This reduction in H3K9me2 is correlated with a loss of HP1 from chromatin, and, indeed, reducing HP1 levels causes loss of muscle integrity and derepression of age-associated recombination of rDNA genes in Drosophila [49]. However, it should be noted that the increase in H3K9me3 levels were observed in the heads of female flies, the reduction in H3K9me2 is seen in the whole bodies of mixed male and female flies [49,50]; thus, it is possible that these differing results are due to cell type- or gender-specific alterations in methylation. Taken together, these data support the model of increasingly open chromatin with age.
Table 1. Age-associated changes in histone methylation.
The results from studies that have analyzed changes in global levels of histone methylation are summarized. HGPS, Hutchinson-Guilford progeria syndrome; this indicates tissues derived from human patients.
| System | Tissue | Modification | Change | Reference |
|---|---|---|---|---|
| Mammals | rat kidney and liver | H4K20me3 | increase | [56] |
| mouse brain | H4K20me1 | decrease | [55] | |
| mouse brain | H3K27me3 | increase | [55] | |
| mouse brain | H3K36me3 | decrease | [55] | |
| mouse brain | H3K79me1/2 | increase | [55] | |
| macaque brain | H3K4me2 | increase | [57] | |
| HGPS | fibroblasts | H4K20me3 | increase | [45] |
| fibroblasts | H3K9me3 | decrease | [45] | |
| fibroblasts | H3K27me3 | decrease | [45,46] | |
| Drosophila | whole animal | H3K4me3 | decrease | [50] |
| whole animal | H3K9me3 | increase | [50] | |
| whole animal | H3K36me3 | decrease | [50] | |
| whole animal | H3K9me2 | decrease | [49] |
Histone methylation has also been assessed in brain tissue from SAMP8 mice, a strain of mice bred under selection for accelerated aging that are subject to premature neurodegeneration [53,54]. In this system, both H3K27me3 and mono- and dimethylated lysine 79 on histone H3 (H3K79me1/2) increase, while monomethylated H4K20 (H4K20me1) and trimethylated lysine 36 of histone H3 (H3K36me3) decrease [55]. Thus, in SAMP8 mice, there is an increase in histone modifications associated with heterochromatin and a decrease in marks indicative of active transcription, which is different from the loss of heterochromatin associated with HGPS. The differences between HGPS and SAMP8 could be caused by 1) different mechanisms by which the modifications assessed accelerate aging, 2) different changes in histone methylation in different tissues, or 3) biological differences in the aging process in humans versus mice. Additionally, as observed in HGPS, an increase in H4K20me3 was observed in livers and kidneys in aged rats [56]. In Drosophila, decreases in H3K36me3 and trimethylated H3K4 (H3K4me3) were seen during aging [50]. Interestingly, an analysis of dimethylated lysine 4 on histone H3 (H3K4me2), which is associated with transcriptional activation, in macaques found that this modification increases at enhancer regions and transcription start sites of genes involved in stress responses during aging [57]; activation of stress response pathways is thought to promote longevity [11].
Although the different histone methylation events studied and methodologies used hinder comparison, several trends emerge about histone methylation and aging. First, it is clear that there are age-associated changes in histone methylation, which raises the possibility that alteration of histone methylation contributes to aging pathologies. Second, despite sometimes conflicting data from different model systems that clearly need to be resolved, there is a trend of increases in “activating” histone marks (H3K4me2/3, H3K36me3) and decreases in “repressive” marks (H3K27me3, H3K9me2/3) indicative of a more actively transcribed, euchromatic genome. This is consistent with previous observations of an open chromatin conformation in aging cells and organisms [38]. Intriguingly, given the observation in macaques [57], it is possible that one role of the open chromatin in aged organisms is to promote the transcription of genes that have anti-aging functions, even though such an open chromatin structure has been associated with detrimental increases in gene expression [37,39,40].
3. Direct evidence that histone methylation regulates aging
3.1. Identification of histone methyltransferases and demethylases that impact lifespan
Although the association studies discussed above clearly demonstrate a misregulation of histone methylation during aging, in many cases it remains uncertain whether these changes are a cause or an effect of the aging process. Recent studies in model organisms suggest that histone methylation plays causative roles in aging; these are summarized in Table 2. The first evidence that altered histone methylation drives the aging process came from RNAi screens performed in C. elegans. These identified putative histone methyltransferases and demethylases that regulate worm lifespan, including the methyltransferases ASH-2, SET-9, SET-15, and SET-26, as well as the demethylases MES-2, JMJD-2, LSD-1, and UTX-1 [42,58–60]. LSD-1 had previously been implicated as the major effector of LiCl-induced lifespan extension in worms [61]. Although these nine enzymes, when knocked down or mutated, regulate organismal lifespan in C. elegans, only UTX-1 levels have been shown to change with age [62]; levels of the other enzymes have not been assessed. The functions of ASH-2 and UTX-1 in regulating lifespan in C. elegans have been well characterized and are discussed in detail below. Of the remaining, only SET-9 and SET-26, a closely-related paralog pair, have been further studied. Extension of lifespan by loss of either SET-9 or SET-26 is partially reduced in a daf-16 mutant background, indicating that both proteins act upstream of the FOXO pathway, though not exclusively [42]. Although no changes in histone methylation were observed in set-26 RNAi animals, there is a slight reduction in the age-dependent decrease in total histone H3 levels [42]. It should be noted that whether SET-6, SET-15, MES-2, JMJD-2, and LSD-1 affect the methylation state of histones, as opposed to other proteins, and whether their catalytic activity is required for their effects on aging, has not been determined. While the mechanisms by which all the identified histone methyltransferases and demethylases regulate lifespan are unknown, significant progress has been made in understanding the functions of the canonical activating H3K4me3 and repressing H3K27me3 modifications during aging.
Table 2. Regulation of aging by histone methyltransferases and demethylases.
Compilation of the literature demonstrating that changes in histone methylation regulate lifespan and aging phenotypes. In all cases, the effect the proteins have on aging was assessed by loss of function mutation and/or siRNA-based knockdown. Blank entries represent missing data.
| System | Protein | Effect | Mark | References |
|---|---|---|---|---|
| C. elegans | UTX-1* | increase lifespan | H3K27me3 | [60,62] |
| ASH-2 | increase lifespan | H3K4me3 | [58] | |
| SET-2 | increase lifespan | H3K4me3 | [58] | |
| WDR-5** | increase lifespan | H3K4me3 | [58] | |
| RPD-2 | decrease lifespan | H3K4me3 | [58] | |
| SET-15 | increase lifespan | [59] | ||
| SET-9 | increase lifespan | none found | [42,59] | |
| SET-26 | increase lifespan | none found | [42] | |
| LSD-1 | increase lifespan | [60,61] | ||
| MES-2 | increase lifespan | [42] | ||
| JMJD-2 | increase lifespan | [42] | ||
| T26A5 | increase lifespan | [60] | ||
| Drosophila | E(z) | increase lifespan | [64] | |
| Esc | increase lifespan | [64] | ||
| Mammals | G9a* | inhibit SASP | [117] | |
| GLP* | inhibit SASP | [117] |
indicates that expression of this enzyme and histone modification are altered during aging; the levels of others have not been assessed.
signifies that WDR-5 is not a histone methyltransferase or demethylase; instead, this enzyme forms a complex with ASH-2 and SET-2 methyltransferases. SASP, senescence-associated secretory phenotype.
