

Abstract
The role of tuberous sclerosis complex (TSC) in the pathogenesis of pancreatic cancers remains largely unknown. The present study shows that neurogenin 3 directed Cre deletion of Tsc1 gene induces the development of pancreatic acinar carcinoma. By cross-breeding the Neurog3-cre mice with Tsc1loxp/loxp mice, we generated the Neurog3-Tsc1−/− transgenic mice in which Tsc1 gene is deleted and mTOR signaling activated in the pancreatic progenitor cells. All Neurog3-Tsc1−/− mice developed notable adenocarcinoma-like lesions in pancreas starting from the age of 100 days old. The tumor lesions are composed of cells with morphological and molecular resemblance to acinar cells. Metastasis of neoplasm to liver and lung was detected in 5% of animals. Inhibition of mTOR signaling by rapamycin significantly attenuated the growth of the neoplasm. Relapse of the neoplasm occurred within 14 days upon cessation of rapamycin treatment. Our studies indicate that activation of mTOR signaling in the pancreatic progenitor cells may trigger the development of acinar carcinoma. Thus, mTOR may serve as a potential target for treatment of pancreatic acinar carcinoma.
Abbreviations: ACC, acinar cell carcinoma; 4EBP-1, 4E binding protein 1; mTOR, mammlian target of rapamycin; Neurog3, neurogenin 3; PDA, pancreatic ductal adenocarcinoma; S6, ribosomal protein S6; TSC, tuberous sclerosis complex
Introduction
Pancreatic cancer is one of the most leading causes of cancer deaths in the United States and many other developed countries [1]. Despite the comprehensive treatment options including surgery resection, radiotherapy, and chemotherapy, the five-year survival rate remains steady at 3% to 5% in the past three decades [2]. The pancreatic cancer is usually silent at the early stage, and most patients have already reached the advanced stage at the time of diagnosis [3]. Most of the pancreatic cancers are ductal adenocarcinoma (PDA). Acinar cell carcinoma (ACC) is rare, accounting for 1% of all pancreatic cancer diagnoses [4]. ACC differs significantly from the more common PDA. The histologic and immunohistochemical (IHC) findings are distinctive and the molecular alterations share little common with PDA [5]. ACC lacks mutations in genes associated with PDA, such as V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS), tumor protein 53 (TP53), deleted in pancreatic cancer 4 (DPC4), and p16 [6]. Instead, ACC may have abnormalities in the adenomatosis polyposis coli gene and β-catenin [5]. Contemporary treatment of pancreatic cancer is often ineffective because current understanding on the risk factors and etiology of pancreatic cancer from environmental and genetic perspective is incomplete. Using the genetically engineered mouse models to recapitulate human pancreatic neoplasia, scientists have gradually uncovered the core signaling pathways and regulatory processes critical for the development of pancreatic cancers [7].
