

Abstract
Objectives
Ischemic preconditioning (IPC) induces cardioprotection against ischemia-reperfusion (IR) injury by inhibiting the mitochondrial permeability transition pore (mPTP). Here, we tested the hypothesis that IPC-induced cardioprotection is mediated by the phosphatase PTEN and phosphodiesterase (PDE) 4.
Methods
Isolated hearts from wildtype mice (WT, n = 110) and myocyte-specific PTEN knockout mice (PKO, n = 94) were exposed to IPC or control (CON), followed by IR. Subcellular fractionation was performed by sucrose gradient ultracentrifugation.
Results
IPC limited myocardial infarct size in WT mice. The PDE4 inhibitor rolipram abolished the protective effect of IPC. However, small infarct size was found in PKO hearts after IR, and IPC did not decrease infarct size but enlarged it in PKO hearts. IPC promoted PDE4D localization to caveolin-3-enriched fractions in WT mice by increasing Akt levels at the caveolae. In PKO hearts, basal PDE4D levels were elevated at the caveolae, and IPC decreased PDE4D levels. Consistent with the subcellular PDE4D protein levels and its activity, elevation of intracellular Ca2+ levels in the ischemic heart and opening of mPTP after IR were inhibited by IPC in WT mice, but not by IPC in PKO mice.
Conclusions
IPC inhibits mPTP opening by regulating the PTEN/PDE4 signaling pathway.
Keywords: Ischemic preconditioning, phosphodiesterase 4, phosphatase and tensin homologue deleted on chromosome ten, Akt, mitochondrial permeability transitional pore
Introduction
Reperfusion following prolonged ischemia causes substantial myocardial injury, contributing to mortality and morbidity of myocardial infarction [1,2]. Ischemic preconditioning (IPC), consisting of one or several brief periods of ischemia and reperfusion, generates powerful protection against ischemia-reperfusion (IR) injury [3]. However, the molecular mechanism underlying IPC is still poorly understood [4]. Reperfusion saves the ischemic myocardium, but it results in calcium overload, which is an important factor in promoting mitochondrial permeability transition pore (mPTP) opening and subsequent cell death [2,5,6]. It has been reported that IPC inhibits mPTP opening through activation of Akt [7].
Akt activity is regulated by phosphatidylinositol (PI) 3-kinase (PI3K) and phosphatase and tensin homologue deleted on chromosome ten (PTEN). PI3K activates Akt by phosphorylating PI-bisphosphate (PIP2) to PIP3 at the plasma membrane, whereas PTEN reverses this process, leading to Akt inactivation [8]. Akt can be localized to caveolae, lipid-rich microdomains of the plasma membrane [9]. Caveolae are essential for induction of IPC [10-12]. However, many studies have suggested that Akt mediates cardioprotection against IR injury by regulating translocation of Bcl-2 family proteins and glycogen synthase kinase-3β from the cytosol to the mitochondria [9,13-16]. Where Akt exerts its protection against IR injury has not been defined.
Caveolae contain many signaling molecules involved in regulation of calcium influx, including L-type calcium channels, beta-adrenoceptors, and cAMP-dependent protein kinase (PKA) [17]. The formation of caveolae is promoted by its structural protein caveolin [18]. There are three isoforms of caveolins, and caveolin 3 is myocyte-specific [19]. Caveolin-3 acts as a scaffolding domain to assemble the signaling complexes [20]. L-type calcium channels are activated by PKA, but local cAMP levels are mainly dependent on phosphodiesterase (PDE) 4 activity [21]. Decreased PDE4 activity has been implicated in the pathogenesis of heart failure and arrhythmias [22]. PDE4D is the major isoform of PDE4 existing in rodent hearts. PDE4D can be recruited to the plasma membrane through β-arrestin [23]. In the present study, we tested the hypothesis that IPC promotes PDE4D localization to the plasma membrane through PTEN inactivation, preventing calcium entry and mPTP opening after IR.
Methods
Animals
All experiments were performed with age-matched male mice. At the time of the experiments, mice were 8-10 weeks old. Myocyte-specific PTEN knockout (PKO) mice (Ptenloxp/loxp;ckm-Cre+/-) were generated by loxp-cre technology. Their phenotypes were described previously [24]. Briefly, PKO mice are characterized by physiological cardiac hypertrophy. All procedures were approved by the Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Mouse Langendorff preparation
Langendorff preparation was performed as described previously [25]. Briefly, after mice were anesthetized with pentobarbital (70 mg/kg), hearts were isolated. The ascending aorta was cannulated with a blunt needle. The heart was perfused at a constant pressure of 100 cm H2O with modified Krebs-Henseleit buffer (in mmol/L, glucose 17, NaCl 120, NaHCO3 25, CaCl2 2.5, KCl 5.9, MgSO4 1.2, and EDTA 0.5), which was maintained at 37°C and bubbled continuously with a mixture of 95% O2 and 5% CO2. Global ischemia was induced by cessation of perfusion, followed by reperfusion. Isolated hearts from WT mice and PKO mice were exposed to IPC, consisting of 10-min ischemia and 5-min reperfusion (I-10/R-5), or an equivalent period of normal perfusion, followed by 30-min ischemia and 120-min reperfusion (I-30/R-120).