3.2. High levels of H3K4 trimethylation reduce lifespan in C. elegans
In eukaryotes, trimethylation of H3K4, a classic mark of transcriptionally active chromatin, is catalyzed by the Trithorax complex. In C. elegans, loss of any of three Trithorax proteins (WDR-5 and the methyltransferases SET-2 and ASH-2) decreases global levels of H3K4me3 and increases lifespan [58]. Similarly, loss of RBR-2, which demethylates H3K4me3, decreases lifespan and increases H3K4me3 levels in C. elegans. Significantly, loss of ASH-2 in rbr-2 mutant animals restores normal levels of H3K4me3 and rescues the observed decrease in lifespan, suggesting that high levels of H3K4me3 are detrimental to lifespan in worms [58]. Although it is unknown how increased H3K4me3 decreases longevity, production of mature eggs during adult life is required for loss of ASH-2 to increase lifespan, which led to the idea that ASH-2 may regulate endocrine signaling from germ cells to soma [58]. Further work showed that the lifespan increase caused by loss of ASH-2, SET-2, or WDR-5 is inherited transgenerationally; that is, that wildtype F3 and F4 descendants of outcrossed ash-2, set-2, or wdr-5 mutants retained increased longevity, though this phenotype was lost one generation later [63]. This increase in lifespan correlates with global differences in transcriptional profiles, though not with global alterations in H3K4me3 levels; indeed, many of the differentially-expressed genes are linked with longevity [63]. However, the H3K4 methylation status of genes with altered expression levels in mutants and their wildtype descendants versus wildtype worms was not specifically analyzed, and, therefore, many of these effects may be indirect. Nevertheless, this study demonstrates that alterations in histone methylation can affect lifespan transgenerationally, though the mechanism by which this occurs remains unclear.
3.3. Lifespan regulation by the repressive H3K27me3 modification
Aging is also regulated at the level of transcription by trimethylation of H3K27. The H3K27me3 demethylase UTX-1 plays a central role in aging in C. elegans through its regulation of the insulin and insulin-like signaling (IIS) pathway. An increase in utx-1 expression in aging worms correlates with a global reduction of H3K27me3 levels and is required for a loss of this mark at the daf-2 locus, which encodes the insulin receptor, causing an age-dependent increase in daf-2 expression [60,62]. Consistent with this, utx-1 knockdown results in a 30% increase in lifespan, and loss of DAF-16 during utx-1 RNAi abrogates the increase in lifespan this treatment incurs. As DAF-16 is required for the increase in lifespan upon loss of DAF-2, this suggests that the primary function of UTX-1 in the context of aging is in regulation of IIS [62]. Intriguingly, knockdown of the UTX-1 ortholog KDM6A in human cells also results in decreased expression of an insulin receptor, IGF1R, and this locus has a decrease in H3K27me3 in aged tissues from both humans and macaques. Thus, it is possible that an increase in IIS mediated by the activity of the H3K27me3 demethylase KDM6A at the insulin receptor locus is a conserved mechanism that promotes aging in animals [62].
Changes in H3K27me3 levels have also been shown to affect lifespan in Drosophila. Mutation of the Polycomb repressive complex 2 (PRC2) components E(z) and Ese both decreases total H3K27me3 and extends lifespan [64]. Furthermore, hypomorphic alleles of trx, which encodes a H3K4 methyltransferase in the Trithorax complex, introduced into E(z) and ese heterozygotes partially reduced lifespan while increasing levels of H3K27me3 [64]. This later phenotype is thought to be due to decreased recruitment of the H3K27 acetyltransferase CBP, as methylation and acetylation on this residue are mutually exclusive [65]. However, Siebold et al. [64] noted that a previous ChIP screen had revealed that E(z) is associated with the Odc1 gene, which encodes an ornithine decarboxylase [66], and thus suggest that PRC2-mediated deposition of H3K27me3 functions to repress expression of genes that promote stress resistance. Indeed, they found that heterozygotes for both E(z) and esc had increased resistance to oxidative stress than did wildtype flies [64].
Additionally, changes in H3K27me3 have been linked to loss of function in mammalian mesenchymal stem cells and muscle satellite cells. In human adipose-derived mesenchymal stem cells, promoters of genes that drive adipogenesis and myogenesis are marked by H3K4me3 and H3K27me3, characteristic of bivalent domains, in both early and late passage cells (young and old, respectively). In young cells, induction of adipocyte differentiation causes a loss of H3K27me3 at promoters of genes involved in this process, while H3K27me3 remains associated with these genes in old cells. The retention of H3K27me3 at these loci is correlated with transcriptional downregulation, and thus inhibition of the adipogenic differentiation program in old cells. This may be caused by an increase in EZH2, which deposits this mark, and increased association of both EZH2 and BMI1 (a PRC1 component) at these loci [67]. In contrast, histone methylation is altered prior to induction of differentiation in muscle satellite (stem) cells in old versus young mice. In old mice, an increase in transcription start site-associated H3K27me3 is observed, though a significant portion (30%) of genes that gain this mark are not expressed in muscle satellite cells or their differentiated progeny. H3K27me3 also accumulates in gene poor regions, suggesting a general accumulation heterochromatin and transcriptional silencing with age [18]. Thus, in both muscle and mesenchymal stem cells, aging is associated with increased levels of H3K27me3 and transcriptional repression.
4. Histone methylation is involved in pathways that regulate aging and lifespan
Although only a few studies show that altering histone methylation regulates lifespan, there is accumulating evidence that histone methylation impinges upon pathways known to affect aging in eukaryotes. In this section, we discuss how histone methylation regulates, and is regulated by, these processes; a summary of the literature is provided in Table 3. Most of the work in this area has focused on how histone methylation impinges upon the cognate process, rather than the effects altered histone methylation have on lifespan. However, two recent studies have identified stress-related changes in histone methylation that affect lifespan and aging phenotypes. Thus, it is possible, and indeed likely, that understanding the cross-regulation between stress response pathways and histone methylation will provide insight into the aging process.
Table 3. Histone methylation contributes to pathways that drive aging.
Recent work in yeast and cultured mammalian cells that implicates changes in histone methylation in cellular processes that drive aging are summarized.
| Pathway | Modification | Change | Enzyme | Effect | References |
|---|---|---|---|---|---|
| Autophagy | H3K9me3 | decrease | G9a, GLP | increase transcription of genes involved in autophagy | [69] |
| DDR | H4K20me2 | increase | MMSET | recruit 53BP to DNA lesions | [80] |
| H3K79me2 | increase | Dot-1 | recruit 53BP to DNA lesions | [77–79] | |
| H3K9me3 | increase | enhance Tip60 activity, which activates ATM | [85] | ||
| H3K4me3 | increase | Set-1 | [89] | ||
| H3K4me3 | increase | recruit tumor suppressor ING | [90] | ||
| H3K27me3 | increase | EZH2 | repress transcription near DNA lesions | [84] | |
| Stress response | H3K4me3 | Set-1 | repress ribosome protein and biogenesis genes | [96] | |
| H3K36me3 | decrease | Rph-1 | activate stress response genes | [97] | |
| H3K36me3 | decrease | Rph-1 | repress sub-telomeric gene expression during ROS-induced hormesis | [99] |
indicates that this occurs in yeast, and also in human cells during growth phases of the cell cycle. DDR, DNA damage response.
4.1. Autophagy
Autophagy is the process by which damaged molecules, macromolecules, and cellular components are degraded into basal metabolites that are reused by the cell to make new ones. A double membrane is formed in the cytoplasm and cellular components are engulfed in this structure, either stochastically or in a directed manner. The autophagosome then fuses with a lysosome, wherein its components are digested. This process is tightly regulated; in particular, the initial fusion of the membrane to form an autophagosome requires the action of the proteins LCB3, DOR, and WIPI1 [68]. During aging, the efficiency of autophagy is decreased, promoting the accumulation of damaged macromolecules and organelles, which contributes to various aging phenotypes. The decrease in autophagy has been linked to an increase in the proportion of damaged mitochondria, and thus to increased ROS, as well as the accumulation of insoluble protein plaques in Alzheimer's and Huntington's diseases. Indeed, abrogating the age-associated decline in autophagy increases longevity [9].
