

Abstract
Metastatic cancer of unknown primary (CUP) accounts for up to 5% of all new cancer cases, with a 5-year survival rate of only 10%. Accurate identification of tissue of origin would allow for directed, personalized therapies to improve clinical outcomes. Our objective was to use transcriptome sequencing (RNA-Seq) to identify lineage-specific biomarker signatures for the cancer types that most commonly metastasize as CUP (colorectum, kidney, liver, lung, ovary, pancreas, prostate, and stomach). RNA-Seq data of 17,471 transcripts from a total of 3,244 cancer samples across 26 different tissue types were compiled from in-house sequencing data and publically available International Cancer Genome Consortium and The Cancer Genome Atlas datasets. Robust cancer biomarker signatures were extracted using a 10-fold cross-validation method of log transformation, quantile normalization, transcript ranking by area under the receiver operating characteristic curve, and stepwise logistic regression. The entire algorithm was then repeated with a new set of randomly generated training and test sets, yielding highly concordant biomarker signatures. External validation of the cancer-specific signatures yielded high sensitivity (92.0% ± 3.15%; mean ± standard deviation) and specificity (97.7% ± 2.99%) for each cancer biomarker signature. The overall performance of this RNA-Seq biomarker-generating algorithm yielded an accuracy of 90.5%. In conclusion, we demonstrate a computational model for producing highly sensitive and specific cancer biomarker signatures from RNA-Seq data, generating signatures for the top eight cancer types responsible for CUP to accurately identify tumor origin.
Introduction
Metastatic cancer of unknown primary origin (CUP) is an important clinical dilemma, comprising 3% to 5% of all new cancer cases [1], [2]. Without a firm histologic diagnosis, the clinical management of these patients varies widely [3], and despite protocol-driven guidelines, outcomes remain poor. Median survival is 6 to 9 months [4], with a 5-year survival rate of only 10% [5], [6]. The role of chemotherapy in the treatment of occult primary tumors is primarily palliative and does not improve long-term survival; the National Comprehensive Cancer Network (NCCN) panel encourages CUP patient enrollment in clinical trials when possible [7]. An accurate method for distinguishing tumor origin to tailor personalized therapies is therefore critical to the successful management of these malignancies.
Thus far, there have been a number of studies focused on identifying unique signatures that distinguish among different cancer types, using immunohistochemistry [8], [9], [10], [11], cytogenetic studies [12], [13], [14], comparative microarray analysis [15], [16], [17], [18], [19], [20], [21], [22], combined microarray and quantitative polymerase chain reaction techniques [23], [24], bead-based miRNA profiling [25], and more recently, limited, high-throughput sequencing data combined with microarray [26]. Currently, only qRT-PCR [23], [24] and microarray-based [19], [22] assays are commercially available for use, with diagnostic accuracies ranging from 74 – 85%.
Compared to traditional microarray technology, transcriptome sequencing (RNA-Seq) possesses a number of advantages, including unlimited genome coverage and discovery potential, a greater than 8000-fold dynamic range for quantifying gene expression levels, and the ability to identify splice variants, unmapped genes, and unrecognized non-coding RNAs [27], [28]. The rapidly decreasing cost of high-throughput sequencing methods has also improved the accessibility of these techniques for clinical application and allowed for the generation of large-scale datasets to robustly interrogate such clinical problems as CUP.
In a review of all published autopsies performed on CUP patients who died from cancer progression from 1944 to 2000, a primary tumor was successfully identified post-mortem in 73% of the 884 cases [29]. The most common tissues of origin were lung (27%), pancreas (24%), kidney (6%), colorectum (6%), stomach (5%), liver (5%), ovary (3%), and prostate (3%) [4], [30], [31], [32], [33], [34], [35], [36], [37], [38]. Our objective was therefore to identify lineage-specific biomarker signatures for each of these cancers, using a large, multi-cancer RNA-Seq database to distinguish tissues of origin from among different cancer types.
Material and Methods
Multi-Cancer RNA-Seq Database
Paired-end RNA-Seq data for 364 cancer samples from 22 different tissue types were used to compile a multi-cancer gene expression dataset as previously described [39]. This dataset was then supplemented with publically available RNA-Seq cancer data accessed from the International Cancer Genome Consortium [40] and The Cancer Genome Atlas [41]. This included four additional cancer types (acute myeloid leukemia, endometrial cancer, head and neck squamous cancer, and lung cancer). The dataset was restricted to those transcripts commonly annotated across all three datasets. The final composite data matrix was comprised of gene expression readouts for 17,471 transcripts from 3,244 cancer samples (139 cancer cell lines and 3,105 tissues) across 26 cancer types.
Model for Deriving an Optimal Biomarker Signature
R language [42] was used to program an algorithm to derive an optimal biomarker signature for a cancer type of interest (Figure 1). The 3,244 samples were randomly allocated to the training or test sets (Table 1). A maximum of 50 samples per tissue type were assigned to the training set (688 samples) and the remainder to the test set (2,556 samples). The training set was used to generate the optimal biomarker signatures for each cancer type, while the test set was reserved for final, external validation. Biomarker signatures were generated for the eight tissue types that account for approximately 80% of CUP cases (colorectum, kidney, liver, lung, ovary, pancreas, prostate, and stomach).
Figure 1.
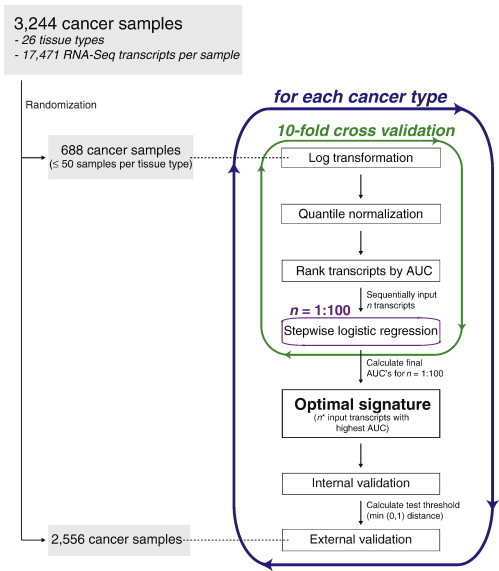
Algorithm for extracting optimal cancer biomarker signatures from RNA-Seq dataset. AUC, area under the receiver operating characteristic curve.
Table 1.