Assessment of myocardial infarct size
Infarct size was measured as described previously [25]. At the end of experiments, mouse hearts were perfused with triphenyltetrazolium chloride (TTC) for 1 min and then incubated with 1% TTC at 37 °C for 15 min. After freezing at -80 °C, hearts were transected into 5 pieces, each of which was weighed. Cardiac sections were incubated with 10% formalin for 30 min. Both sides (A and B) of each section were photographed. Viable myocardium stains red and infarcted tissue was white. The areas of red and white color in the left ventricle (LV) and the muscle area of cardiac sections were measured by computerized planimetry (Image J, NIH, Bethesda, Maryland, USA). Total infarct weight of LV was determined by the equation: [(S1/C1 × W1) + (S2/C2 × W1) + (S2/C2 × W2) + (S3/C3 × W3) + (S4/C4 × W4) + (S5/C5 × W5)], where S is the area of LV infarction for the slice represented by the subscript, C is the muscle area of the cardiac section, and W is the weight of that respective section. Total weight of viable myocardium in LV was calculated in a similar fashion. Infarct size (IS) was calculated as a percentage of LV as follows: IS/LV = total infarct weight/(total infarct weight + total weight of viable myocardium) × 100%.
Subcellular fractionation
Enrichment of caveolae membranes with detergent-free reagents was performed as previously described [26]. Briefly, hearts were homogenized in 0.5 M sodium carbonate (pH 11). The homogenate was suspended in an equal volume of 80% sucrose prepared in Mes-buffered saline. The solution was placed on the bottom of an ultracentrifuge tube, overlaid with a 5-35 % discontinuous sucrose gradient, followed by 16 h of centrifugation at 38, 000 rpm. After centrifugation, 12 1-ml gradient fractions were concentrated by precipitation with trichloroacetic acid. The pellets of the 12 fractions (from top to bottom) were washed and dissolved in sample buffer and loaded onto 12 lanes (from left to right, respectively) of one SDS-PAGE gel.
Measurement of PDE4 activity
PDE4 activity was measured by PDE-GloTM phosphodiesterase assay kit (Promega Corp., WI, USA). PDE4 negatively regulates cAMP levels. cAMP binds the inactive PKA holoenzyme. Released catalytic subunits of PKA consume ATP. The level of remaining ATP was measured by luminescence assay using the luciferase-based Kinase-Glu Reagent. PDE4 activity is directly proportional to the remaining ATP levels. According to the manufacturer's instructions, 10 μg of caveolin-3-enriched membrane protein was incubated with cAMP (1 μM) to initiate the PDE reaction. The PDE-GloTM termination buffer containing the PDE4 inhibitor (Rolipram, 100 μM) and detection solution were added and mixed well. The Kinase-Glo reagent was added and incubated for 10 min at room temperature. Luminescence was measured using a plate-reading luminometer.
Assessment of intracellular Ca2+ levels
Intracellular free Ca2+ [Ca2+] levels were measured with a rhod-2 AM fluorescent probe (Anaspec Inc., Freemont, CA, USA) as previously described with modifications [27]. Rhod-2 AM is membrane permeable and becomes a Ca2+-sensitive fluorescence probe when trapped in the cytosol and de-esterified to rhod-2. Rhod-2 dye (25 μg/ml in 2 ml) was added without recirculation through a parallel infusion line just above the aortic cannula. During loading, only the bolus line was opened. Dye loading was followed by a 10-min washout period with the perfusion buffer to remove extracellular dye. Rolipram was added into the perfusion line during the washout. At the end of the experiment, heart tissues were homogenized in Cai2+-free KH buffer. Equal amounts (100 μg) of protein from each sample were loaded into each well. Plates were read at excitation 531 nm/emission 593 nm.
Measurement of mPTP opening in isolated hearts
mPTP opening in mouse hearts was assessed as previously described, with modifications [28]. Briefly, isolated hearts were balanced with the perfusion buffer and loaded with 1 μM calcein-AM (Anaspec Inc., Freemont, CA, USA) for 15 min. During the last 5-min of calcein-AM perfusion, CoCl2 (200 μM) was added into the perfusion line to quench the cytosolic calcein signal. Remaining calcein in the extracellular space was washed out with a 10-min perfusion in the absence or presence of inhibitors, followed by I-30/R-15. At the end of the experiment, heart tissues were homogenized on ice in calcium-free buffer, followed by centrifugation at 12,000 rpm for 10 min. Supernatants (cytosolic fractions) were collected for measurement of protein concentration. Calcein fluorescence was measured at 484-nm excitation and 520-nm emission in 100 μg of protein. Without IR, low fluorescence intensity was detected in the cytosolic fractions while high fluorescence intensity was found in the mitochondrial fractions (from pellets after centrifugation), suggesting that mPTP were closed. Inhibitors were added into the perfusion line 5 min before 30-min ischemia.
Immuoprecipitation and immunoblotting assay
Cardiac tissues were homogenized in lysis buffer (in mM: pH 7.5 Tris 20, NaCl 150, EDTA 1, EGTA 1, PMSF 1, Na3VO4 1, 1% Triton). Heart lysates were incubated with primary antibody overnight at 4 °C. The target protein was pulled down following 2 h incubation with G protein agarose beads. Proteins were detected by using primary antibodies, followed by horseradish peroxidase-conjugated secondary antibody and enhanced chemiluminescence. Antibodies against PTEN, β-arrestin, p-Akt (S-473), and total Akt were purchased from Cell Signaling Technology (Danvers, Massachusetts, USA). PDE4D antibody was from Santa Cruz Biotechnology (Santa Cruz, California, USA).