Expression of LCB3, DOR, and WIPI1 are regulated at the level of histone methylation in human cell lines [69]. The histone methyltransferase G9a, which mono- and dimethylates lysine 9 on histone H3 (H3K9me1/2), associates with the promoters of these genes and represses their expression. Significantly, induction of autophagy by starvation causes G9a to dissociate from the promoters of these genes, concomitant with a reduction in H3K9 methylation and increased gene expression. Inhibition or knockdown of G9a in cultured cells also promotes formation of autophagosomes, which further suggests that regulation of LCB3, DOR, and WIPI1, among others, at the level of transcription by histone methylation is a key mechanism by which autophagy is controlled [69]. It is therefore possible that age-associated changes in histone methylation patterns may regulate cellular pathways that contribute to aging by altering the transcription of genes whose products are necessary parts of the pathway. Interestingly, in human HGPS cells and aged Drosophila, H3K9 methylation is reduced [45,49]. Thus, it is possible that particular loci become hypermethylated at H3K9 during aging, despite a global decrease in H3K9 methylation.
4.2. DNA damage response
As organisms age, their cells accumulate DNA damage, both in the form of mutations from improperly repaired DNA and from irreparable lesions, particularly double stranded DNA breaks (DSBs), that continuously elicit the DNA damage response (DDR). Indeed, γH2A.X foci, which mark DSBs, are more prevalent in old cells and organisms than they are in young [70]. The DNA damage could be caused by an age-dependent decrease in a cell's ability to effect repairs; likewise, continued activation of the DDR pathway may contribute to aging pathologies [71]. In non-homologous end joining (NHEJ), DSBs induce the local phosphorylation of H2A.X, causing formation of γH2A.X. γH2A.X recruits Mcd1, which, in turn, promotes the association of the MRN complex with DSBs. MRN serves as a platform for the assembly for ATM kinase and mediator proteins, including 53BP1, which helps activate the DDR pathway downstream of ATM [72,73]. ATM phosphorylates many substrates, including Chk1 and Chk2, which mediate cell cycle arrest and activate damage repair proteins or induce apoptosis, respectively [74]. In alternate NHEJ, PARP1 is recruited to DSBs, where it catalyzes the formation of poly-ADP ribose chains, which recruit various enzymes required for DNA repair [75]. Histone methylation is both required for the recruitment and activation of DDR components and is altered in response to DNA damage (discussed below), suggesting that histone methylation may both regulate efficacy of the DDR and, potentially, mediate its effects on aging.
H4K20me2 or H3K79me2 is required for the recruitment of 53BP1 to DNA lesions in human cells and yeast [76–79]. In cultured cells, the methyltransferase MMSET is recruited to DSBs by MDC1 where it causes a local increase in H4K20me2, thus permitting binding of 53BP1 [80]. However, during the growth phases of the cell cycle, H4K20me1/2 levels are low in mammals [81], and H3K79me2 mediates recruitment of 53BP1 to DSBs, though how prevalent this mechanism is varies between cell types [79]. Indeed, H3K79 methylation is not required for 53BP1 recruitment using a chicken system [82], suggesting divergence in the role of histone methylation in DNA damage responses among eukaryotes. In budding yeast, in which H4K20me1 was only recently confirmed to exist and regulates telomere silencing [83], recruitment of Rad9, the 53BP1 ortholog, to DSBs occurs through interaction with H3K79me2 [78]. In mammalian cells and yeast, the local increase in H3K79me2 is mediated by the methyltranferase DOT1L (Dot1 in yeast); this enzyme is required for repair of DSBs [77,78]. Thus, methylation of H4K20 and H3K79 are critical for the initiation of the DNA damage response, and any changes in the activities of enzymes regulating these marks may contribute to reduced DDR and thus accelerate the aging process.
H3K9me3, a classic constitutive heterochromatin mark, is also required early during DDR activation in cultured human and mouse cells. H3K9me3 levels increase at the site of DNA damage, though global levels remain unchanged [84,85]. H3K9me3 accessibility may also be regulated during the DDR, as in the context of normal cells, members of the HP1 family of proteins associate with this mark [51,52]. Upon DNA damage, HP1β is phosphorylated, causing it to dissociate from H3K9me3, exposing H3K9me3 in the vicinity of DSBs [85]. The acetyltransferase Tip60 is then able to bind H3K9me3, which enhances its enzymatic activity [85-87]. When DNA damage occurs, Tip60 acetylates ATM kinase, thus further activating it to promote the DDR [85]. It has also recently been shown that a global reduction in H3K9me3 levels promotes the resolution of DSB repair, consistent with the idea that a more open chromatin structure enhances DNA accessibility to repair factors [88].
Interestingly, both H3K4me3 and H3K27me3 are associated with DSBs. In yeast, H3K4me3 is deposited by Set1, which is recruited to DSBs and is essential for NHEJ in the absence of the yeast MRN ortholog [89]. In cultured cells, H3K4me3 is required to recruit the tumor suppressor ING to DSBs [90], which mediates cell cycle arrest until repair is accomplished or senescence induced [91]. The methyltransferase EZH2, which trimethylates H3K27, is also recruited to DSBs, where it is thought to inhibit transcription in the vicinity of DNA damage [84]. Studies in cultured cells and C. elegans confirmed that knockdown or knockout of EZH2 sensitizes cells and worms to DNA damage [92]. It is further demonstrated that the EZH2 is recruited to DSBs independently of γH2A.X by PARP1 and that it functions to repress transcription in the vicinity of DNA damage [92], consistent with the idea that transcription must be repressed in the presence of DNA lesions [93].
4.3. Environmental stress responses
Cells must respond to numerous exogenous and endogenous stresses, including ROS, heat, osmotic imbalances, unfolded proteins, and damaged organelles [94]. As discussed above, many of these stresses increase during aging, and stress response pathways tend to be activated in aged organisms. Consistent with this, activating stress response pathways promotes longevity [26]. Indeed, hormesis, in which low levels of exposure to a particular stressor extends lifespan and confers resistance to more extreme levels of stress [95], may very well utilize epigenetic changes as a form of cellular memory. Recent work in yeast has shown that altered histone methylation is critical for stress responses, during both acute stress responses and hormesis. Thus, regulation of stress response genes is a powerful mechanism by which histone methylation my impact aging.
Recent work in yeast has suggested that chromatin modifications regulate the dynamics of transcriptional changes in response to oxidative stress [96]. In a high throughput screen, H3K4me3 was found to correlate with repression of ribosomal protein and biogenesis genes under stress conditions. Deletion of SET1, which encodes the H3K4 methyltransferase, results in upregulation of ribosomal protein and biogenesis genes in cells exposed to oxidative stress, heat shock, or rapamycin treatment, further confirming this result [96]. In the case of the ribosome biogenesis genes only, deletion of members of the RPD3L histone deacetylase complex causes transcriptional upregulation under oxidative stress conditions. It is suggested that H3K4me3 recruits the RPD3L complex to ribosome biogenesis genes to silence their expression during stress responses, and, further, that a distinct mechanism must be responsible for repression of the ribosomal protein genes [96]. Changes in H3K4me3 levels (discussed in Section 2) or distribution (see Section 4.4.3) during aging may therefore affect lifespan by altering the stress response.