One of the important signaling pathways implicated in the development of pancreatic cancers is the tuberous sclerosis complex (TSC)-mechanistic target of rapamycin (mTOR) pathway [8]. As a member of the phakomatoses and tumor suppressor genes, TSC is associated with benign tumors, most notably in the central nervous system. In addition, functional links between the TSC genes and other tumor suppressors responsible for Cowden's disease (PTEN), Peutz-Jeghers syndrome (LKB1), and familial polyposis (APC) suggest that TSC may contribute to the development of pancreatic cancers [9]. As the inhibitors of the mTOR signaling pathway, TSC1 and TSC2 form as the heterodimer to inhibit mTOR signaling and subsequently attenuate cell growth, proliferation, division, and survival [10]. Expression analyses have indicated that the expression of genes in the mTOR pathway is altered frequently in pancreatic neuroendocrine tumors (15%), but not in pancreatic ductal adenocarcinomas (0.80%) [11]. However, Pten is lost in a large number of PDAC tumors and mTOR might play a role in PDAC stem cells [12], [13]. Animal studies have demonstrated that deletion of ribosomal protein (rp) S6 renders the mice resistant to pancreatic cancer precursor lesions induced by either 7,12-Dimethylbenz(a)anthracene (DMBA) or mutation of Kras [14]. Furthermore, inhibition of mTOR signaling attenuates the growth of pancreatic cancer cell lines both in vitro and in vivo [15], [16], [17], [18]. Everolimus, a selective inhibitor of mTORC1, has been shown to increase progression-free survival in a subset of pancreatic neuroendocrine tumors patients with advanced disease [19]. All these data suggest that the TSC-mTOR signaling pathway is associated with the development of pancreatic cancers. Nonetheless, two important questions remain to be unraveled. Does activation of mTOR signaling lead to the development of pancreatic cancer? Which type of pancreatic cancer, ie the neuroendocrine, the ductal adenocarcinoma, or acinar cell carcinoma is caused by activation of mTOR signaling. To address these questions, we generated a new genetically engineered mouse model Neurog3-Tsc1−/− mice by cross-breeding the Neurog3-cre mice with Tsc1loxp/loxp mice. The functional domain of Tsc1 gene was deleted and mTOR signaling thus activated in pancreatic progenitor cells expressing neurogenin 3 in the transgenic mice. Adenocarcinoma-like lesions were observed starting from 100 days in Neurog3-Tsc1−/− mice. The tumor lesions are malignant epithelial neoplasm composed of cells with morphological resemblance to acinar cells and with evidence of α-amylase production, a typical pancreatic exocrine enzyme. Metastasis to liver and lung was observed in 5% of animals. The neoplasm was significantly attenuated by rapamycin, a selective inhibitor of mTOR signaling. Relapse of the neoplasm occurred within 14 days upon cessation of the rapamycin treatment. All these data suggest that activation of mTOR signaling in the pancreatic progenitor cells may trigger the development of acinar carcinoma.
Materials and Methods
Materials
Rabbit anti-S6, rabbit anti-phospho-S6 (ser235, 236), rabbit anti-4EBP-1, rabbit anti-phospho-4EBP-1 (Thr37, 46), rabbit anti-β-Catenin (species cross-reactivity: human, mouse, and rat) and mouse anti-proliferating cell nuclear antigen (PCNA) were from Cell Signaling Technology (Beverly, MA). Rabbit anti-α-Amylase was from Sigma Chemical Co. (St. Louis, MO). Mouse anti-CA19-9 was from Origene (Beijing, China). Goat anti-insulin A (C-12) (species cross-reactivity: human, mouse, and rat), Goat anti-glucagon (N-17) (species cross-reactivity: human, mouse, and rat), rabbit anti-somatostatin (FL-116) (species cross-reactivity: human, mouse, and rat), goat anti-rabbit fluoresceinisothiocyanate-conjugated IgG, donkey anti-goat fluoresceinisothiocyanate- conjugated IgG, donkey anti-goat Texas Red-conjugated IgG, goat anti-mouse Texas Red-conjugated IgG, and chicken anti-rabbit fluoresceinisothiocyanate-conjugated IgG and rapamycin were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Dimethylsulfoxide was from Sigma Chemical Co. (St. Louis, MO). Aprotinin was purchased from Amersham Biosciences (Pittsburgh, PA). IRDye-conjugated affinity purified anti-rabbit, anti-mouse IgGs were purchased from Rockland (Gilbertsville, PA).