Statistical analysis
Data are presented as mean ± standard error of the mean. The difference among groups was analyzed using ANOVA with the Tukey post hoc test. Differences were considered significant if p < 0.05.
Results
PTEN and PDE4 are required for IPC-induced cardioprotection against IR injury
WE previously reported that PTEN inactivation confers cardioprotection against IR injury in isolated mouse hearts [25]. In the present study, IPC decreased infarct size in WT (IPC/WT) mice. Small infarct size was seen in CON/PKO mice. Surprisingly, IPC increased infarct size in PKO mice to levels similar to those seen in WT/CON mice (Fig. 1A and 1B), suggesting that IPC abolishes the native protection in PKO mice. Since PDE4 plays an important role in regulating local cAMP levels and L-type calcium channel activity, we wondered whether PDE4 is involved in cardioprotection induced by IPC and PTEN inactivation. Hearts from IPC/WT and CON/PKO were treated with rolipram (Ro), a PDE4 inhibitor, followed by I-30/R-120. Ro inhibited the infarct-limiting effect in these two groups (Fig. 1A and 1B). These results suggest that PTEN and PDE4 are necessary for IPC cardioprotection against IR injury.
Fig. 1.
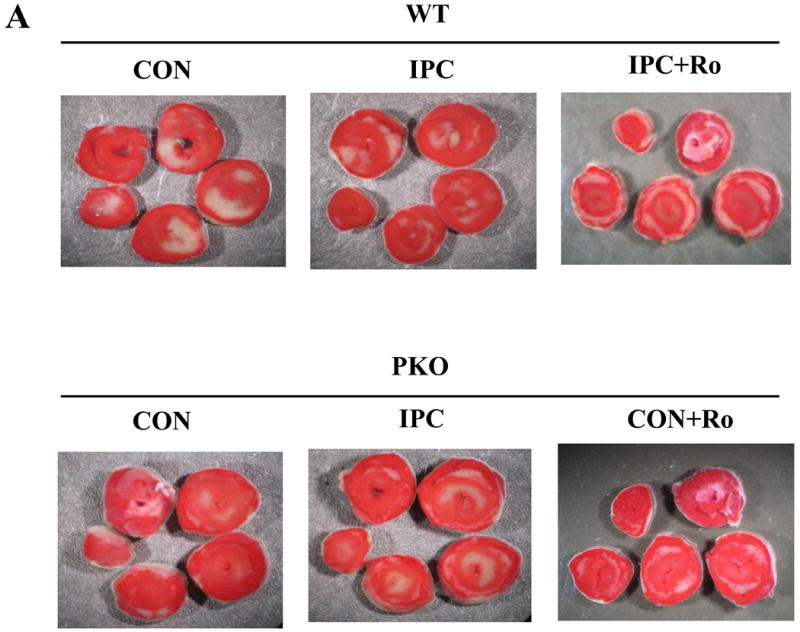

Myocardial infarct size after IR. A: Cardiac sections of one representative heart from each group. Isolated hearts from WT and PKO mice were exposed to CON [30-min ischemia and 120-min reperfusion (I-30/R-120)], or IPC [10-min ischemia and 5-min reperfusion (I-10/R-5) plus I-30/R-120] in the absence or presence of rolipram (Ro) (10 μM). After IR, the heart was cut into 5 sections and stained by TTC. Each group contains 6-8 hearts. B: Analysis of infarct size. Infarcted area of myocardium was measured by Image J software. N = 6-8/group. *: p<0.01 vs. CON/WT; #: p<0.01 vs. IPC/WT. †: p<0.01 vs. CON/PKO; §: p<0.01 vs. CON/PKO.
IPC promotes localization of Akt and PDE4D to the plasma membrane by regulating PTEN/Akt
Many studies have reported that PI3K/Akt signaling pathway plays an important role in IPC cardioprotection [6,8]. This signaling pathway cross-talks with PTEN at the plasma membrane [8,9]. To determine whether PTEN inactivation affects Akt localization to the plasma membrane, subcellular fractionations were performed after exposure of isolated hearts to CON or IPC. The same volumes of samples from 12 fractions were loaded onto SDS-PAGE gels for protein analysis. Cav-3-enriched fractions were identified in lane 4 (Fig. 2A). Only small amounts of Akt were localized at the subcellular domain (Fig. 2B). To compare Akt protein levels, the same amounts of proteins from the Cav-3-enriched fractions were loaded onto SDS-PAGE gels. IPC increased Akt protein levels in WT mice (Fig. 2C). The PI3K inhibitor LY20094 blocked this effect (Fig. 2C). However, in PKO mice, there were high basal levels of Akt protein at the Cav-3-enriched fractions, and IPC decreased Akt protein levels when compared with CON (Fig. 2C), suggesting that PTEN is required for Akt localization to the caveolae at the plasma membrane in IPC.
Fig. 2.