Also in yeast, the H3K36me3 demethylase Rph1 is a critical player in the environmental stress response [97]. Under normal conditions, Rph1 is associated with the promoters of stress response genes, where it functions to repress transcription. However, in response to DNA damage and oxidative stress, the kinase Rad53 (ortholog of mammalian CHK2) phosphorylates Rph1, causing it to dissociate from these promoters, thus increasing transcription of target genes, possibly by the stress response transcription factor Msn2/4 [97]. Approximately 26% of genes upregulated by loss of Rph1 under oxidative stress conditions have increased H3K36me3 levels at their promoters, which suggests that at least part of Rph1's repression of stress response genes is mediated by its demethylase activity. Interestingly, the Rph1 DNA binding site is substantially similar to that of Msn2/4, raising the possibility that this demethylase may also function to repress stress responses under normal conditions by excluding transcriptional activators from their promoters [97]. This mechanism (exclusion of other proteins) may be used by histone methyltransferase and demethylases in other contexts, and, indeed, could explain how SET-9 and SET-26 affect lifespan in C. elegans without altering histone methylation [42].
Rph1 has also been implicated in extending chronological lifespan in yeast during hormetic adaptation to low levels of oxidative stress. Transiently increasing mitochondrial ROS production has been shown to promote longevity in yeast [98], and the H3K36me3 demethylase Rph1 is required for this process [99]. As in acute oxidative stress, when yeast are subject to low levels of oxidative stress, Rph1 is phosphorylated by Rad53, causing its dissociation from DNA. However, during hormetic (low level) oxidative stress, Rad53 is activated in the absence of DNA damage by the Tel1 (ATM) kinase [99]. In this context, Rad53 activation causes dissociation of Rph1 from sub-telomeric chromosomal regions, concomitant with an increase in H3K36me3 at these loci. Unlike during acute stress response, H3K36me3 accumulation functions to repress transcription by enhancing the binding of silencing protein Sir3 to these loci. It is suggested that the Rph1 demethylase functions to transmit a signal from stressed mitochondria to the nucleus during oxidative hormesis [99]. Thus, altered histone methylation may be a common mechanism by which hormesis functions to regulate lifespan.
4.4. Cellular senescence in mammals
As mammals age, their cells undergo a process termed cellular senescence, which is characterized by irreversible cell cycle exit and the secretion of various pro-inflamatory cytokines (the senescence-associated secretory phenotype, SASP) [100,101]. Both aspects of senescence contribute to aging phenotypes, and both are thought to be anti-cancer adaptations. Although the SASP can affect most cells, a loss of proliferation (canonical cellular senescence) has a particularly pronounced effect on stem cell function. When stem cells become senescent, they are no longer able to divide, and thus tissue regeneration and homeostasis are lessened, which contributes to tissue degeneration and immune deficiencies [102]. Additionally, cytokines secreted by senescent cells contribute to chronic inflammation and, paradoxically, may even promote cancer [101]. Both cell cycle arrest and the SASP are regulated by histone methylation.
4.4.1. Transcriptional control of the cell cycle
Canonically, cell cycle arrest is mediated by transcription of the INK4/ARF locus, which encodes the p15INK4b, ARF, and p16INK4a tumor suppressor genes [13]. Expression of the INK4/ARF locus is increased in aged mammalian tissues and senescent cultured cells [13] and was recently shown to be upregulated in old mice [103]. In addition to regulation by histone acetylation [104,105], this locus is subject to Polycomb-mediated repression in young cells, which is relieved in old cells, as seen by a loss of H3K27me3 at the promoter [106–109]. The alteration of H3K27me3 levels is mediated by a decrease in expression of the histone methyltransferase EZH2 and increased JMJD3 demethylase expression [106–109].
The complexity of transcriptional control of cell cycle arrest during senescence is only beginning to be appreciated. Polycomb-mediated repression of INK4/ARF expression is regulated by both signaling pathways and histone ubiquitination. In aged mouse pancreas, there is a reduction in PTEN, which inhibits the ATK/PI3K pathway; this correlates with an increase in EZH2 and BMI1 expression and a decrease in p16INK4a expression. Likewise, in MEFs, loss of PTEN causes an increase in EZH2 and H3K27me3 at the INK4/ARF locus [110]. In human cells, expression of JMJD3, which encodes an H3K27me3 demethylase, is activated by ERK signaling and the transcription factor AP1 [111]. Thus, repression of INK4/ARF is regulated by the opposing activities of ATK and ERK signaling. The displacement of PRC1 and 2 from the INK4/ARF locus, and thus transcriptional activation of this locus, is further regulated by ZRF1, which binds monoubiquitinated lysine 119 of histone H2A (H2AK119ub), in human and mouse cell lines [112]. Furthermore, despite the predominance of this locus in studies on cell cycle arrest, the loss of H3K4me3 at additional loci required for cell cycle progression, mediated by JARID1B and Rb, has been implicated in the loss of cell proliferation [113]. Additionally, though the mechanism is unknown, a decrease in H3K79me3 is seen during aging of human cell cultures, and loss of the H3K79 methyltransferase DOT1L can induce senescence in this system [114]. Histone methylation is thus a highly regulated and important regulator of cellular senescence in mammals.
4.4.2. Broad redistribution of H3K4me3 and H3K27me3 in senescent cells
Numerous studies have assessed the status of H3K4me3 and H3K27me3 at specific cell cycle-related loci in cultured mammalian cells. However, until recently, the genome-wide distribution of these marks has not been examined during senescence. Shah et al. [115] discovered that the distribution of these marks is drastically altered in senescent vs. proliferating human cells. Particularly, broad regions of the genome are enriched for each of these marks specifically in senescent cells (termed mesas), and the H3K4me3 and H3K27me3 mesas significantly overlap. These occur mainly in gene-poor regions that are highly associated with Lamin B1 in proliferating cells. In addition distinct genomic regions are found depleted for H3K27me3 (canyons), which are much less frequently associated with Lamin B1 binding domains. Instead, the H3K27me3 canyons are enriched for gene bodies and enhancers; indeed, a number of genes upregulated during senescence, including many of those in the senescence-associated secretory phenotype (SASP), are located within these canyons. It is further discovered that in HGPS, in which Lamin B1 is mutated, the senescence-associated H3K27me3 canyons are present even in proliferating cells. These results suggest that at least some of the changes in histone methylation observed during human aging result from previous alterations in nuclear architecture [115].
4.4.3. SASP is regulated by changes in histone methylation induced by DNA damage
Unrepaired double stranded DNA breaks, a hallmark of old cells, constantly activate the DNA damage response [116]. One effect of this is the activation of the APC/C ubiquitin ligase by Cdh1. APC/C targets the histone methyltransferases G9a and GLP, which catalyze deposition of H3K9me1/2, for proteosome-mediated degradation [117]. In non-senescent human cells, G9a and GLP are associated with the promoters of IL-6 and IL-8/Gro-α, two cytokines upregulated as part of the SASP. In senescing cells with unrepaired DNA lesions, both G9a and GLP are reduced at the IL-6 and IL-8 promoters, which correlates with a decrease in repressive H3K9me1/2 at these loci and transcriptional upregulation of the genes. It is further showed that reduction of G9a/GLP and an increase in IL-6 and IL-8/Gro-α expression occur during DNA damage-induced senescence in skin papillomas and in tissues from aged mice that accumulate DNA damage. This suggests that the reduction of H3K9me1/2 by DNA damage-induced G9a/GLP degradation contributes to the physiology of aging [117], and shows a clear link between aging pathologies, DNA damage, and histone methylation.