Allocation of Cancer Samples to RNA-Seq Training and Test Sets
| Cancer Type | All | Training Set | Test Set |
|---|---|---|---|
| Adrenal gland | 3 | 3 | 0 |
| Acute myeloid leukemia | 174 | 50 | 124 |
| Bladder | 70 | 50 | 20 |
| Breast | 864 | 50 | 814 |
| Cervix | 8 | 8 | 0 |
| Colorectum | 244 | 50 | 194 |
| Endometrium | 333 | 50 | 283 |
| Germ cell | 1 | 1 | 0 |
| Kidney | 24 | 24 | 0 |
| Liver | 15 | 15 | 0 |
| Head and neck | 263 | 50 | 213 |
| Lung | 348 | 50 | 298 |
| Lymphoma | 11 | 11 | 0 |
| Medulloblastoma | 1 | 1 | 0 |
| Melanoma | 136 | 50 | 86 |
| Merkel cell | 3 | 3 | 0 |
| Myeloproliferative neoplasm | 9 | 9 | 0 |
| Neuroblastoma | 2 | 2 | 0 |
| Neuroepithelioma | 1 | 1 | 0 |
| Oropharynx | 4 | 4 | 0 |
| Ovary | 418 | 50 | 368 |
| Pancreas | 76 | 50 | 26 |
| Prostate | 154 | 50 | 104 |
| Rhabdomyosarcoma | 1 | 1 | 0 |
| Salivary gland | 4 | 4 | 0 |
| Stomach | 77 | 50 | 27 |
| Total | 3244 | 688 | 2556 |
Transcript normalization
Transcript reads were normalized with log transformation followed by quantile normalization to account for variations between and within datasets, such as differences in the amount of starting material and reported transcript units. The entire 688-sample training set was then divided into 10 randomly generated subsets, each with an equal proportion of samples of the cancer type of interest. A 10-fold cross-validation method was used to train the model on 9-fold and test each signature on the remaining 1-fold.
Univariate transcript analysis
Within each 9-fold training subset, area under the receiver operating characteristic (ROC) curve values (AUC) [43] were calculated for each of the 17,471 transcripts. The transcripts were then sorted by decreasing AUC.
Stepwise logistic regression
To generate an optimal signature, an iterative approach was used, rather than inputting all 17,471 transcripts into the model, which would increase computational burden and the likelihood of overfitting. The top 100 transcripts, based on univariate AUC rank, were introduced into a stepwise logistic regression model, to determine the optimal signature at each iteration for n, between 1 and 100 input transcripts. Logistic regression was performed in both directions to optimize the Akaike information criterion (AIC) [44], [45], [46] so that at each step, it was calculated whether the current signature would be improved not only by adding the next variable but also by discarding any of the variables present within the currently optimized signature. The final signatures were used to calculate the predicted likelihood of each sample in the remaining 1-fold being of that cancer type, given n input transcripts.
Biomarker signature selection
A final “cross-validated AUC” was determined for each signature generated from n transcripts, based on the calculated predictions for each sample compared to their true identities. The optimal biomarker signature was determined to be the one generated from the top n* number of transcripts that yielded the highest cross-validated AUC. The entire 688-sample training set was then used as the input training set to generate a final, optimal biomarker signature based on the top n* transcripts.
Internal validation
Each cancer biomarker signature was internally validated by using the entire 688-sample training set as the input. Each sample received a predicted value, m, between 0 and 1, indicating likelihood of the sample being the cancer type of interest. The predicted values were then used to generate ROC curves for each signature. Optimal score thresholds, k, (above which was defined as “positive” for that cancer type and below which was “negative”) were calculated by selecting the point on the ROC curve with the minimum distance from (0,1), which represents a perfect test of 100% sensitivity and specificity [47].
External validation
Each cancer biomarker signature was then externally validated against the reserved 2,556-sample test set using the optimal score thresholds. Overall sensitivity and specificity were calculated for each cancer signature.
Duplicate cancer predictions
Each of the 2,556 cancer samples in the reserved test set was tested using each of the eight cancer biomarker signatures. Those samples that predicted positive for more than one cancer type were assigned the cancer type that had the highest relative predicted value, defined as [m – k]/[1 – k].
Additional analysis
Graphs were plotted using GraphPad Prism. The heat map was generated with Cluster 3.0 [48] and visualized using TreeView [49]. Statistical analysis was performed using R and GraphPad Prism.
Results
The results of our biomarker-generating model are shown in Figure 2. For all eight cancer types, the maximum, cross-validated AUC was obtained within the first 100 input transcripts. Interestingly, cross-validated AUC plots of the colorectal, lung, pancreas, and stomach cancer samples yielded prominent peaks, beyond which the inclusion of additional transcripts worsened the biomarker signature’s accuracy. Conversely, liver and ovarian cancer samples yielded relatively flat curves of near-perfect cross-validated AUC’s, suggesting that these cancer types have such unique biomarker profiles that many highly accurate signatures may be generated. Optimal signatures for each tissue type were objectively determined by selecting the number of input transcripts, n*, that corresponded with the maximum cross-validated AUC (Figure 2, red points).
Figure 2.
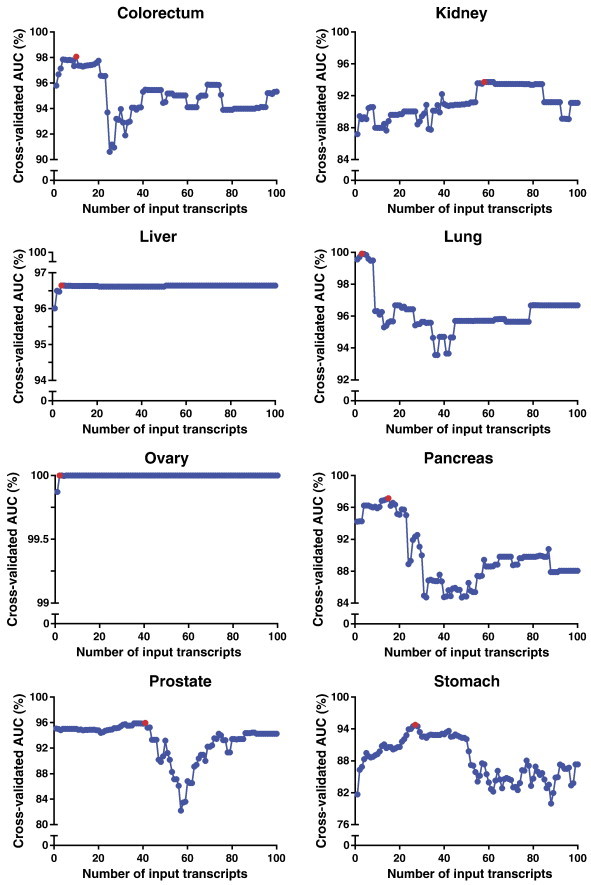
Scatter plots of 10-fold cross-validation method to determine optimal biomarker signatures for 8 different cancer types. Points highlighted in red indicate the highest, cross-validated AUC for each cancer type. AUC, area under the receiver operating characteristic curve.
Next, using the entire 688-sample training set as the input test set, the final list of transcripts was generated for each cancer biomarker signature, by performing stepwise logistic regression of the top n* transcripts. The entire model was then run again using a new, random allocation of training and test samples. The final biomarker signatures for each cancer type were concordant with the signatures generated from the first randomization (Table 2), with an overall cosine similarity measurement of 0.53 [50].
Table 2.