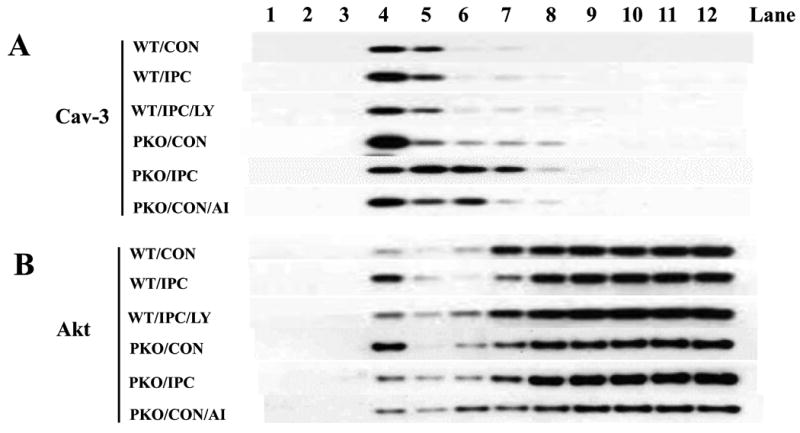
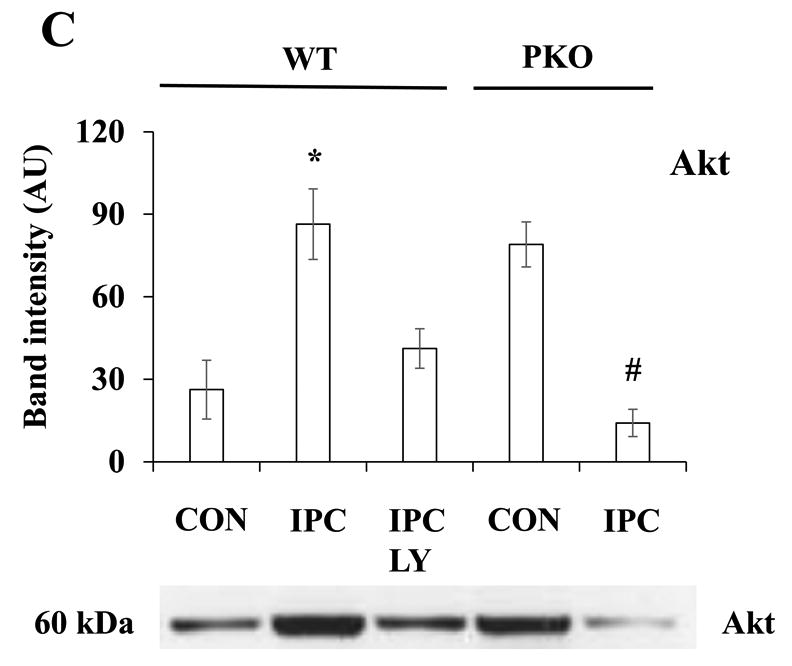
IPC increases Akt localization to the plasma membrane through PTEN signaling pathway. A-B: Distribution of Caveolin (Cav)-3 and Akt in subcellular fractions. Isolated hearts from WT or PKO mice were treated with no ischemia (CON), I-10/R-5 (IPC), IPC+LY (LY20094, 10 μM), or CON+AI (Akt Inhibitor, 20 μM). Subcellular fractionations were performed with sucrose gradient centrifugation. Equal volumes of cell lysates were loaded onto SDS PAGE gels. Protein expression of caveolin (Cav)-3 was measured. Representative blots from at least three independent experiments. C: Akt protein levels in Cav-3-enriched fractions. Equal amounts of protein from Cav-3-enriched fractions were loaded. N = 4/group, *: p<0.01 vs. CON/WT or IPC/LY; #: p<0.01 vs. CON/PKO.
To determine whether Akt regulates PDE4D localization, PDE4D protein levels in 12 subcellular fractions and Cav-3-enriched fractions were measured. Under basal conditions, few amounts of PDE4D were detected in Cav-3-enriched fractions, and PDE4D protein levels at the microdomain were regulated by the treatments (Fig. 3A). IPC increased PDE4D protein levels compared with CON in WT mice, and this effect was inhibited by LY (Fig. 3B). This suggests that PDE4D localization to the caveolae requires Akt activation. Consistent with Akt protein levels, PDE4D protein levels were elevated under basal conditions in PKO mice (Fig. 3B). IPC decreased PDE4D protein levels when compared with CON/PKO (Fig. 3B). In a similar way, IPC increased PDE4 activity in Cav-3-enriched fractions in WT mice, and LY inhibited this effect; In PKO mice, PDE4 activity at the microdomain was increased under basal conditions; IPC decreased its activity (Fig. 3C). Taken together, these results suggest that activated Akt modulates PDE4D protein levels at the caveolea.
Fig. 3.