5. Concluding remarks
The role of histone methylation in the regulation of lifespan and aging phenotypes is just beginning to be studied. While there is ample evidence for global changes in levels of histone methylation during aging, and even for a genome-wide redistribution of particular methylated histones (summarized in Table 1), the mechanisms by which these are achieved and by which they impact aging are unknown. Similarly, while loss of particular histone demethylases and methyltransferases promote longevity in invertebrate model systems (see Table 2), in most cases, the processes affected by a reduction in these enzymes are unknown. For example, knockdown of UTX-1 in C. elegans, which likely globally affects H3K27me3 levels, mediates lifespan extension primarily through its effects at the insulin receptor locus [60,62]. As the field progresses, it will be critical to determine 1) which loci are subject to altered histone methylation during aging, 2) whether and how the change in histone methylation at a particular locus affects lifespan and aging phenotypes, and 3) how altered histone methylation is accomplished, i.e., by changes in levels and activities of histone methyltransferases/demethylases or redistribution of these enzymes along the chromatin and what drives these processes.
The available evidence strongly implicates histone methylation in the transcriptional regulation of both longevity and pathways that contribute to aging phenotypes. In addition to changes in histone methylation, both globally and at specific loci, during the aging process, the loss of several histone methyltransferases and demethylases increases lifespan in C. elegans. As the molecular pathways that underlie aging pathologies are highly conserved, it is thus likely that histone methylation also regulates lifespan in other organisms. Furthermore, expression of genes involved in insulin and insulin-like signaling (IIS); the senescence-associated secretory phenotype (SASP); tumor suppression; autophagy; and, particularly, stress response are regulated by histone methylation (Figure 1A); thus, histone methylation must regulate the aging process, at least insofar as these pathways contribute to it. Indeed, as these processes, and probably many others, are regulated at the level of histone methylation, any changes in histone methylation may result in their misregulation, as is beautifully exemplified by the increase in SASP gene expression induced by chromatin changes that result from chronic DNA damage [117]. Furthermore, as repair of DNA damage requires novel histone methylation, the observed age-associated alterations in histone methylation may affect the ability of aged cells and organisms to execute the DDR (Figure 1B). Thus histone methylation, and alteration thereof, is likely to impact many different facets of the aging process.
Figure 1. Impact of differential histone methylation on aging processes.
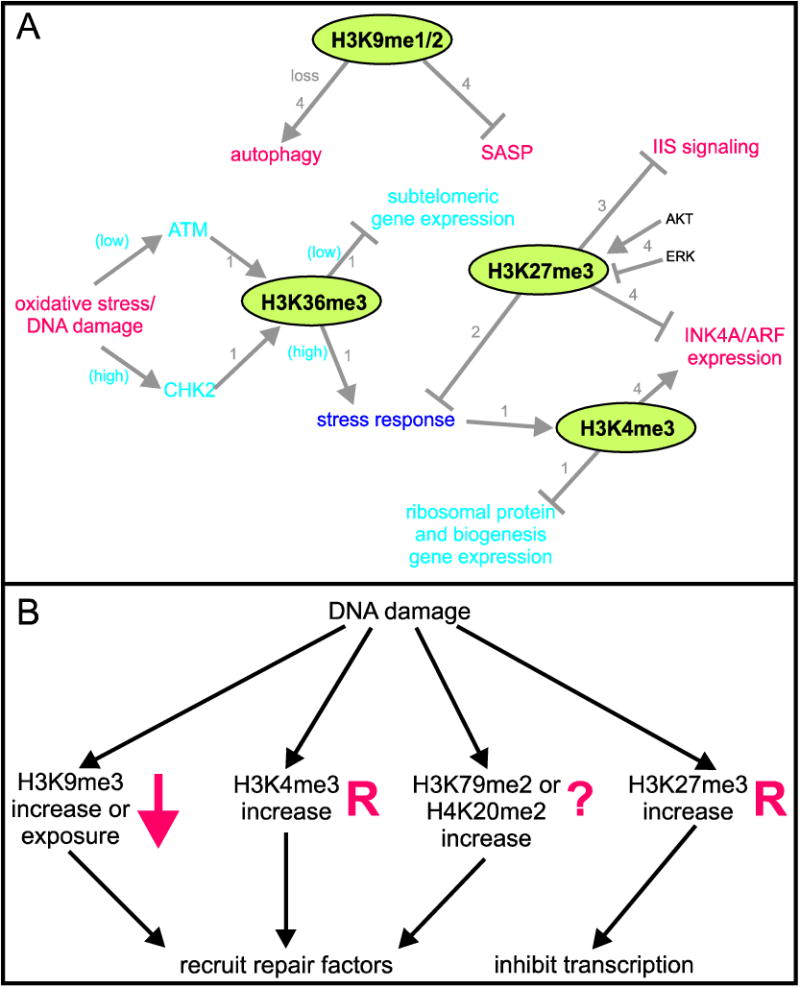
(A) A depiction of how histone methylation regulates and is regulated by major pathways that affect aging. Histone modifications are circled, and aging processes shown in large text. Red indicates a process that promotes or is caused by aging, while purple text highlights processes that promote longevity. Arrows indicate positive regulation (promotion) and lines ending in bars signify inhibition of the terminal process. Yeast-specific responses are in blue text. Numbers next to lines indicate from what system this data was derived. 1) yeast, 2) Drosophila, 3) C. elegans, 4) mammals. The diagram is based on data from references [60,62,64,96,97,99,106-109,117]. (B) Histone methylation necessary for DNA damage repair may be altered during aging. Numerous chromatin changes occur following DNA damage that promote repair of lesions, as indicated; several of these modifications are known to be altered during aging, highlighted in red. Down arrow, decrease; R, significant redistribution; ?, uncharacterized, though different methylation events at the same residue are known to be altered, e.g., H4K20me3 increases. Supporting data from references [45,115].
Nevertheless, histone methylation is potentially a powerful target for treating, slowing, or reversing age-associated physiological changes. Like other epigenetic modifications, histone methylation is reversible, and thus by altering the activity of enzyme that regulate these modifications it may be possible to revert chromatin in aged patients to a younger state. As at least IIS and the SASP are implicated as being direct controlled by chromatin state, it is theoretically possible to disrupt these aspects of aging by potentiating or inactivating histone methyltransferase/demethylase activity. Indeed, numerous small molecule inhibitors of histone deacetylases (HDACi) have been identified, and several have proven effective in improving the efficacy of cancer treatment regimens [118,119]. Thus, in principles, regulating the function of chromatin modifying enzymes may be a powerful means to combat aging and other diseases to which epigenetic dysregulation contributes. In particular, recent advances in the development of small molecule inhibitors of methyltransferases and demethylases have yielded compounds that inhibit the function of specific enzymes without significant cytotoxicity [120]. Thus, as additional histone methyltransferase and demethylase inhibitors are discovered, and the roles of histone methylation in the aging process are elucidated, it may be possible to develop novel treatments to delay or prevent the onset of age-associated pathologies.
Highlights.