Animals and Animal Care
Neuroge3-Cre mice that express the Cre recombinase gene under the control of the Neurogenin3 gene promoter, as well as Tsc1loxP/loxP mice, in which exons 17 and 18 of the Tsc1 gene are flanked by loxP sites by homologous recombination, were purchased from the Jackson Laboratory (Bar Harbor, ME). For Tsc1loxp/loxp mice, a targeting vector containing a loxP-flanked neomycin resistance-thymidine kinase gene cassette preceding exon 17 and a third loxP site downstream of exon 18 was electroporated into 129S4/SvJae derived J1 embryonic stem (ES) cells. Correctly targeted ES cells were injected into C57BL/6 J blastocysts. Chimeric mice were backcrossed for germ-line transmission to 129/SvJae mice. For Neurogenin3-cre mice, a bacterial artificial chromosome (BAC) encoding the mouse neurogenin 3 sequence was modified by the insertion of a Cre recombinase gene, preceded by nuclear localization sequence, into exon 1 of the neurogenin 3 gene at the initiator methionine. The BAC was injected into FVB/N embryos which were next implanted into pseudopregnant CD1 females. Founder line C1 was obtained and backcrossed to CD1 mice for 7 generations after which mice were intercrossed. The Neurog3-Tsc1−/− mice were generated by breeding Tsc1loxP/loxP mice with Neurog3-Cre mice. Control experiments were performed using littermate Tsc1loxP/loxP animals. Deletion of the Tsc1 gene was validated by the absence of its mRNA and protein, and the increased phosphorylation of 4EBP-1 and S6, its downstream signaling molecules, in pancreas tissues. Mice were housed on a 12 h–12 h light–dark cycle. Normal chow and water were available ad libitum.
Genotyping
For genotyping, genomic DNA was extracted from tail cuttings. Polymerase chain reactions were carried out for each animal to test for the presence of the TSC1 and/or the deletion of its exons 17 and 18 using LoxP primers, and Cre constructs using cre-specific primers, respectively.
Immunostaining
Mice were deeply anesthetized using sodium pentobarbital (40 mg/kg, ip), perfused transcardially with 20 ml 0.1 M PBS (pH 7.4), followed by 20 ml 4% paraformaldehyde in PBS. The whole pancreas was quickly removed and fixed in 4% paraformaldehyde, followed by dehydrated, embedded in wax, and sectioned at 6 μm. Tissue sections were de-paraffinized and hydrated by graded washes with xylene and ethanol. Antigen retrieval was accomplished by boiling in 10 mM sodium citrate acid buffer (pH 6.0) for 15 minutes. Endogenous peroxidase activity was blocked by incubation of slides in 3% H2O2 for 10 minutes. Nonspecific binding was blocked with 1% bovine serum albumin. Sections were then incubated with primary antibodies overnight at 4°C. For immunohistochemistry, secondary antibody staining was performed with a poly-HRP detection system for rabbit or mouse primary antibody (polink-2 plus HRP, Golden Bridge International, USA) for 1 hour at room temperature. Immunoreactivity was detected using diaminobenzidine substrate (peroxide substrate kit, SK-4100; Vector Laboratories) for 2–5 minutes. Slides were then counterstained with Mayer’s hematoxylin before dehydration and mounting. For immunofluorescence, secondary antibodies used included chicken anti-rabbit fluoresceinisothiocyanate-conjugated IgG (1:100), goat anti-mouse Texas Red-conjugated IgG (1:100 dilution), goat anti-rabbit fluoresceinisothiocyanate-conjugated IgG (1:100 dilution), donkey anti-goat fluoresceinisothiocyanate-conjugated IgG (1:100 dilution) and donkey anti-goat Texas Red-conjugated IgG (1:100 dilution). Pancreatic tissues were incubated with secondary antibody for 1 hour at room temperature. Fluorescent signals were observed and photomicrographs taken under a confocal laser scanning microscope (Leica, Germany). Controls included substituting primary antibodies with relevant IgGs.