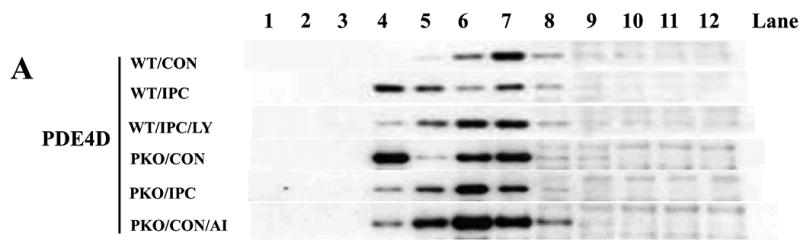
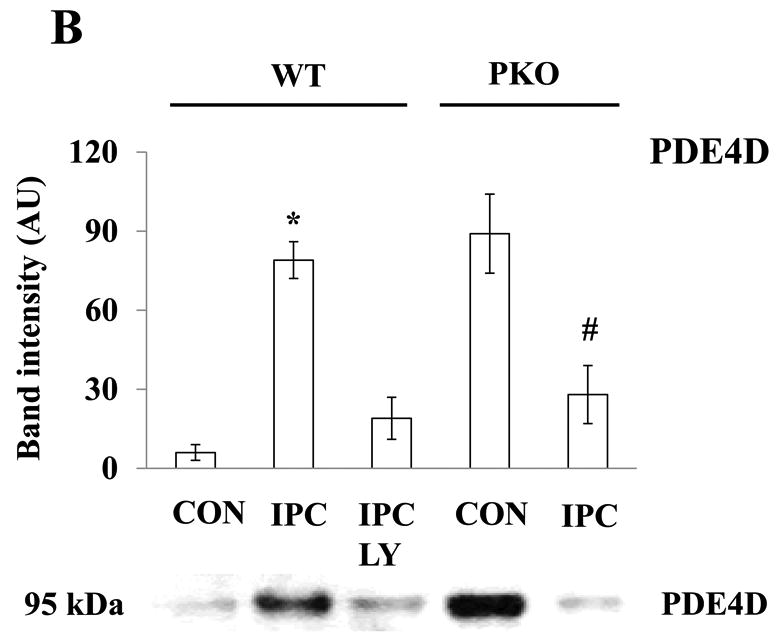

Akt activation increases PDE4D localization to the plasma membrane. A: Distribution of PDE4D in subcellular fractions. Isolated hearts from WT and PKO mice were exposed to CON, IPC, IPC+LY, or CON+AI, and followed by subcellular fractionation. Representative blots from at least three independent experiments. B: PDE4D protein levels in Cav-3-enriched fractions. N = 4/group, *: p<0.01 vs. CON/WT or IPC/LY; #: p<0.01 vs. CON/PKO. C: PDE4 activity in Cav-3-enriched fractions. N = 6/group, *: p<0.05 vs. CON/WT or IPC/LY; #: p<0.05 vs. CON/PKO.
IPC promotes the association between Akt, β-arrestin, and PDE4D at the plasma membrane
To determine whether the adaptor β-arrestin interacts with PDE4D and Akt, the association between β-arrestin, PDE4D, and Akt was measured. Akt or PDE4D was pulled down from Cav-3-enriched fractions generated from WT/IPC hearts. The membrane was immunoblotted with β-arrestin antibody. β-arrestin was found in the immunoprecipitates (Fig. 4). In a reverse experiment, β-arrestin was pulled down from the same lysates and followed by immunoblotting assay of Akt or PDE4D. Akt and PDE4D were present in the immunoprecipitates (Fig. 4). These results suggest that Akt is associated with β-arrestin and PDE4D at the plasma membrane.
Fig. 4.

IPC promotes associations between Akt, β-arrestin, and PDE4D in Cav-3-enriched fractions. Cav-3 enriched fractions from IPC/WT hearts were immunoprecipitated with an antibody against Akt or PDE4D, followed by immunoblotting with antibodies against β-arrestin (Arr) (upper panel). In a reverse experiment, cell lysates were pulled down with β-arrestin antibody, followed by immunoblotting with antibodies against Akt or PDE4D (lower panel). Representative blots from at least three independent experiments.
IPC inhibits Ca2+ overload in the ischemic heart through PDE4
To determine whether PDE4D localization to the plasma membrane inhibits intracellular Ca2+ overload in the ischemic heart, we exposed isolated hearts from WT mice to non-ischemia (NIS), 30-min ischemia (CON), I-10/R-5 plus 30-min ischemia in the absence or presence of Ro (IPC or IPC/Ro) (Fig. 5A). IPC decreased intracellular Ca2+ levels at the end of ischemia compared with CON/WT, and Ro abolished the inhibitory effect of IPC in the ischemic heart (Fig. 5B). However, PKO mice, there was no significant difference between NIS and CON (Fig. 5B). Without PTEN, IPC increased intracellular Ca2+ levels in the ischemic heart compared with CON; moreover, Ro had a similar effect (Fig. 5B).
Fig. 5.


Intracellular Ca2+ levels in ischemic hearts. A: Experimental protocols. Isolated hearts from WT and PKO mice were loaded with the fluorescent dye Rod-2 (Rd), followed by 10-min washout (Wo) and 30-min ischemia (I). Ro was added into the perfusion line during washout. Preconditioning with 10-min ischemia and 5 min-reperfusion (R) followed 5-min washout. B: Intracellular Ca2+ levels in WT and PKO hearts. N = 4/group, *: p<0.05 vs. NIS/WT or IPC/WT; #: p<0.05 vs. IPC/WT; †: p>0.05 vs. NIS/PKO; §: p<0.05 vs. CON/PKO; **: p<0.01 vs. CON/PKO.
IPC inhibits mPTP opening in the heart through PDE4
Increased intracellular Ca2+ levels is directly associated with opening of mPTP, leading to cell death. To determine whether IPC inhibits mPTP opening after IR, we exposed isolated hearts from WT mice to NIS, I-30/R-15 (CON), cyclosporine A+CON (CON/CsA), Verapamil+CON (CON/Ve), I-10/R-5+CON (IPC), Rolipram+IPC (IPC/Ro) (Fig. 6A). CON caused a significant increase in fluorescence intensity when compared with NIS, indicating mPTP opening (Fig. 6B). CsA or Ve inhibited the increase (Fig. 6B), suggesting that the assay of mPTP opening was accurate. Consistent with the intracellular Ca2+ levels in the ischemic heart, IPC inhibited mPTP opening in WT mice, and this effect was abolished by Ro (Fig. 6B). Moreover, hearts from PKO mice were exposed to NIS, CON, IPC, IPC/Ve, and CON/Ro. CON did not cause mPTP opening in PKO mice; however, the protection was lost after IPC (Fig. 6C). Ve reversed the detrimental effect of IPC, and Ro resulted in mPTP opening in CON (Fig. 6C). These results suggest that mPTP opening is dependent on PDE4 and L-type calcium channel activities in the heart exposed to IR.