Cancer Biomarker Signatures Generated from Two Separate Randomizations
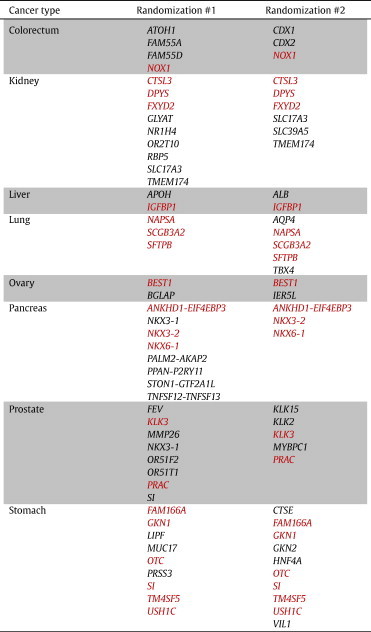
Transcripts highlighted in red are common between the two signatures.
ROC curves were then generated for each biomarker signature, yielding high AUC’s (Figure 3); for comparison, lines of identity are shown, representing a random test that has no prognostic value. From each cancer signature ROC curve, threshold cut-offs minimizing the distance to (0,1) were calculated to use in subsequent external validation testing (Figure 3, red points).
Figure 3.
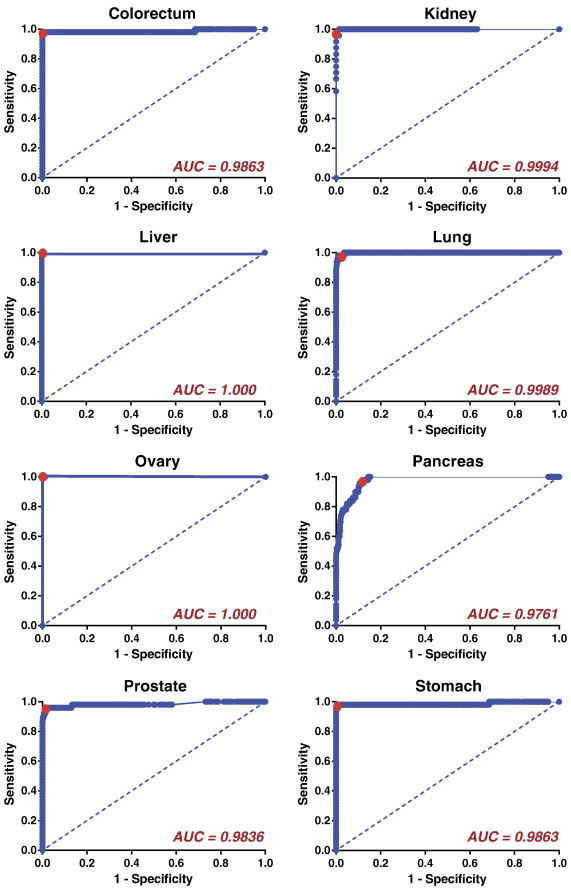
Internal validation of eight cancer-specific biomarker signatures yields high area under the ROC curve values. The entire 688-sample RNA-Seq training set was used as the test set for each cancer signature. Dotted lines indicate lines of identity. Points of minimum distance to (0,1) are highlighted in red. ROC, receiver operating characteristic; AUC, area under the ROC curve.
The cancer biomarker signatures were then externally validated using the reserved 2,556-sample RNA-Seq test set. Each sample was tested against each of the 8 biomarker signatures and predicted to be positive or negative for that cancer type based on the threshold cut-offs calculated previously. The true identities of each of the 2,556 samples were then compared to the predicted identities, and high sensitivity (92.0% ± 3.15%; mean ± standard deviation) and specificity (97.7% ± 2.99%) were demonstrated for each biomarker signature (Table 3). A heat map representation of the 43 transcripts comprising the 8 biomarker signatures for all 3,244 samples illustrated the strength with which each cancer signature successfully distinguished samples from among the different cancer types (Figure 4). Despite similarities in expression patterns across different cancers, the transcript signatures were nonetheless highly specific, requiring elevated expression across all the biomarker transcripts for a given cancer type (Table 3). Of the 3,244 samples, 2.9% had positive predictions for more than one cancer. These samples with duplicate predictions were assigned the final identity of the cancer type with the highest relative predicted value, yielding an overall accuracy of 90.5% (Table 4).
Table 3.
External Validation of Eight Cancer-Specific Biomarker Signatures Using 2,556-Sample RNA-Seq Test Set
| Colorectum | Kidney | Liver | Lung | Ovary | Pancreas | Prostate | Stomach | |
|---|---|---|---|---|---|---|---|---|
| TP | 174 | 0 | 0 | 274 | 360 | 24 | 95 | 24 |
| TN | 2355 | 2533 | 2552 | 2222 | 2188 | 2389 | 2424 | 2305 |
| FP | 7 | 23 | 4 | 36 | 0 | 141 | 28 | 224 |
| FN | 20 | 0 | 0 | 24 | 8 | 2 | 9 | 3 |
| Sensitivity (%) | 89.7 | - | - | 91.9 | 97.8 | 92.3 | 91.3 | 88.9 |
| Specificity (%) | 99.7 | 99.1 | 99.8 | 98.4 | 100 | 94.4 | 98.9 | 91.1 |
TP, true positive; TN, true negative; FP, false positive; FN, false negative.
Figure 4.
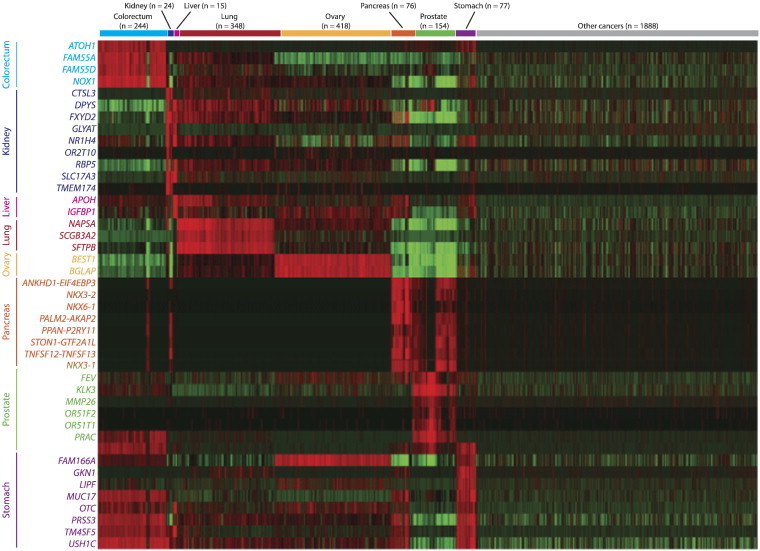
RNA-Seq heat map of 8 cancer-specific biomarker signatures (rows) across all 3,244 cancer samples (columns).
Table 4.