Western Blot Analysis
Tissues extracts were immunoblotted with TSC1 (1:600 dilution), S6 (1:1000 dilution), pS6 (1:1000 dilution), β-actin(1:1000 dilution), 4EBP-1 (1:1000 dilution), p4EBP-1 (1:1000 dilution), β-Catenin (1:1000 dilution) as described previously [20], [21]. Briefly, pancreatic tissues were isolated and homogenized in lysis buffer. Proteins were subjected to SDS-PAGE with a 10% running gel, then transferred to a polyvinylidene fluoride membrane. Membranes were incubated for 1 hour at room temperature with 5% fat-free milk in Tris buffered saline containing Tween 20, followed by incubation overnight at 4°C with primary antibodies. Specific reaction was detected using IRDye-conjugated second antibody and visualized using the Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE).
Rapamycin Treatment
Rapamycin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was initially dissolved in 100% DMSO, stored at − 20 °C, and further diluted in DMSO immediately before use. Animals bearing the neoplasm as confirmed by ultrasound imaging were treated daily by intraperitoneal injection with rapamycin at the dose of 1 mg/kg for consecutive 28 days. The neoplasm was monitored by ultrasound at the times indicated before, during, and after treatment with rapamycin.
Ultrasound Imaging
Image of pancreatic neoplasm was examined by an experienced hepatic-biliary-pancreatic radiologist using a Philips iU22 machine (Philips Ultrasound, USA) according to the manufacturer’s guidelines. Ultrasound images of the lesions were recorded using a high-frequency probe (5 to 12 MHz) prior and subsequent to rapamycin treatment at the times indicated.
Statistical Analysis
Data were expressed as mean ± SEM and analyzed by repeated-measures analysis of variance, one-way ANOVA, Student–Newman–Keuls test (comparisons between multiple groups), unpaired Student’s t test (between two groups) and non-parametric test as appropriate, using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA). A value of P < .05 denotes statistical significance.
Study Approval
The animals used in this study were reviewed and approved by the Animal Care and Use Committee of Peking University in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85–23, revised 1996).
Results
Generation of Neurog3-Tsc1−/− Mouse Strain and Validation of mTOR Activation in Pancreas
By cross-breeding the Neurog3-Cre mice with Tsc1loxp-loxp mice, we have generated a mouse strain Neurog3-Tsc1−/−. Analysis of pancreatic TSC1 and its related mTOR signaling molecules validated the deletion of Tsc1 gene in the pancreas. As shown in Figure 1, TSC1 expression was detected at a relative high level in the pancreas in the wild-type littermate control (WT), whereas Neurog3-Tsc1−/− (TN) mice demonstrated a significant reduction of TSC1 immunoreactivity. Since TSC1 has been recognized as the negative upstream regulator of mTOR signaling in many cell types, we next examined the mTOR activity in the pancreas. Phosphorylation of S6 ribosomal protein (pS6) and 4E binding protein 1 (p4EBP1), the downstream targets of mTOR [10], and proliferating cell nuclear antigen (PCNA) was significantly increased in pancreas of the Neurog3-Tsc1−/− (TN) mice relative to the control littermates (WT). These data indicate that deletion of TSC1 results in activation of mTOR signaling in the pancreas of Neurog3-Tsc1−/−(TN) mice. Thus, we have successfully generated a mouse strain with pancreatic deletion of TSC1 and subsequent activation of mTOR signaling.
Figure 1.
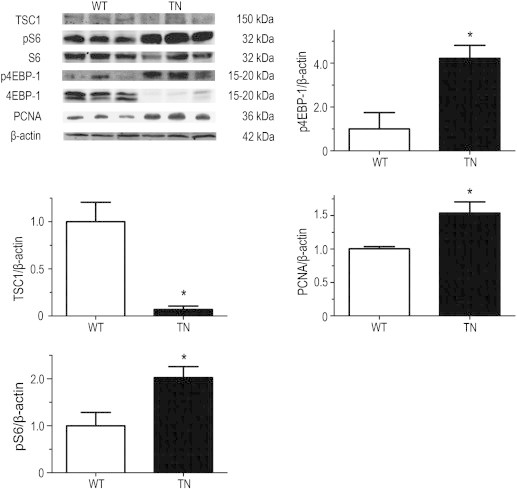
Deletion of TSC1 and activation of mTOR signaling in the pancreas of Neurog3-TSC1 −/− transgenic mice. The Neurog3-Tsc1−/− mice were generated by breeding Tsc1loxP/loxP mice with Neurog3-Cre mice. TSC1, phospho-S6 (pS6), and phospho-4EBP-1 in pancreatic tissue with tumor nodules were examined by Western blotting using specific antibodies. S6, 4EBP-1, and β-actin were used as loading controls. Shown are representative results from 6 experiments. Signal intensity of TSC1, pS6, p4EBP-1, and PCNA were analyzed and expressed as mean ± SEM. n = 6. *P < .05 versus wild-type mice.