Fig. 6.


mPTP opening after IR. A: Experimental protocols. Isolated hearts from WT and PKO mice were loaded with the fluorescent dye cacein (Cn) for 15-min, followed by total 25-min washout (W). Rolipram (Ro), cyclosporine A (CsA, 1μM), or Verapamil (Ve, 10 μM) was added during washout. B: mPTP opening in WT after IR. N = 5-8/group, *: p<0.01 vs. NIS or IPC or CON/Ve or CON/CsA; #: p<0.01 vs. IPC. C: mPTP opening in PKO after IR. N = 6-7/group, *: p>0.05 vs. NIS and p<0.01 vs. IPC; #: p<0.01 vs. IPC; §: p<0.01 vs. CON.
Discussion
In the present study, we report three important findings. First, we demonstrate that PTEN is required for induction of IPC. In PKO mice, IPC did not reduce myocardial infarct size after IR. Second, we show that IPC promotes localization of PDE4D to the caveolae in the heart by regulating PTEN/Akt pathway. Akt inhibition blocked PDE4D localization to the plasma membrane induced by IPC and PTEN inactivation. Third, we demonstrate that PDE4 plays an important role in controlling calcium overload and mPTP opening. PDE4 inhibition abolished the inhibition of calcium overload and mPTP opening in IPC/WT mice. Therefore, the present study has demonstrated that PTEN plays a critical role in response to ischemia and turning on the Akt/PDE4 signaling pathway at the plasma membrane. This may lead to inhibition of calcium overload and mPTP opening after IR.
Many studies have reported that IPC activates the PI3K/Akt signaling pathway and protects the heart from IR injury [13,16]. However, the role of PTEN in IPC is not fully understood. In the present study, we showed that IPC inhibits Akt phosphorylation in PTEN-deficient hearts. It suggests that Akt requires PI3K and PTEN for its activation. IPC increases Akt phosphorylation in the myocardium of wild-type rats [13]. However, brief ischemia without reperfusion has been shown to cause a modest increase in Akt phosphorylation [29]. Ischemia decreases PTEN protein levels in the heart. PTEN inactivation itself can be sufficient to increase Akt phosphorylation if PI3K activity remains unchanged. As a protein enzyme, PI3K is likely impaired by ischemia. Early reperfusion further suppresses PTEN activity through reactive oxygen species [29]. Although PI3K is essential for production of PIP3, which causes Akt activation, the present study suggests PI3K itself is insufficient to activate Akt in the preconditioned heart. Therefore, despite the importance of PI3K, PTEN inactivation is the key factor in Akt activation and IPC induction. PI3K and PTEN may also be involved in activation of other reperfusion injury salvage kinases such as p42/p44 [29-31]. However, their role in IPC is less certain [32,33].
PDE4D plays an important role in regulating the spatiotemporal dynamics of cAMP signals in rodent cardiomyocytes [20]. At the plasma membrane, L-type Ca2+ channel activity is associated with compartmentalized cAMP signaling through cAMP-dependent protein kinase (PKA) [20,21]. L-type Ca2+ channels, PKA, β-arrestin and Akt have been identified in the caveolae of cardiac myocytes [21,34]. β-arrestin may be the key for recruiting PDE4D to the plasma membrane. It has been reported that β-arrestin is involved in modulation of β2 adrenoceptor signaling and induction of IPC [23,35-37]. In the present study, we demonstrate that β-arrestin is associated with PDE4D and Akt in preconditioned membrane fractions. Akt has been shown to form a complex with PDE4D through β-arrestin at the plasma membrane, promoting localization of PDE4D [38]. Consistent with the finding, the present study showed that IPC-induced Akt activation increases localization of PDE4D to the caveolae, suggesting that PDE4D may mediates the compartmentalization of cAMP at the plasma membrane and limit later increase in L-type Ca2+ channel activity by reducing local PKA activity in the preconditioned heart.
IPC has been shown to inhibit necrosis and apoptosis in the infarcted heart through the PI3K/PTEN/Akt signaling pathway [13,14,39]. However, necrotic cell death is critically related to calcium overload and subsequent mPTP opening while apoptotic cell death is promoted by release of cytochrome c [40]. Akt regulates Bcl-2 family proteins and block the release of cytochrome c from the mitochondria [40,41]. In the present study, our results suggest that Akt may inhibit calcium overload in the ischemic heart by increasing PDE4D localization to the plasma membrane. This provides new insights into the molecular mechanism by which IPC inhibits both necrosis and apoptosis. Our observations are consistent with previous reports, in which caveolin-3 deficiency or a defect in the plasma membrane resulted in loss of IPC [11,42]. GSK-3β has been implicated in inhibition of mPTP opening, and GSK-3β is a substrate of Akt [43]. Further study is needed to determine whether Akt regulates GSK-3β at the plasma membrane. Based on the present study and previous observations, a hypothesis of IPC-induced cardioprotection is proposed (Fig. 7).