Overall Performance of RNA-Seq Biomarker Generating Algorithm for Predicting Tissue of Origin in 2,556 Cancer Samples
| Samples | % | ||
|---|---|---|---|
| TP | 946 | Sensitivity | 95.0 |
| TN | 1366 | Specificity | 87.6 |
| FP | 194 | PPV | 83.0 |
| FN | 50 | NPV | 96.5 |
| Accuracy | 90.5 |
Samples with duplicate cancer predictions were assigned the identity with the highest predicted value. TP, true positive; TN, true negative; FP, false positive; FN, false negative; PPV, positive predictive value; NPV, negative predictive value.
Discussion
In this study, we demonstrate an effective and efficient model for extracting highly sensitive and specific cancer biomarker signatures from a large RNA-Seq dataset. This technique yielded transcript signatures for the top eight cancer types that cause metastatic CUP. The robustness of the final signatures was demonstrated by external validation, through testing of a large test set randomly allocated a priori. By reserving these 2,556 samples solely for external validation, this test set represents a large dataset of “clinical unknowns” (i.e., identities unknown to the training model). The high sensitivities and specificities achieved with each of the cancer biomarker signatures therefore represent a realistic approximation of the accuracy with which the signatures may be used to predict tissue origin of a CUP sample in the clinical setting.
Many of the cancer biomarkers identified in this study have previously been well-characterized in their respective cancer types. The prostate cancer signature includes KLK3, which is responsible for encoding prostate-specific antigen, the serine protease used as a serum marker in prostate cancer screening and disease monitoring [51], as well as PRAC, which is known to be highly expressed in prostate cancers [52], [53]. Similarly, NKX3-1 is an androgen-regulated homeobox gene, which transcriptionally regulates oxidative damage response and is required for prostate stem cell maintenance; aberrant expression has been found to correlate strongly with prostate cancer progression [54], [55], [56], [57], [58], [59]. In addition, a recent study demonstrated that immunohistochemical staining with the kidney biomarker FXYD2, a Na-K-ATPase regulator, is highly sensitive and specific for renal cell carcinoma [60]. Similarly, NOX1 was highly expressed in our colorectal cancer samples, as confirmed in prior studies, which stimulates mitogenesis and angiogenesis though a ROS-mediated mechanism; NOX1 expression has also been found to correlate strongly with activating KRAS mutations, which are present in approximately 50% of colorectal tumors [61], [62]. IGFBP1, is a hepatocyte-derived secreted protein required for normal liver regeneration by inhibiting proapoptotic signals [63], with overexpression previously identified in hepatocellular cancers [64], as well as in our study. The lung biomarker NAPSA is a well characterized proteinase expressed in type II pneumocytes [65], [66], [67], whose expression has high sensitivity and specificity for distinguishing primary lung adenocarcinoma from metastatic pulmonary lesions from other primaries [67]. The pancreas biomarker NKX6-1 is a transcriptional regulator that has been shown to play an important role in beta cell differentiation during pancreatic development [68], [69], [70], [71]. Similarly, GKN1 is highly expressed in the gastric epithelium, providing protection to the antral mucosa and promoting healing after injury; it also acts as a tumor suppressor and is down-regulated compared to normal gastric tissues [72], [73], [74], [75], [76] but in our model was still significantly overexpressed as compared to other cancer types.
Our cancer signatures also identified transcripts that have not previously been associated with the cancer types of interest. Although not yet characterized in ovarian cancer, BEST1, which forms calcium-activated chloride channels across epithelial cells to promote cell proliferation [77], has been shown to be up-regulated in colon cancers [78]. Similarly, hypermethylation of DPYS, a gene important in pyrimidine metabolism, has been identified in prostate, colon, and breast cancers, as well as melanomas [79], [80], [81], but in our study, high expression was most sensitive and specific for kidney cancer. Interestingly, the transmembrane glycoprotein SI was highly expressed in both prostate and gastric cancer samples. Mutations in SI have previously been identified in head and neck, colorectal, and ovarian cancers, and in a recent study, SI mutations resulted in significant gene enrichment in oxidative phosphorylation, glycolysis/gluconeogenesis, and B-cell receptor signaling pathways, for promoting malignant progression in chronic lymphocytic leukemia [82].
In our pancreatic cancer signature, our model also identified several gene fusions not previously associated with this disease, including ANKHD1-EIF4EBP3, a readthrough transcript of the neighboring cell survival scaffolding gene ANKHD1 and the downstream translational repressor EIF4EBP3, both of which are effectors of the RAS/MAPK pathway [83], [84], which is known to play a critical role in the development and progression of pancreatic cancer [85], [86], [87], [88]. The prostate biomarker NKX3-1 and related family member NKX3-2 also comprised the pancreas signature. While the role of NKX3-2 in pancreatic cancer has not yet been characterized, its role in chondreogenesis and skeletal development has been well studied, acting as a transcriptional repressor downstream of SHH through interactions with the signal transduction protein SMAD4 [89], [90], [91], [92]. SHH and its related hedgehog-signaling pathways are well-known mediators of pancreatic carcinogenesis and are the targets of many new therapeutics [88], [93], [94], [95]. Similarly, inactivation of the tumor suppressor SMAD4 plays a critical role in the development of pancreatic cancer and correlates with increased tumor aggressiveness and poor prognosis [96], [97], [98], [99]. It is important to note that a common difficulty encountered in the analysis of pancreatic adenocarcinoma tissues is frequent contamination by a dense, desmoplastic stroma that characteristically surround these tumor cells and can occupy up to 90% of a tumor sample’s content [100]. However, in our study of 76 pancreatic cancer samples, we were nonetheless able to extract an 8-transcript signature to distinguish pancreatic samples from other cancer types with high sensitivity and specificity.
As compared to other studies focused on distinguishing tissue of origin for CUP, our study has multiple strengths. We analyzed a large number of cancer samples from 26 different tissue types (3,244 samples as compared to the previous studies analyzing fewer than 800 samples) [15], [16], [17], [18], [19], [23], [24], [25], [26]. In addition, we used multiple validation methods to strengthen our biomarker signatures, specifically reserving 2,556 samples for external validation testing, to yield an overall accuracy of 90.5%. This is as compared to previously reported classification accuracies of 76% to 89% [15], [16], [17], [19], [23], [24]. Finally, the use of RNA-Seq expression data has a number of potential advantages over microarray techniques, as previously outlined, including wide genome coverage, which allowed us to identify several new biomarkers, such as BEST1 in ovarian cancer and the gene fusion ANKHD1-EIF4EBP3 in pancreatic cancer. To our knowledge, this is the first CUP study using large-scale RNA-Seq data for both training and validation to demonstrate a highly accurate model for cancer prediction.
One of the limitations of our study is that, although RNA-Seq allows for the capability to detect unmapped genes, in this proof-of-concept study, we limited our analysis to only annotated transcripts. While computationally more intensive, a dataset comprised of chromosomal positions rather than annotated genes would allow for additional discovery of novel biomarkers and could potentially improve the accuracy. In addition, in our dataset, there were insufficient kidney and liver samples to allocate to our test set for external validation; however, the kidney and liver biomarker signatures nonetheless yielded strong specificities of > 99%.