Review role of histone methylation in aging of various model systems
Levels and distribution of histone methylation change during aging
Altered histone methylation may impact repair of DNA damage
Histone methylation transcriptionally regulates many aging-related processes
Role of histone methylation in aging is only beginning to be understood
Acknowledgments
The authors would like to thank two anonymous reviewers for helpful comments and critiques. This work was supported by NIH grant R00AG037646. W.D. is a CPRIT scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The Hallmarks of Aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rajawat YS, Hilioti Z, Bossis I. Aging: Central role for autophagy and the lysosomal degradative system. Ageing Res Rev. 2009;8:199–213. doi: 10.1016/j.arr.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Seo AY, Joseph AM, Dutta D, Hwang JCY, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010;123:2533–2542. doi: 10.1242/jcs.070490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freitas AA, de Magalhães JP. A review and appraisal of the DNA damage theory of ageing. Mutat Res Mutat Res. 2011;728:12–22. doi: 10.1016/j.mrrev.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Löw P. The role of ubiquitin–proteasome system in ageing. Gen Comp Endocrinol. 2011;172:39–43. doi: 10.1016/j.ygcen.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Back P, Braeckman BP, Matthijssens F. ROS in Aging Caenorhabditis elegans: Damage or Signaling? Oxid Med Cell Longev. 2012;2012 doi: 10.1155/2012/608478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lagouge M, Larsson NG. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J Intern Med. 2013;273:529–543. doi: 10.1111/joim.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegde ML, Mantha AK, Hazra TK, Bhakat KK, Mitra S, Szczesny B. Oxidative genome damage and its repair: Implications in aging and neurodegenerative diseases. Mech Ageing Dev. 2012;133:157–168. doi: 10.1016/j.mad.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lionaki E, Markaki M, Tavernarakis N. Autophagy and ageing: Insights from invertebrate model organisms. Ageing Res Rev. 2013;12:413–428. doi: 10.1016/j.arr.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Hegde AN, Upadhya SC. Role of ubiquitin–proteasome-mediated proteolysis in nervous system disease. Biochim Biophys Acta BBA - Gene Regul Mech. 2011;1809:128–140. doi: 10.1016/j.bbagrm.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kourtis N, Tavernarakis N. Cellular stress response pathways and ageing: intricate molecular relationships. EMBO J. 2011;30:2520–2531. doi: 10.1038/emboj.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008;20:150–155. doi: 10.1016/j.ceb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 13.Gil J, Peters G. Regulation of the INK4b–ARF–INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–677. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- 14.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 15.Conboy IM, Rando TA. Aging, Stem Cells and Tissue Regeneration: Lessons from Muscle. Cell Cycle. 2005;4:407–410. doi: 10.4161/cc.4.3.1518. [DOI] [PubMed] [Google Scholar]
- 16.Morrison SJ, Uchida N, Weissman IL. The Biology of Hematopoietic Stem Cells, Annu. Rev. Cell Dev Biol. 1995;11:35–71. doi: 10.1146/annurev.cb.11.110195.000343. [DOI] [PubMed] [Google Scholar]
- 17.Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. Unique and Independent Roles for MLL in Adult Hematopoietic Stem Cells and Progenitors. Cell Stem Cell. 2007;1:324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L, Cheung TH, Charville GW, Hurgo BMC, Leavitt T, Shih J, et al. Chromatin Modifications as Determinants of Muscle Stem Cell Quiescence and Chronological Aging. Cell Rep. 2013;4:189–204. doi: 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maslov AY, Barone TA, Plunkett RJ, Pruitt SC. Neural Stem Cell Detection, Characterization, and Age-Related Changes in the Subventricular Zone of Mice. J Neurosci. 2004;24:1726–1733. doi: 10.1523/JNEUROSCI.4608-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMahon KA, Hiew SYL, Hadjur S, Veiga-Fernandes H, Menzel U, Price AJ, et al. Mll Has a Critical Role in Fetal and Adult Hematopoietic Stem Cell Self-Renewal. Cell Stem Cell. 2007;1:338–345. doi: 10.1016/j.stem.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahlenius H, Visan V, Kokaia M, Lindvall O, Kokaia Z. Neural Stem and Progenitor Cells Retain Their Potential for Proliferation and Differentiation into Functional Neurons Despite Lower Number in Aged Brain. J Neurosci. 2009;29:4408–4419. doi: 10.1523/JNEUROSCI.6003-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinkraus KA, Kaeberlein M, Kennedy BK. Replicative Aging in Yeast: The Means to the End. Annu Rev Cell Dev Biol. 2008;24:29–54. doi: 10.1146/annurev.cellbio.23.090506.123509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, et al. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- 25.Grotewiel MS, Martin I, Bhandari P, Cook-Wiens E. Functional senescence in Drosophila melanogaster. Ageing Res Rev. 2005;4:372–397. doi: 10.1016/j.arr.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 27.Masoro EJ. Overview of caloric restriction and ageing. Mech Ageing Dev. 2005;126:913–922. doi: 10.1016/j.mad.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 28.Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an Insulin Receptor-Like Gene That Regulates Longevity and Diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 29.Fontana L, Partridge L, Longo VD. Extending Healthy Life Span—From Yeast to Humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhan M, Yamaza H, Sun Y, Sinclair J, Li H, Zou S. Temporal and spatial transcriptional profiles of aging in Drosophila melanogaster. Genome Res. 2007;17:1236–1243. doi: 10.1101/gr.6216607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wood SH, Craig T, Li Y, Merry B, de Magalhães JP. Whole transcriptome sequencing of the aging rat brain reveals dynamic RNA changes in the dark matter of the genome. AGE. 2013;35:763–776. doi: 10.1007/s11357-012-9410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dierick JF, Pascal T, Chainiaux F, Eliaers F, Remacle J, Larsen PM, et al. Transcriptome and Proteome Analysis in Human Senescent Fibroblasts and Fibroblasts Undergoing Premature Senescence Induced by Repeated Sublethal Stresses. Ann N Y Acad Sci. 2000;908:302–305. doi: 10.1111/j.1749-6632.2000.tb06659.x. [DOI] [PubMed] [Google Scholar]
- 33.Lesur I, Campbell JL. The transcriptome of prematurely aging yeast cells is similar to that of telomerase-deficient cells. Mol Biol Cell. 2004;15:1297–1312. doi: 10.1091/mbc.E03-10-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato M, Chen X, Inukai S, Zhao H, Slack FJ. Age-associated changes in expression of small, noncoding RNAs, including microRNAs, in C. elegans. RNA. 2011;17:1804–1820. doi: 10.1261/rna.2714411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruzanov P, Riddle DL. Deep SAGE analysis of the Caenorhabditis elegans transcriptome. Nucleic Acids Res. 2010;38:3252–3262. doi: 10.1093/nar/gkq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feser J, Tyler J. Chromatin structure as a mediator of aging. FEBS Lett. 2011;585:2041–2048. doi: 10.1016/j.febslet.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459:802–807. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsurumi A, Li WX. Global heterochromatin loss: A unifying theory of aging? Epigenetics. 2012;7:680–688. doi: 10.4161/epi.20540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawahara TLA, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 Links Histone H3 Lysine 9 Deacetylation to NF-κB-Dependent Gene Expression and Organismal Life Span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee S, Jung JW, Park SB, Roh K, Lee SY, Kim JH, et al. Histone deacetylase regulates high mobility group A2-targeting microRNAs in human cord blood-derived multipotent stem cell aging. Cell Mol Life Sci. 2011;68:325–336. doi: 10.1007/s00018-010-0457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, et al. Elevated Histone Expression Promotes Life Span Extension. Mol Cell. 2010;39:724–735. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ni Z, Ebata A, Alipanahiramandi E, Lee SS. Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell. 2012;11:315–325. doi: 10.1111/j.1474-9726.2011.00785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Djuric U, Ellis J. Epigenetics of induced pluripotency, the seven-headed dragon. Stem Cell Res Ther. 2010;1:3. doi: 10.1186/scrt3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mattout A, Biran A, Meshorer E. Global epigenetic changes during somatic cell reprogramming to iPS cells. J Mol Cell Biol. 2011;3:341–350. doi: 10.1093/jmcb/mjr028. [DOI] [PubMed] [Google Scholar]