Fig. 7.

A hypothesis of IPC-induced cardioprotection. IPC inactivates PTEN and decreases dephosphorylation of PIP3, which is produced by PI3K. PIP3 elevation causes Akt activation. Activated Akt promotes the localization of PDE4D to the caveolae at the plasma membrane through β-arrestin and thereby induces the compartmentalization of cAMP signaling, limiting later increase in PKA activity and L-type calcium channel opening in response to lethal IR. This prevents activation of caspases and cell death from calcium overload and mPTP opening in reperfused hearts. Arr, β-arrestin; Cav, caveolin-3; LC, L-type calcium channel; PM, plasma membrane; Mt, mitochondria.
In conclusion, IPC inactivates PTEN and promotes PDE4D localization to the plasma membrane through Akt, leading to inhibition of calcium overload in ischemic hearts and mPTP opening after IR.
Acknowledgments
This work was supported by Public Health Service grants HL88071 and HL65608 from the National Heart, Lung and Blood Institute, National Institutes of Health.
Abbreviations
- IPC
ischemic preconditioning
- IR
ischemia-reperfusion
- IS
infarct size
- LY
LY294002
- mPTP
mitochondrial permeability transitional pore
- PDE4D
phosphodiesterase 4D
- PI
phosphatidylinositol
- PI3K
phosphatidylinositol-3-kinase
- PKO
myocyte-specific PTEN knockout
- PTEN
phosphatase and tensin homologue deleted on chromosome ten
Footnotes
Conflict of interest: None
References
- 1.American Heart Association Heart disease and stroke statistics--2011 update: A report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 3.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 4.Hausenloy DJ, Yellon DM. The therapeutic potential of ischemic conditioning: an update. Nat Rev Cardiol. 2011;8:619–29. doi: 10.1038/nrcardio.2011.85. [DOI] [PubMed] [Google Scholar]
- 5.Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol. 2003;549:513–24. doi: 10.1113/jphysiol.2003.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60(3):617–25. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 7.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–49. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oudit GY, Sun H, Kerfant BG, Crackower MA, Penninger JM, Backx PH. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J Mol Cell Cardiol. 2004;37:449–71. doi: 10.1016/j.yjmcc.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 9.Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell. 2002;111:293–303. doi: 10.1016/s0092-8674(02)01083-8. [DOI] [PubMed] [Google Scholar]
- 10.Patel HH, Head BP, Petersen HN, Niesman IR, Huang D, Gross GJ, Insel PA, Roth DM. Protection of adult rat cardiac myocytes from ischemic cell death: role of caveolar microdomains and delta-opioid receptors. Am J Physiol. 2006;291:H344–50. doi: 10.1152/ajpheart.01100.2005. [DOI] [PubMed] [Google Scholar]
- 11.Tsutsumi YM, Horikawa YT, Jennings MM, Kidd MW, Niesman IR, Yokoyama U, Head BP, Hagiwara Y, Ishikawa Y, Miyanohara A, Patel PM, Insel PA, Patel HH, Roth DM. Cardiac-specific overexpression of caveolin-3 induces endogenous cardiac protection by mimicking ischemic preconditioning. Circulation. 2008;118:1979–88. doi: 10.1161/CIRCULATIONAHA.108.788331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun J, Murphy E. Calcium-sensing receptor: a sensor and mediator of ischemic preconditioning in the heart. Am J Physiol. 2010;299:H1309–17. doi: 10.1152/ajpheart.00373.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tong H, Chen W, Steenbergen C, Murphy E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ Res. 2000;87:309–315. doi: 10.1161/01.res.87.4.309. [DOI] [PubMed] [Google Scholar]
- 14.Cai ZP, Shen Z, Van Kaer L, Becker LC. Ischemic preconditioning-induced cardioprotection is lost in mice with immunoproteasome subunit low molecular mass polypeptide-2 deficiency. FASEB J. 2008;22:4248–4257. doi: 10.1096/fj.08-105940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krieg T, Qin Q, Philipp S, Alexeyev MF, Cohen MV, Downey JM. Acetylcholine and bradykinin trigger preconditioning in the heart through a pathway that includes Akt and NOS. Am J Physiol. 2004;287:H2606–2611. doi: 10.1152/ajpheart.00600.2004. [DOI] [PubMed] [Google Scholar]
- 16.Matsui T, Rosenzweig A. Convergent signal transduction pathways controlling cardiomyocyte survival and function: the role of PI 3-kinase and Akt. J Mol Cell Cardiol. 2005;38:63–71. doi: 10.1016/j.yjmcc.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L-type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc Natl Acad Sci U S A. 2006;103:7500–5. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gratton JP, Bernatchez P, Sessa WC. Caveolae and caveolins in the cardiovascular system. Circ Res. 2004;94:1408–17. doi: 10.1161/01.RES.0000129178.56294.17. [DOI] [PubMed] [Google Scholar]
- 19.Tang Z, Scherer PE, Okamoto T, Song K, Chu C, Kohtz DS, Nishimoto I, Lodish HF, Lisanti MP. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem. 1996;271:2255–61. doi: 10.1074/jbc.271.4.2255. [DOI] [PubMed] [Google Scholar]