We have demonstrated the strength of this model in its ability to accurately and efficiently distinguish samples of one type (i.e., cancer type of interest) from another (i.e., heterogeneous group of other cancer types). While this study focused specifically on deriving lineage-specific cancer signatures by RNA-Seq, this model may be applied to any large dataset to query other clinical questions, such as deriving a signature that distinguishes premalignant and cancerous lesions from inflammatory and other benign conditions. Similarly, given a dataset of patients with known clinical outcomes, our model could be used to derive biomarker signatures that identify patients who would respond to a given therapy or identify patients with worse outcomes compared to clinically and histologically-matched cohorts who should be targeted for aggressive treatment and surveillance.
Conclusions
In this study, we introduced a computational model that successfully extracted accurate, lineage-specific cancer signatures for the top eight tissue types that contribute to CUP using RNA-Seq. Through external validation of a large dataset, we have shown how these signatures may be used to accurately identify tumors of unknown origin, demonstrating the translational potential of not only our cancer biomarker signatures but also the model itself, which may be applied to other clinical queries.
Footnotes
Conflict of Interests: None.
References
- 1.Pimiento J.M., Teso D., Malkan A., Dudrick S.J., Palesty J.A. Cancer of unknown primary origin: a decade of experience in a community-based hospital. Am J Surg. 2007;194:833–837. doi: 10.1016/j.amjsurg.2007.08.039. [discussion 837–838] [DOI] [PubMed] [Google Scholar]
- 2.Pavlidis N., Pentheroudakis G. Cancer of unknown primary site: 20 questions to be answered. Ann Oncol. 2010;21(Suppl. 7):vii303–vii307. doi: 10.1093/annonc/mdq278. [DOI] [PubMed] [Google Scholar]
- 3.Shaw P.H., Adams R., Jordan C., Crosby T.D. A clinical review of the investigation and management of carcinoma of unknown primary in a single cancer network. Clin Oncol (R Coll Radiol) 2007;19:87–95. doi: 10.1016/j.clon.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Pavlidis N., Briasoulis E., Hainsworth J., Greco F.A. Diagnostic and therapeutic management of cancer of an unknown primary. Eur J Cancer. 2003;39:1990–2005. doi: 10.1016/s0959-8049(03)00547-1. [DOI] [PubMed] [Google Scholar]
- 5.Hainsworth J.D., Greco F.A. Treatment of patients with cancer of an unknown primary site. N Engl J Med. 1993;329:257–263. doi: 10.1056/NEJM199307223290407. [DOI] [PubMed] [Google Scholar]
- 6.Blaszyk H., Hartmann A., Bjornsson J. Cancer of unknown primary: clinicopathologic correlations. APMIS. 2003;111:1089–1094. doi: 10.1111/j.1600-0463.2003.apm1111203.x. [DOI] [PubMed] [Google Scholar]
- 7.Occult Primary (Cancer of Unknown Primary [CUP]). Version 3. 2014. National Comprehensive Cancer Network clinical Practice Gidelines in Oncology. [nccn.org] [Google Scholar]
- 8.Werling R.W., Yaziji H., Bacchi C.E., Gown A.M. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: An immunohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27:303–310. doi: 10.1097/00000478-200303000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Kaufmann O., Dietel M. Thyroid transcription factor-1 is the superior immunohistochemical marker for pulmonary adenocarcinomas and large cell carcinomas compared to surfactant proteins A and B. Histopathology. 2000;36:8–16. doi: 10.1046/j.1365-2559.2000.00801.x. [DOI] [PubMed] [Google Scholar]
- 10.Dennis J.L., Hvidsten T.R., Wit E.C., Komorowski J., Bell A.K., Downie I., Mooney J., Verbeke C., Bellamy C., Keith W.N. Markers of adenocarcinoma characteristic of the site of origin: development of a diagnostic algorithm. Clin Cancer Res. 2005;11:3766–3772. doi: 10.1158/1078-0432.CCR-04-2236. [DOI] [PubMed] [Google Scholar]
- 11.Kaufmann O., Fietze E., Mengs J., Dietel M. Value of p63 and cytokeratin 5/6 as immunohistochemical markers for the differential diagnosis of poorly differentiated and undifferentiated carcinomas. Am J Clin Pathol. 2001;116:823–830. doi: 10.1309/21TW-2NDG-JRK4-PFJX. [DOI] [PubMed] [Google Scholar]
- 12.Motzer R.J., Rodriguez E., Reuter V.E., Bosl G.J., Mazumdar M., Chaganti R.S. Molecular and cytogenetic studies in the diagnosis of patients with poorly differentiated carcinomas of unknown primary site. J Clin Oncol. 1995;13:274–282. doi: 10.1200/JCO.1995.13.1.274. [DOI] [PubMed] [Google Scholar]
- 13.Atkin N.B., Baker M.C. Specific chromosome change, i(12p), in testicular tumours? Lancet. 1982;2:1349. doi: 10.1016/s0140-6736(82)91557-4. [DOI] [PubMed] [Google Scholar]
- 14.Ilson D.H., Motzer R.J., Rodriguez E., Chaganti R.S., Bosl G.J. Genetic analysis in the diagnosis of neoplasms of unknown primary tumor site. Semin Oncol. 1993;20:229–237. [PubMed] [Google Scholar]
- 15.Bloom G., Yang I.V., Boulware D., Kwong K.Y., Coppola D., Eschrich S., Quackenbush J., Yeatman T.J. Multi-platform, multi-site, microarray-based human tumor classification. Am J Pathol. 2004;164:9–16. doi: 10.1016/S0002-9440(10)63090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramaswamy S., Tamayo P., Rifkin R., Mukherjee S., Yeang C.H., Angelo M., Ladd C., Reich M., Latulippe E., Mesirov J.P. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci U S A. 2001;98:15149–15154. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shedden K.A., Taylor J.M., Giordano T.J., Kuick R., Misek D.E., Rennert G., Schwartz D.R., Gruber S.B., Logsdon C., Simeone D. Accurate molecular classification of human cancers based on gene expression using a simple classifier with a pathological tree-based framework. Am J Pathol. 2003;163:1985–1995. doi: 10.1016/S0002-9440(10)63557-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tothill R.W., Kowalczyk A., Rischin D., Bousioutas A., Haviv I., van Laar R.K., Waring P.M., Zalcberg J., Ward R., Biankin A.V. An expression-based site of origin diagnostic method designed for clinical application to cancer of unknown origin. Cancer Res. 2005;65:4031–4040. doi: 10.1158/0008-5472.CAN-04-3617. [DOI] [PubMed] [Google Scholar]