Development of Pancreatic Neoplasm in Neurog3-Tsc1−/− Mice
As a critical molecule controlling the cell proliferation, mTOR influences tumor development and growth in many tissues [22], [23], [24]. We thus explored the occurrence of pancreatic neoplasm in the Neurog3-Tsc1−/− mice. As shown in Figure 2A, abdominal swelling appeared and palpable mass was found in Neurog3-Tsc1−/− mice starting from 100 days old. By the age of 300 days old, all Neurog3-Tsc1−/− animals (n = 25) developed palpable mass in the pancreas (Figure 2B). Analysis of survival rate demonstrated that all Neurog3-Tsc1−/− mice died by the age of 13 months, whereas 100% of wild-type littermates survived at least 16 months (Figure 2C). Autopsies revealed multinodular pancreatic tumors ranging from 3 to 25 mm in diameter of individual nodule and 3 to 4 grams in total weight of tumor nodules (Figure 2, D and E). No tumor was observed in wild-type littermates up to the age of 16 months.
Figure 2.
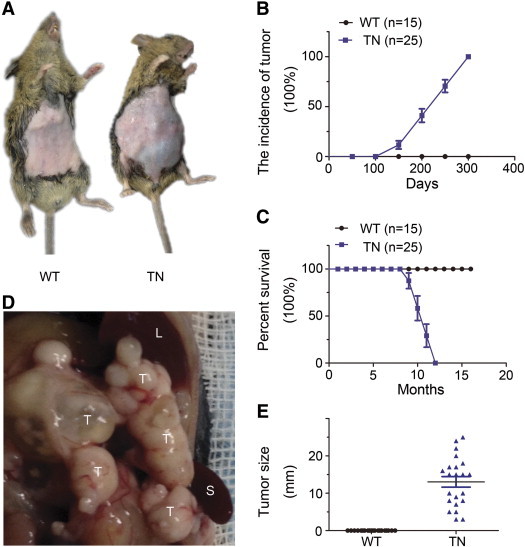
Pancreatic neoplasms inNeurog3-Tsc1−/− mice. (A) Abdominal swelling and palpable mass were observed in Neurog3-Tsc1−/− mice (TN), but not in wild-type littermates (WT). (B) Pancreatic tumor incidence and survival rate (C) for Neurog3-Tsc1−/− (n = 25) and wildtypemice (n = 15) analyzed by Kaplan-Meier. (D) Gross photograph of multi-nodular pancreatic tumors. T, L, and S indicate tumor, liver, and spleen, respectively. (E) Tumor size. Tumor size was measured at the week of 45.
Recapitulation of the Pathologic Features of Human Pancreatic Acinar Adenocarcinoma in Neurog3-Tsc1−/− Mice
Both morphological and immunoreactivity analysis of the pancreatic tumor in Neurog3-Tsc1 −/− mice revealed the hallmarks of human pancreatic acinar adenocarcinomas (Figure 3). Tumor lesions are composed of well circumscribed nodules with necrotic foci (Figure 3A). Microscopic examination revealed distinct epithelial neoplasm with evidence of low-grade malignancy. Tumors are highly cellular with gross lobules separated by scant fibrous stroma. Tumor cells are arranged predominantly in acinar pattern mixed with trabecular or solid structure (Figure 3B). Nuclei are somewhat irregular in size and polarized in acinar formations (Figure 3B). Nuclear atypia and mitoses are rare. Hemorrhage and necrosis are common.