- 45.Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci. 2006;103:8703–8708. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, et al. Correlated alterations in genome organization, histone methylation, and DNA–lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013;23:260–269. doi: 10.1101/gr.138032.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brandt A, Krohne G, Großhans J. The farnesylated nuclear proteins KUGELKERN and LAMIN B promote aging-like phenotypes in Drosophila flies. Aging Cell. 2008;7:541–551. doi: 10.1111/j.1474-9726.2008.00406.x. [DOI] [PubMed] [Google Scholar]
- 48.Haithcock E, Dayani Y, Neufeld E, Zahand AJ, Feinstein N, Mattout A, et al. Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2005;102:16690–16695. doi: 10.1073/pnas.0506955102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larson K, Yan SJ, Tsurumi A, Liu J, Zhou J, Gaur K, et al. Heterochromatin Formation Promotes Longevity and Represses Ribosomal RNA Synthesis. PLoS Genet. 2012;8:e1002473. doi: 10.1371/journal.pgen.1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wood JG, Hillenmeyer S, Lawrence C, Chang C, Hosier S, Lightfoot W, et al. Chromatin remodeling in the aging genome of Drosophila. Aging Cell. 2010;9:971–978. doi: 10.1111/j.1474-9726.2010.00624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 52.Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 53.Miyamoto M, Kiyota Y, Yamazaki N, Nagaoka A, Matsuo T, Nagawa Y, et al. Age-related changes in learning and memory in the senescence-accelerated mouse (SAM) Physiol Behav. 1986;38:399–406. doi: 10.1016/0031-9384(86)90112-5. [DOI] [PubMed] [Google Scholar]
- 54.Takeda T, Hosokawa M, Takeshita S, Irino M, Higuchi K, Matsushita T, et al. A new murine model of accelerated senescence. Mech Ageing Dev. 1981;17:183–194. doi: 10.1016/0047-6374(81)90084-1. [DOI] [PubMed] [Google Scholar]
- 55.Wang CM, Tsai SN, Yew TW, Kwan YW, Ngai SM. Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology. 2010;11:87–102. doi: 10.1007/s10522-009-9231-5. [DOI] [PubMed] [Google Scholar]
- 56.Sarg B, Koutzamani E, Helliger W, Rundquist I, Lindner HH. Postsynthetic Trimethylation of Histone H4 at Lysine 20 in Mammalian Tissues Is Associated with Aging. J Biol Chem. 2002;277:39195–39201. doi: 10.1074/jbc.M205166200. [DOI] [PubMed] [Google Scholar]
- 57.Han Y, Han D, Yan Z, Boyd-Kirkup JD, Green CD, Khaitovich P, et al. Stress-associated H3K4 methylation accumulates during postnatal development and aging of rhesus macaque brain. Aging Cell. 2012;11:1055–1064. doi: 10.1111/acel.12007. [DOI] [PubMed] [Google Scholar]
- 58.Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, et al. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature. 2010;466:383–387. doi: 10.1038/nature09195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, et al. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005;19:1544–1555. doi: 10.1101/gad.1308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maures TJ, Greer EL, Hauswirth AG, Brunet A. The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell. 2011;10:980–990. doi: 10.1111/j.1474-9726.2011.00738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McColl G, Killilea DW, Hubbard AE, Vantipalli MC, Melov S, Lithgow GJ. Pharmacogenetic Analysis of Lithium-induced Delayed Aging in Caenorhabditis elegans. J Biol Chem. 2008;283:350–357. doi: 10.1074/jbc.M705028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jin C, Li J, Green CD, Yu X, Tang X, Han D, et al. Histone Demethylase UTX-1 Regulates C. elegans Life Span by Targeting the Insulin/IGF-1 Signaling Pathway. Cell Metab. 2011;14:161–172. doi: 10.1016/j.cmet.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 63.Greer EL, Maures TJ, Ucar D, Hauswirth AG, Mancini E, Lim JP, et al. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature. 2011;479:365–371. doi: 10.1038/nature10572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siebold AP, Banerjee R, Tie F, Kiss DL, Moskowitz J, Harte PJ. Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Proc Natl Acad Sci. 2010;107:169–174. doi: 10.1073/pnas.0907739107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A, et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009;136:3131–3141. doi: 10.1242/dev.037127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schwartz YB, Kahn TG, Nix DA, Li XY, Bourgon R, Biggin M, et al. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat Genet. 2006;38:700–705. doi: 10.1038/ng1817. [DOI] [PubMed] [Google Scholar]
- 67.Noer A, Lindeman LC, Collas P. Histone H3 Modifications Associated With Differentiation and Long-Term Culture of Mesenchymal Adipose Stem Cells. Stem Cells Dev. 2009;18:725–736. doi: 10.1089/scd.2008.0189. [DOI] [PubMed] [Google Scholar]
- 68.Kroemer G, Mariño G, Levine B. Autophagy and the Integrated Stress Response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.ArtalMartinez de Narvajas A, Gomez TS, Zhang JS, Mann AO, Taoda Y, Gorman JA, et al. Epigenetic Regulation of Autophagy by the Methyltransferase G9a. Mol Cell Biol. 2013 doi: 10.1128/MCB.00813-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mah LJ, El-Osta A, Karagiannis TC. γH2AX as a molecular marker of aging and disease. Epigenetics. 2010;5:129–136. doi: 10.4161/epi.5.2.11080. [DOI] [PubMed] [Google Scholar]
- 71.Burgess RC, Misteli T, Oberdoerffer P. DNA damage, chromatin, and transcription: the trinity of aging. Curr Opin Cell Biol. 2012;24:724–730. doi: 10.1016/j.ceb.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Noon AT, Goodarzi AA. 53BP1-mediated DNA double strand break repair: Insert bad pun here. DNA Repair. 2011;10:1071–1076. doi: 10.1016/j.dnarep.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 73.DiTullio RA, Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, et al. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol. 2002;4:998–1002. doi: 10.1038/ncb892. [DOI] [PubMed] [Google Scholar]
- 74.Cha HJ, Yim H. The accumulation of DNA repair defects is the molecular origin of carcinogenesis. Tumor Biol (nd) :1–10. doi: 10.1007/s13277-013-1038-y. [DOI] [PubMed] [Google Scholar]
- 75.Huber A, Bai P, de Murcia JM, de Murcia G. PARP-1, PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA Repair. 2004;3:1103–1108. doi: 10.1016/j.dnarep.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 76.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, et al. Structural Basis for the Methylation State-Specific Recognition of Histone H4-K20 by 53BP1 and Crb2 in DNA Repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huyen Y, Zgheib O, D RA, Jr, Gorgoulis VG, Zacharatos P, Petty TJ, et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 78.Wysocki R, Javaheri A, Allard S, Sha F, Côté J, Kron SJ. Role of Dot1-Dependent Histone H3 Methylation in G1 and S Phase DNA Damage Checkpoint Functions of Rad9. Mol Cell Biol. 2005;25:8430–8443. doi: 10.1128/MCB.25.19.8430-8443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wakeman TP, Wang Q, Feng J, Wang XF. Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53BP1 foci at G1/G2 cell-cycle phases. EMBO J. 2012;31:2169–2181. doi: 10.1038/emboj.2012.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470:124–128. doi: 10.1038/nature09658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karachentsev D, Druzhinina M, Steward R. Free and chromatin-associated mono-, di-, and trimethylation of histone H4-lysine 20 during development and cell cycle progression. Dev Biol. 2007;304:46–52. doi: 10.1016/j.ydbio.2006.