- 19.Rosenfeld N., Aharonov R., Meiri E., Rosenwald S., Spector Y., Zepeniuk M., Benjamin H., Shabes N., Tabak S., Levy A. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26:462–469. doi: 10.1038/nbt1392. [DOI] [PubMed] [Google Scholar]
- 20.Ojala K.A., Kilpinen S.K., Kallioniemi O.P. Classification of unknown primary tumors with a data-driven method based on a large microarray reference database. Genome Med. 2011;3:63. doi: 10.1186/gm279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ross D.T., Scherf U., Eisen M.B., Perou C.M., Rees C., Spellman P., Iyer V., Jeffrey S.S., Van de Rijn M., Waltham M. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 22.Horlings H.M., van Laar R.K., Kerst J.M., Helgason H.H., Wesseling J., van der Hoeven J.J., Warmoes M.O., Floore A., Witteveen A., Lahti-Domenici J. Gene expression profiling to identify the histogenetic origin of metastatic adenocarcinomas of unknown primary. J Clin Oncol. 2008;26:4435–4441. doi: 10.1200/JCO.2007.14.6969. [DOI] [PubMed] [Google Scholar]
- 23.Ma X.J., Patel R., Wang X., Salunga R., Murage J., Desai R., Tuggle J.T., Wang W., Chu S., Stecker K. Molecular classification of human cancers using a 92-gene real-time quantitative polymerase chain reaction assay. Arch Pathol Lab Med. 2006;130:465–473. doi: 10.5858/2006-130-465-MCOHCU. [DOI] [PubMed] [Google Scholar]
- 24.Talantov D., Baden J., Jatkoe T., Hahn K., Yu J., Rajpurohit Y., Jiang Y., Choi C., Ross J.S., Atkins D. A quantitative reverse transcriptase-polymerase chain reaction assay to identify metastatic carcinoma tissue of origin. J Mol Diagn. 2006;8:320–329. doi: 10.2353/jmoldx.2006.050136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu J., Getz G., Miska E.A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B.L., Mak R.H., Ferrando A.A. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 26.Quon G., Morris Q. ISOLATE: a computational strategy for identifying the primary origin of cancers using high-throughput sequencing. Bioinformatics. 2009;25:2882–2889. doi: 10.1093/bioinformatics/btp378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sultan M., Schulz M.H., Richard H., Magen A., Klingenhoff A., Scherf M., Seifert M., Borodina T., Soldatov A., Parkhomchuk D. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pentheroudakis G., Golfinopoulos V., Pavlidis N. Switching benchmarks in cancer of unknown primary: from autopsy to microarray. Eur J Cancer. 2007;43:2026–2036. doi: 10.1016/j.ejca.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 30.Briasoulis E., Pavlidis N. Cancer of unknown primary origin. Oncologist. 1997;2:142–152. [PubMed] [Google Scholar]
- 31.Abbruzzese J.L., Abbruzzese M.C., Lenzi R., Hess K.R., Raber M.N. Analysis of a diagnostic strategy for patients with suspected tumors of unknown origin. J Clin Oncol. 1995;13:2094–2103. doi: 10.1200/JCO.1995.13.8.2094. [DOI] [PubMed] [Google Scholar]
- 32.Stewart J.F., Tattersall M.H., Woods R.L., Fox R.M. Unknown primary adenocarcinoma: incidence of overinvestigation and natural history. Br Med J. 1979;1:1530–1533. doi: 10.1136/bmj.1.6177.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Chevalier T., Cvitkovic E., Caille P., Harvey J., Contesso G., Spielmann M., Rouesse J. Early metastatic cancer of unknown primary origin at presentation. A clinical study of 302 consecutive autopsied patients. Arch Intern Med. 1988;148:2035–2039. [PubMed] [Google Scholar]
- 34.Hamilton C.S., Langlands A.O. ACUPS (adenocarcinoma of unknown primary site): a clinical and cost benefit analysis. Int J Radiat Oncol Biol Phys. 1987;13:1497–1503. doi: 10.1016/0360-3016(87)90317-8. [DOI] [PubMed] [Google Scholar]
- 35.Kirsten F., Chi C.H., Leary J.A., Ng A.B., Hedley D.W., Tattersall M.H. Metastatic adeno or undifferentiated carcinoma from an unknown primary site–natural history and guidelines for identification of treatable subsets. Q J Med. 1987;62:143–161. [PubMed] [Google Scholar]
- 36.Nystrom J.S., Weiner J.M., Wolf R.M., Bateman J.R., Viola M.V. Identifying the primary site in metastatic cancer of unknown origin. Inadequacy of roentgenographic procedures. JAMA. 1979;241:381–383. [PubMed] [Google Scholar]
- 37.Moertel C.G., Reitemeier R.J., Schutt A.J., Hahn R.G. Treatment of the patient with adenocarcinoma of unknown origin. Cancer. 1972;30:1469–1472. doi: 10.1002/1097-0142(197212)30:6<1469::aid-cncr2820300609>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 38.Osteen R.T., Kopf G., Wilson R.E. In pursuit of the unknown primary. Am J Surg. 1978;135:494–497. doi: 10.1016/0002-9610(78)90026-0. [DOI] [PubMed] [Google Scholar]
- 39.Kothari V., Wei I., Shankar S., Kalyana-Sundaram S., Wang L., Ma L.W., Vats P., Grasso C.S., Robinson D.R., Wu Y.M. Outlier kinase expression by RNA sequencing as targets for precision therapy. Cancer Discov. 2013;3:280–293. doi: 10.1158/2159-8290.CD-12-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.International Cancer Genome Consortium. 2014. http://www.icgc.org. [Google Scholar]
- 41.The Cancer Genome Atlas. 2014. http://cancergenome.nih.gov. [Google Scholar]
- 42.R language. 2014. http://www.R-project.org. [Google Scholar]
- 43.Sing T., Sander O., Beerenwinkel N., Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- 44.Everitt B.S., Hothorn T. CRC PressINC; 2010. A handbook of statistical analyses using R. [Vol.] [Google Scholar]
- 45.Akaike H. A new look at the statistical model identification. IEEE Trans Autom Control. 1974;19:716–723. [Google Scholar]
- 46.McCullagh P., Nelder J.A. Chapman & Hall; 1989. Generalized linear model. [Vol.] [Google Scholar]
- 47.Perkins N.J., Schisterman E.F. The inconsistency of “optimal” cutpoints obtained using two criteria based on the receiver operating characteristic curve. Am J Epidemiol. 2006;163:670–675. doi: 10.1093/aje/kwj063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Hoon M.J., Imoto S., Nolan J., Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 49.Eisen M. TreeView. 2002. http://rana.lbl.gov/EisenSoftware.htm. [Google Scholar]