Figure 3.
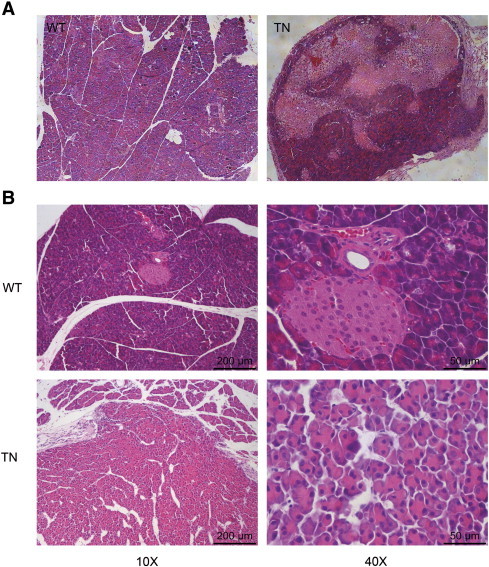
Histology of the pancreas from WT and TN mice. (A) Cross section of normal pancreas (left) and pancreatic neoplasm (right) from representative of 8-month-old wild-type littermates (WT) and Neurog3-Tsc1−/− (TN) mice respectively. (B) Micrograph of normal pancreas (upper) and neoplasm (lower). Hematoxylin and eosin (H&E) staining showed the distinct epithelial neoplasm with cellular and structure resemblance of pancreatic acini.
The abundant eosinophilic granular apical cytoplasm in the tumor cells suggests the morphological resemblance to acinar cells. We thus examined the presence of zymogen granules by immunostaining to detect the expression of α-amylase, a pancreatic exocrine enzyme. As shown in Figure 4, moderate α-amylase immunoreactivity was detected in tumor cells of Neurog3-Tsc1−/− mice, while surrounding normal acinar cells demonstrated a strong signal (Figure 4A, red). Unlike normal pancreas in Neurog3-Tsc1−/− transgenic mice and wild-type littermates, the acinar lesions demonstrated negative immunoreactivity for carbohydrate antigen 19–9 (CA19-9) (Figure 4A, green), a selective marker for pancreatic duct epithelial cells. Furthermore, immunoreactivity for chromogranin A, an endocrine cell marker, was not detected in the tumor cells, whereas surrounding islet cells showed strong signal (Figure 4A). Significant increases in levels of amylase mRNA (Figure 4C) and protein (Figure 4B) were detected in the pancreatic tumor of Neurog3-Tsc1−/− mice relative to wild-type littermates.
Figure 4.
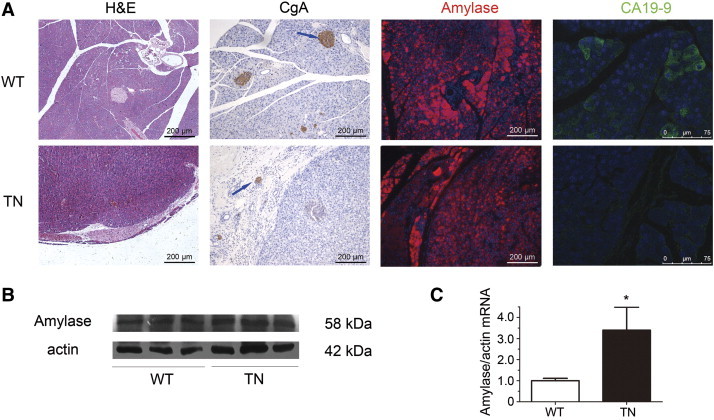
Histological and immunoreactivity analysis. (A) Shown are the representative of normal pancreas and neoplasm from 8-month-old wild-type (WT) (upper panel) and Neurog3-Tsc1−/− (TN) mice (lower panel) respectively. (1) H&E staining shows solid, well-demarcated pancreatic adenocarcinoma in TN mice. (2) Chromogranin A immunoreactivity (CgA, arrow) was observed in pancreatic islets but not the neoplasm. (3) Amylase immunoreactivity was strong in pancreatic acinar cells, while neoplasm demonstrated modest staining. (4) Ductal cells stained positive for CA19-9, a specific marker for ductal cells, while neoplasm had no staining. (B) Amylase in pancreas with tumor nodules was examined by western blot. Shown are representative results from 6 experiments. (C) Expression of Amylase mRNA in pancreas with tumor nodules was examined by qPCR. n = 5. *P < .05 versus wild type mice.