- 20.Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechêne P, Mazet JL, Conti M, Fischmeister R, Vandecasteele G. Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res. 2008;102:1091–100. doi: 10.1161/CIRCRESAHA.107.167817. [DOI] [PubMed] [Google Scholar]
- 21.Verde I, Vandecasteele G, Lezoualc'h F, Fischmeister R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol. 1999;127:65–74. doi: 10.1038/sj.bjp.0702506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science. 2002;298:834–6. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- 24.Zu L, Bedja D, Fox-Talbot K, Gabrielson KL, Van Kaer L, Becker LC, Cai ZP. Evidence for a role of immunoproteasomes in regulating cardiac muscle mass in diabetic mice. J Mol Cell Cardiol. 2010;49:5–15. doi: 10.1016/j.yjmcc.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zu L, Zheng X, Wang B, Parajuli N, Steenbergen C, Becker LC, Cai ZP. Ischemic preconditioning attenuates mitochondrial localization of PTEN induced by ischemia-reperfusion. Am J Physiol. 2011;300:H2177–86. doi: 10.1152/ajpheart.01138.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oka N, Asai K, Kudej RK, Edwards JG, Toya Y, Schwencke C, Vatner DE, Vatner SF, Ishikawa Y. Downregulation of caveolin by chronic beta-adrenergic receptor stimulation in mice. Am J Physiol. 1997;273:C1957–62. doi: 10.1152/ajpcell.1997.273.6.C1957. [DOI] [PubMed] [Google Scholar]
- 27.Talukder MA, Kalyanasundaram A, Zhao X, Zuo L, Bhupathy P, Babu GJ, Cardounel AJ, Periasamy M, Zweier JL. Expression of SERCA isoform with faster Ca2+ transport properties improves postischemic cardiac function and Ca2+ handling and decreases myocardial infarction. Am J Physiol. 2007;293:H2418–28. doi: 10.1152/ajpheart.00663.2007. [DOI] [PubMed] [Google Scholar]
- 28.Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76:725–34. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cai Z, Semenza GL. PTEN activity is modulated during ischemia and reperfusion: involvement in the induction and decay of preconditioning. Circ Res. 2005;97:1351–9. doi: 10.1161/01.RES.0000195656.52760.30. [DOI] [PubMed] [Google Scholar]
- 30.Ruan H, Li J, Ren S, Gao J, Li G, Kim R, Wu H, Wang Y. Inducible and cardiac specific PTEN inactivation protects ischemia/reperfusion injury. J Mol Cell Cardiol. 2009;46:193–200. doi: 10.1016/j.yjmcc.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 31.Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM. Regulation of myocardial contractility and cell size bydistinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–49. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 32.Kunuthur SP, Mocanu MM, Hemmings BA, Hausenloy DJ, Yellon DM. The Akt1 isoform is an essential mediator of ischaemic preconditioning. J Cell Mol Med. 2012;16:1739–49. doi: 10.1111/j.1582-4934.2011.01491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mocanu MM, Bell RM, Yellon DM. PI3 kinase and not p42/p44 appears to be implicated in the protection conferred by ischemic preconditioning. J Mol Cell Cardiol. 2002;34:661–8. doi: 10.1006/jmcc.2002.2006. [DOI] [PubMed] [Google Scholar]
- 34.Weiss S, Oz S, Benmocha A, Dascal N. Regulation of cardiac L-type Ca2+ channel CaV1.2 via the β-adrenergic-cAMP-protein kinase A pathway: old dogmas, advances, and new uncertainties. Circ Res. 2013;113:617–31. doi: 10.1161/CIRCRESAHA.113.301781. [DOI] [PubMed] [Google Scholar]
- 35.Nichols CB, Rossow CF, Navedo MF, Westenbroek RE, Catterall WA, Santana LF, McKnight GS. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ Res. 2010;107:747–56. doi: 10.1161/CIRCRESAHA.109.216127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiang Y, Naro F, Zoudilova M, Jin SL, Conti M, Kobilka B. Phosphodiesterase 4D is required for beta2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc Natl Acad Sci U S A. 2005;102:909–14. doi: 10.1073/pnas.0405263102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tong H, Bernstein D, Murphy E, Steenbergen C. The role of beta-adrenergic receptor signaling in cardioprotection. FASEB J. 2005;19:983–5. doi: 10.1096/fj.04-3067fje. [DOI] [PubMed] [Google Scholar]
- 38.Bjørgo E, Solheim SA, Abrahamsen H, Baillie GS, Brown KM, Berge T, Okkenhaug K, Houslay MD, Taskén K. Cross talk between phosphatidylinositol 3-kinase and cyclic AMP (cAMP)-protein kinase a signaling pathways at the level of a protein kinase B/beta-arrestin/cAMP phosphodiesterase 4 complex. Mol Cell Biol. 2010;30:1660–72. doi: 10.1128/MCB.00696-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol. 2005;288:H971–6. doi: 10.1152/ajpheart.00374.2004. [DOI] [PubMed] [Google Scholar]
- 40.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–7. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao CM, Zhang Y, Weisleder N, Ferrante C, Wang X, Lv F, Zhang Y, Song R, Hwang M, Jin L, Guo J, Peng W, Li G, Nishi M, Takeshima H, Ma J, Xiao RP. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation. 2010;121:2565–74. doi: 10.1161/CIRCULATIONAHA.110.954628. [DOI] [PubMed] [Google Scholar]
- 43.Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res. 2009;104:1240–52. doi: 10.1161/CIRCRESAHA.109.197996. [DOI] [PMC free article] [PubMed] [Google Scholar]