- 50.Pesquita C., Faria D., Falcao A.O., Lord P., Couto F.M. Semantic similarity in biomedical ontologies. PLoS Comput Biol. 2009;5:e1000443. doi: 10.1371/journal.pcbi.1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eeles R.A., Kote-Jarai Z., Giles G.G., Olama A.A.A., Guy M., Jugurnauth S.K., Mulholland S., Leongamornlert D.A., Edwards S.M., Morrison J. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–321. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 52.Edwards S., Campbell C., Flohr P., Shipley J., Giddings I., Te-Poele R., Dodson A., Foster C., Clark J., Jhavar S. Expression analysis onto microarrays of randomly selected cDNA clones highlights HOXB13 as a marker of human prostate cancer. Br J Cancer. 2005;92:376–381. doi: 10.1038/sj.bjc.6602261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu X.F., Olsson P., Wolfgang C.D., Bera T.K., Duray P., Lee B., Pastan I. PRAC: A novel small nuclear protein that is specifically expressed in human prostate and colon. Prostate. 2001;47:125–131. doi: 10.1002/pros.1055. [DOI] [PubMed] [Google Scholar]
- 54.Bhatia-Gaur R., Donjacour A.A., Sciavolino P.J., Kim M., Desai N., Young P., Norton C.R., Gridley T., Cardiff R.D., Cunha G.R. Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 1999;13:966–977. doi: 10.1101/gad.13.8.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bowen C., Bubendorf L., Voeller H.J., Slack R., Willi N., Sauter G., Gasser T.C., Koivisto P., Lack E.E., Kononen J. Loss of NKX3.1 expression in human prostate cancers correlates with tumor progression. Cancer Res. 2000;60:6111–6115. [PubMed] [Google Scholar]
- 56.Ellwood-Yen K., Graeber T.G., Wongvipat J., Iruela-Arispe M.L., Zhang J., Matusik R., Thomas G.V., Sawyers C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 57.He W.W., Sciavolino P.J., Wing J., Augustus M., Hudson P., Meissner P.S., Curtis R.T., Shell B.K., Bostwick D.G., Tindall D.J. A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics. 1997;43:69–77. doi: 10.1006/geno.1997.4715. [DOI] [PubMed] [Google Scholar]
- 58.Nelson W.G., De Marzo A.M., Isaacs W.B. Prostate cancer. N Engl J Med. 2003;349:366–381. doi: 10.1056/NEJMra021562. [DOI] [PubMed] [Google Scholar]
- 59.Wang X., Julio M.K.D., Economides K.D., Walker D., Yu H., Halili M.V., Hu Y.P., Price S.M., Abate-Shen C., Shen M.M. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaut J.P., Crimmins D.L., Lockwood C.M., McQuillan J.J., Ladenson J.H. Expression of the Na +/K+− transporting ATPase gamma subunit FXYD2 in renal tumors. Mod Pathol. 2013;26:716–724. doi: 10.1038/modpathol.2012.202. [DOI] [PubMed] [Google Scholar]
- 61.Laurent E., McCoy Iii J.W., Macina R.A., Liu W., Cheng G., Robine S., Papkoff J., Lambeth J.D. Nox1 is over-expressed in human colon cancers and correlates with activating mutations in K-Ras. Int J Cancer. 2008;123:100–107. doi: 10.1002/ijc.23423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kamata T. Roles of Nox1 and other Nox isoforms in cancer development. Cancer Sci. 2009;100:1382–1388. doi: 10.1111/j.1349-7006.2009.01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leu J.I., Crissey M.A.S., Taub R. Massive hepatic apoptosis associated with TGF-β1 activation after Fas ligand treatment of IGF binding protein-1–deficient mice. J Clin Invest. 2003;111:129–139. doi: 10.1172/JCI16712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Borlak J., Meier T., Halter R., Spanel R., Spanel-Borowski K. Epidermal growth factor-induced hepatocellular carcinoma: Gene expression profiles in precursor lesions, early stage and solitary tumours. Oncogene. 2005;24:1809–1819. doi: 10.1038/sj.onc.1208196. [DOI] [PubMed] [Google Scholar]
- 65.Chuman Y., Bergman A., Ueno T., Saito S., Sakaguchi K., Alaiya A.A., Franzen B., Bergman T., Arnott D., Auer G. Napsin A, a member of the aspartic protease family, is abundantly expressed in normal lung and kidney tissue and is expressed in lung adenocarcinomas. FEBS Lett. 1999;462:129–134. doi: 10.1016/s0014-5793(99)01493-3. [DOI] [PubMed] [Google Scholar]
- 66.Hirano T., Auer G., Maeda M., Hagiwara Y., Okada S., Ohira T., Okuzawa K., Fujioka K., Franzen B., Hibi N. Human tissue distribution of TA02, which is homologous with a new type of aspartic proteinase, napsin A. Jpn J Cancer Res. 2000;91:1015–1021. doi: 10.1111/j.1349-7006.2000.tb00879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ueno T., Linder S., Elmberger G. Aspartic proteinase napsin is a useful marker for diagnosis of primary lung adenocarcinoma. Br J Cancer. 2003;88:1229–1233. doi: 10.1038/sj.bjc.6600879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Habener J.F., Kemp D.M., Thomas M.K. Minireview: transcriptional regulation in pancreatic development. Endocrinology. 2005;146:1025–1034. doi: 10.1210/en.2004-1576. [DOI] [PubMed] [Google Scholar]
- 69.Sander N., Sussel L., Conners J., Scheel D., Kalamaras J., Dela Cruz F., Schwitzgebel V., Hayes-Jordan A., German M. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of β-cell formation in the pancreas. Development. 2000;127:5533–5540. doi: 10.1242/dev.127.24.5533. [DOI] [PubMed] [Google Scholar]
- 70.Schwitzgebel V.M., Scheel D.W., Conners J.R., Kalamaras J., Lee J.E., Anderson D.J., Sussel L., Johnson J.D., German M.S. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development. 2000;127:3533–3542. doi: 10.1242/dev.127.16.3533. [DOI] [PubMed] [Google Scholar]
- 71.Yang L., Li S., Hatch H., Ahrens K., Cornelius J.G., Petersen B.E., Peck A.B. In vitro trans-differentiation of adult hepatic stem cells into pancreatic endocrine hormone-producing cells. Proc Natl Acad Sci U S A. 2002;99:8078–8083. doi: 10.1073/pnas.122210699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moss S.F., Lee J.W., Sabo E., Rubin A.K., Rommel J., Westley B.R., May F.E.B., Gao J., Meitner P.A., Tavares R. Decreased expression of gastrokine 1 and the trefoil factor interacting protein TFIZ1/GKIM2 in gastric cancer: influence of tumor histology and relationship to prognosis. Clin Cancer Res. 2008;14:4161–4167. doi: 10.1158/1078-0432.CCR-07-4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oien K.A., McGregor F., Butler S., Ferrier R.K., Downie I., Bryce S., Burns S., Keith W.N. Gastrokine I is abundantly and specifically expressed in superficial gastric epithelium, down-regulated in gastric carcinoma, and shows high evolutionary conservation. J Pathol. 2004;203:789–797. doi: 10.1002/path.1583. [DOI] [PubMed] [Google Scholar]