Metastasis of the Pancreatic Neoplasm in Neurog3-Tsc1−/− Mice
Given the invasive behavior of the pancreatic acinar adenocarcinomas, we conducted a histologic survey of organs for evidence of metastasis. This survey revealed metastasis of the pancreatic neoplasm to the liver (Figure 5A) and the lungs (Figure 5B) in 2 (5%) of 40 Neuroge3-Tsc1−/− mice. The metastasis lesions in liver was histologically confirmed to be of pancreatic origin by the staining of α-amylase (Figure 5C), although lymph nodes metastasis was not detected.
Figure 5.
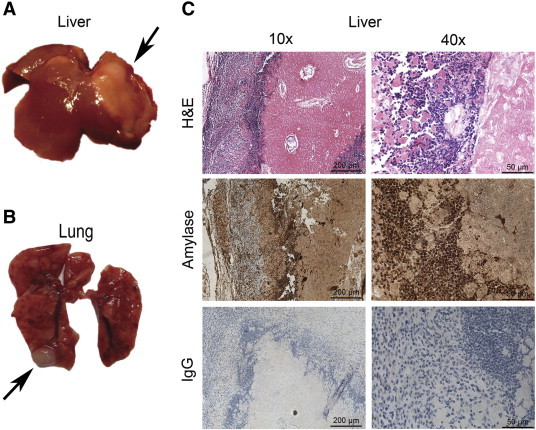
Metastasis of the pancreatic neoplasm in Neurog3-TSC1−/− mice. (A) Hepatic metastases (arrows). (B) Pulmonary metastases (arrows). (C) The hepatic lesion was demonstrated to be of pancreatic origin by histological and immunoreactivity examination. Cells of the metastatic neoplasm resembled the original pancreatic lesion and were positive for α-amylase.
Attenuation of the Pancreatic Neoplasm by Rapamycin in Neuroge3-Tsc1−/− mice
To further demonstrate that Tsc1-mTOR signaling is critical for the development of pancreatic acinar adenocarcinomas in Neuroge3-Tsc1−/− mice, we used rapamycin to block the mTOR signaling and evaluated its effect on the tumor growth. The neoplasm was monitored by transabdominal ultrasonography which was chosen because of its non-invasive, non-radioactive nature and the comparative difficulty in imaging the mouse pancreas with other technologies [25]. Prior to rapamycin treatment (defined as day 0), the existence of pancreatic neoplasm was confirmed and recorded by B-mode ultrasonic examination. Abdominal mass with well-defined heterogeneous internal echoes mass was depicted by B-mode ultrasonic examination. Signature of lower or even non-echoes was also detected, suggesting the presence of different degrees of necrosis in tumor lesions. As shown in Figure 6, daily intraperitoneal injection of rapamycin at a dose of 1 mg/kg significantly reduced the pancreatic neoplasm in Neuroge3-Tsc1−/− mice. The diameter of lesions was significantly reduced from 0.89 ± 0.04 cm at day 0 to 0.58 ± 0.02 cm and 0.40 ± 0.03 cm at day 14 and 28 respectively (P < 0.05, n = 6). Interestingly, relapse of tumors occurred upon cessation of rapamycin treatment with tumor size returned to the level prior to treatment (0.83 ± 0.06 cm versus 0.89 ± 0.04, P < .05).