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.FitzGerald J, Moureau S, Drogaris P, O'Connell E, Abshiru N, Verreault A, et al. Regulation of the DNA Damage Response and Gene Expression by the Dot1L Histone Methyltransferase and the 53Bp1 Tumour Suppressor. PLoS ONE. 2011;6:e14714. doi: 10.1371/journal.pone.0014714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Edwards CR, Dang W, Berger SL. Histone H4 Lysine 20 of Saccharomyces cerevisiae Is Monomethylated and Functions in Subtelomeric Silencing. Biochemistry (Mosc) 2011;50:10473–10483. doi: 10.1021/bi201120q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.O'Hagan HM, Mohammad HP, Baylin SB. Double Strand Breaks Can Initiate Gene Silencing and SIRT1-Dependent Onset of DNA Methylation in an Exogenous Promoter CpG Island. PLoS Genet. 2008;4:e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11:1376–1382. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun Y, Xu Y, Roy K, Price BD. DNA Damage-Induced Acetylation of Lysine 3016 of ATM Activates ATM Kinase Activity. Mol Cell Biol. 2007;27:8502–8509. doi: 10.1128/MCB.01382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Young LC, McDonald DW, Hendzel MJ. Kdm4b Histone Demethylase Is a DNA Damage Response Protein and Confers a Survival Advantage following γ-Irradiation. J Biol Chem. 2013;288:21376–21388. doi: 10.1074/jbc.M113.491514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Faucher D, Wellinger RJ. Methylated H3K4, a Transcription-Associated Histone Modification, Is Involved in the DNA Damage Response Pathway. PLoS Genet. 2010;6:e1001082. doi: 10.1371/journal.pgen.1001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peña PV, Hom RA, Hung T, Lin H, Kuo AJ, Wong RPC, et al. Histone H3K4me3 Binding Is Required for the DNA Repair and Apoptotic Activities of ING1 Tumor Suppressor. J Mol Biol. 2008;380:303–312. doi: 10.1016/j.jmb.2008.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Feng X, Hara Y, Riabowol K. Different HATS of the ING1 gene family. Trends Cell Biol. 2002;12:532–538. doi: 10.1016/s0962-8924(02)02391-7. [DOI] [PubMed] [Google Scholar]
- 92.Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Khobta A, Anderhub S, Kitsera N, Epe B. Gene silencing induced by oxidative DNA base damage: association with local decrease of histone H4 acetylation in the promoter region. Nucleic Acids Res. 2010;38:4285–4295. doi: 10.1093/nar/gkq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kültz D. Evolution of the cellular stress proteome: from monophyletic origin to ubiquitous function. J Exp Biol. 2003;206:3119–3124. doi: 10.1242/jeb.00549. [DOI] [PubMed] [Google Scholar]
- 95.Rattan SIS. Aging, anti-aging, and hormesis. Mech Ageing Dev. 2004;125:285–289. doi: 10.1016/j.mad.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 96.Weiner A, Chen HV, Liu CL, Rahat A, Klien A, Soares L, et al. Systematic Dissection of Roles for Chromatin Regulators in a Yeast Stress Response. PLoS Biol. 2012;10:e1001369. doi: 10.1371/journal.pbio.1001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liang CY, Wang LC, Lo WS. Dissociation of the H3K36 demethylase Rph1 from chromatin mediates derepression of environmental stress-response genes under genotoxic stress in Saccharomyces cerevisiae. Mol Biol Cell. 2013 doi: 10.1091/mbc.E12-11-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pan Y, Schroeder EA, Ocampo A, Barrientos A, Shadel GS. Regulation of Yeast Chronological Life Span by TORC1 via Adaptive Mitochondrial ROS Signaling. Cell Metab. 2011;13:668–678. doi: 10.1016/j.cmet.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schroeder EA, Raimundo N, Shadel GS. Epigenetic Silencing Mediates Mitochondria Stress-Induced Longevity. Cell Metab. 2013;17:954–964. doi: 10.1016/j.cmet.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jeyapalan JC, Sedivy JM. Cellular senescence and organismal aging. Mech Ageing Dev. 2008;129:467–474. doi: 10.1016/j.mad.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Coppé JP, Desprez PY, Krtolica A, Campisi J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu Rev Pathol Mech Dis. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Signer RAJ, Morrison SJ. Mechanisms that Regulate Stem Cell Aging and Life Span. Cell Stem Cell. 2013;12:152–165. doi: 10.1016/j.stem.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, et al. Monitoring Tumorigenesis and Senescence In Vivo with a p16INK4a-Luciferase Model. Cell. 2013;152:340–351. doi: 10.1016/j.cell.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Feng Y, Wang X, Xu L, Pan H, Zhu S, Liang Q, et al. The transcription factor ZBP-89 suppresses p16 expression through a histone modification mechanism to affect cell senescence. FEBS J. 2009;276:4197–4206. doi: 10.1111/j.1742-4658.2009.07128.x. [DOI] [PubMed] [Google Scholar]
- 105.Place RF, Noonan EJ, Giardina C. HDACs and the senescent phenotype of WI-38 cells. BMC Cell Biol. 2005;6:37. doi: 10.1186/1471-2121-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Agherbi H, Gaussmann-Wenger A, Verthuy C, Chasson L, Serrano M, Djabali M. Polycomb Mediated Epigenetic Silencing and Replication Timing at the INK4a/ARF Locus during Senescence. PLoS ONE. 2009;4:e5622. doi: 10.1371/journal.pone.0005622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Agger K, Cloos PAC, Rudkjær L, Williams K, Andersen G, Christensen J, et al. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A–ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009;23:1171–1176. doi: 10.1101/gad.510809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–530. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Barradas M, Anderton E, Acosta JC, Li S, Banito A, Rodriguez-Niedenführ M, et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009;23:1177–1182. doi: 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zeng N, Yang KT, Bayan JA, He L, Aggarwal R, Stiles JW, et al. PTEN controls β-cell regeneration in aged mice by regulating cell cycle inhibitor p16ink4a. Aging Cell. 2013:n/a–n/a. doi: 10.1111/acel.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lin TY, Cheng YC, Yang HC, Lin WC, Wang CC, Lai PL, et al. Loss of the candidate tumor suppressor BTG3 triggers acute cellular senescence via the ERK– JMJD3–p16INK4a signaling axis. Oncogene. 2012;31:3287–3297. doi: 10.1038/onc.2011.491. [DOI] [PubMed] [Google Scholar]
- 112.Ribeiro JD, Morey L, Mas A, Gutierrez A, Luis NM, Mejetta S, et al. ZRF1 controls oncogene-induced senescence through the INK4-ARF locus. Oncogene. 2013;32:2161–2168. doi: 10.1038/onc.2012.241. [DOI] [PubMed] [Google Scholar]
- 113.Nijwening JH, Geutjes EJ, Bernards R, Beijersbergen RL. The Histone Demethylase Jarid1b (Kdm5b) Is a Novel Component of the Rb Pathway and Associates with E2f-Target Genes in MEFs during Senescence. PLoS ONE. 2011;6:e25235. doi: 10.1371/journal.pone.0025235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim W, Kim R, Park G, Park JW, Kim JE. Deficiency of H3K79 Histone Methyltransferase Dot1-like Protein (DOT1L) Inhibits Cell Proliferation. J Biol Chem. 2012;287:5588–5599. doi: 10.1074/jbc.M111.328138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787–1799. doi: 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, Von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8:311–323. doi: 10.1111/j.1474-9726.2009.00481.x. [DOI] [PubMed] [Google Scholar]
- 117.Takahashi A, Imai Y, Yamakoshi K, Kuninaka S, Ohtani N, Yoshimoto S, et al. DNA Damage Signaling Triggers Degradation of Histone Methyltransferases through APC/CCdh1 in Senescent Cells. Mol Cell. 2012;45:123–131. doi: 10.1016/j.molcel.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 118.New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: Can we interpret the code? Mol Oncol. 2012;6:637–656. doi: 10.1016/j.molonc.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nebbioso A, Carafa V, Benedetti R, Altucci L. Trials with “epigenetic” drugs: An update. Mol Oncol. 2012;6:657–682. doi: 10.1016/j.molonc.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hoffmann I, Roatsch M, Schmitt ML, Carlino L, Pippel M, Sippl W, et al. The role of histone demethylases in cancer therapy. Mol Oncol. 2012;6:683–703. doi: 10.1016/j.molonc.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]