- 74.Xing R., Li W., Cui J., Zhang J., Kang B., Wang Y., Wang Z., Liu S., Lu Y. Gastrokine 1 induces senescence through p16/Rb pathway activation in gastric cancer cells. Gut. 2012;61:43–52. doi: 10.1136/gut.2010.230623. [DOI] [PubMed] [Google Scholar]
- 75.Yoon J.H., Kang Y.H., Choi Y.J., Park I.S., Nam S.W., Lee J.Y., Lee Y.S., Park W.S. Gastrokine 1 functions as a tumor suppressor by inhibition of epithelial-mesenchymal transition in gastric cancers. J Cancer Res Clin Oncol. 2011;137:1697–1704. doi: 10.1007/s00432-011-1051-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoon J.H., Song J.H., Zhang C., Jin M., Kang Y.H., Nam S.W., Lee J.Y., Park W.S. Inactivation of the Gastrokine 1 gene in gastric adenomas and carcinomas. J Pathol. 2011;223:618–625. doi: 10.1002/path.2838. [DOI] [PubMed] [Google Scholar]
- 77.Kunzelmann K., Kongsuphol P., Aldehni F., Tian Y., Ousingsawat J., Warth R., Schreiber R. Bestrophin and TMEM16-Ca2 + activated Cl − channels with different functions. Cell Calcium. 2009;46:233–241. doi: 10.1016/j.ceca.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 78.Spitzner M., Martins J.R., Soria R.B., Ousingsawat J., Scheidt K., Schreiber R., Kunzelmann K. Eag1 and bestrophin 1 are up-regulated in fast-growing colonic cancer cells. J Biol Chem. 2008;283:7421–7428. doi: 10.1074/jbc.M703758200. [DOI] [PubMed] [Google Scholar]
- 79.Chung W., Kwabi-Addo B., Ittmann M., Jelinek J., Shen L., Yu Y., Issa J.P.J. Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling. PLoS One. 2008:3. doi: 10.1371/journal.pone.0002079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Furuta J., Nobeyama Y., Umebayashi Y., Otsuka F., Kikuchi K., Ushijima T. Silencing of peroxiredoxin 2 and aberrant methylation of 33 CpG islands in putative promoter regions in human malignant melanomas. Cancer Res. 2006;66:6080–6086. doi: 10.1158/0008-5472.CAN-06-0157. [DOI] [PubMed] [Google Scholar]
- 81.Vasiljević N., Wu K., Brentnall A.R., Kim D.C., Thorat M.A., Kudahetti S.C., Mao X., Xue L., Yu Y., Shaw G.L. Absolute quantitation of DNA methylation of 28 candidate genes in prostate cancer using pyrosequencing. Dis Markers. 2011;30:151–161. doi: 10.3233/DMA-2011-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rodríguez D., Ramsay A.J., Quesada V., Garabaya C., Campo E., Freije J.M.P., López-Otín C. Functional analysis of sucrase-isomaltase mutations from chronic lymphocytic leukemia patients. Hum Mol Genet. 2013;22:2273–2282. doi: 10.1093/hmg/ddt078. [DOI] [PubMed] [Google Scholar]
- 83.Prakash T., Sharma V.K., Adati N., Ozawa R., Kumar N., Nishida Y., Fujikake T., Takeda T., Taylor T.D. Expression of conjoined genes: another mechanism for gene regulation in eukaryotes. PLoS One. 2010;5:e13284. doi: 10.1371/journal.pone.0013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Poulin F., Brueschke A., Sonenberg N. Gene fusion and overlapping reading frames in the mammalian genes for 4E-BP3 and MASK. J Biol Chem. 2003;278:52290–52297. doi: 10.1074/jbc.M310761200. [DOI] [PubMed] [Google Scholar]
- 85.Almoguera C., Shibata D., Forrester K., Martin J., Arnheim N., Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 86.Bos J.L. ras Oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 87.Hingorani S.R., Petricoin Iii E.F., Maitra A., Rajapakse V., King C., Jacobetz M.A., Ross S., Conrads T.P., Veenstra T.D., Hitt B.A. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 88.Morton J.P., Mongeau M.E., Klimstra D.S., Morris J.P., Yie C.L., Kawaguchi Y., Wright C.V.E., Hebrok M., Lewis B.C. Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis. Proc Natl Acad Sci U S A. 2007;104:5103–5108. doi: 10.1073/pnas.0701158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim D.W., Lassar A.B. Smad-dependent recruitment of a histone deacetylase/Sin3A complex modulates the bone morphogenetic protein-dependent transcriptional repressor activity of Nkx3.2. Mol Cell Biol. 2003;23:8704–8717. doi: 10.1128/MCB.23.23.8704-8717.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lefebvre V., Smits P. Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res C Embryo Today. 2005;75:200–212. doi: 10.1002/bdrc.20048. [DOI] [PubMed] [Google Scholar]
- 91.Murtaugh L.C., Zeng L., Chyung J.H., Lassar A.B. The chick transcriptional repressor Nkx3.2 acts downstream of Shh to promote BMP-dependent axial chondrogenesis. Dev Cell. 2001;1:411–422. doi: 10.1016/s1534-5807(01)00039-9. [DOI] [PubMed] [Google Scholar]
- 92.Zeng L., Kempf H., Murtaugh L.C., Sato M.E., Lassar A.B. Shh establishes an Nkx3.2/Sox9 autoregulatory loop that is maintained by BMP signals to induce somitic chondrogenesis. Genes Dev. 2002;16:1990–2005. doi: 10.1101/gad.1008002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Katoh Y., Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009;9:873–886. doi: 10.2174/156652409789105570. [DOI] [PubMed] [Google Scholar]
- 94.Lee C.J., Dosch J., Simeone D.M. Pancreatic cancer stem cells. J Clin Oncol. 2008;26:2806–2812. doi: 10.1200/JCO.2008.16.6702. [DOI] [PubMed] [Google Scholar]
- 95.Thayer S.P., Di Magliano M.P., Heiser P.W., Nielsen C.M., Roberts D.J., Lauwers G.Y., Qi Y.P., Gysin S., Fernández-del Castillo C., Yajnik V. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hahn S.A., Schutte M., Shamsul Hoque A.T.M., Moskaluk C.A., Da Costa L.T., Rozenblum E., Weinstein C.L., Fischer A., Yeo C.J., Hruban R.H. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 97.Hezel A.F., Kimmelman A.C., Stanger B.Z., Bardeesy N., DePinho R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 98.Rozenblum E., Schutte M., Goggins M., Hahn S.A., Panzer S., Zahurak M., Goodman S.N., Sohn T.A., Hruban R.H., Yeo C.J. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57:1731–1734. [PubMed] [Google Scholar]
- 99.Wilentz R.E., Iacobuzio-Donahue C.A., Argani P., McCarthy D.M., Parsons J.L., Yeo C.J., Kern S.E., Hruban R.H. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002–2006. [PubMed] [Google Scholar]
- 100.Mahadevan D., Von Hoff D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6:1186–1197. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]