Figure 6.
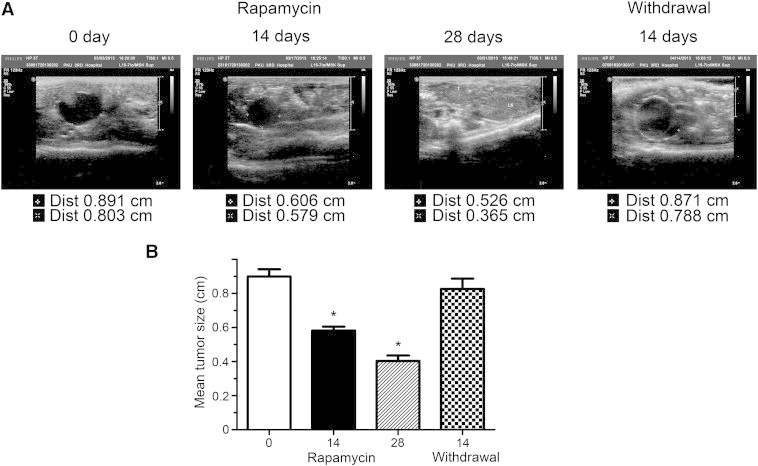
Ultrasound imaging of the pancreatic lesions in Neurog3-TSC1−/− mice. (A) Representative ultrasound images of the pancreatic neoplasms before, during, and after treatment with rapamycin. Mice were treated intraperitoneally with 1 mg/kg rapamycin for 28 consecutive days. The pancreatic lesions were examined on day 0, 14, 28 during treatment, and 14 days after stoppage of rapamycin treatment. (B) The diameter of pancreatic lesions was measured and expressed as mean ± SEM in cm. Asterisk denotes P < .01. n = 5.
Discussion
The present study demonstrates that activation of mTOR signaling by deletion of Tsc1 gene in cells expressing neurogenin 3 results in the development of pancreatic acinar carcinoma. This general conclusion is supported by the following distinct observations: (1) level of TSC1 is significantly reduced, whereas mTOR activity increased in the pancreas of Neuroge3-Tsc1−/− mice relative to wild-type littermates; (2) hundred percent of Neuroge3-Tsc1−/− mice develops pancreatic neoplasm by the age of 300 days; (3) no Neuroge3-Tsc1−/− mouse survives over 13 months; (4) the pancreatic neoplasm resembles low grade acinar adenocarcinoma with tumor cells arranged in acinar pattern and evidence of pancreatic exocrine enzyme production; (5) metastasis to liver and lungs occurs in 5% of Neuroge3-Tsc1−/− mice; (6) inhibition of mTOR signaling by rapamycin attenuates the growth of the pancreatic neoplasm.
Genetically engineered mouse models are invaluable for the understanding of the pathobiology of pancreatic neoplasia, and thus provide an indispensible tool for the development of novel therapeutic drugs for this deadly lesion. Most of the mouse transgenic models of pancreatic neoplasms were developed using the elastase 1 (EL-1) promoter, an enzyme with the highest specificity for the pancreas [26], [27], [28]. Dependent on the oncogenes used, three general types of exocrine pancreatic neoplasms have been reported: pure acinar cell neoplasms, dominantly acinar cell neoplasms with conspicuous ductal metaplasia, and duct-like neoplasms. Since elastase I is solely expressed in the differentiated acinar cells, the presence of duct cell neoplasms in the transgenic mice such as EL-1-myc, EL-1-Kras, and EL-1-Hras suggests the background changes of acinar to ductal cell metaplasia. Similar changes of acinar to endocrine cell neoplasms have also been observed in EL-1-Tag mice which develop significant incidence of islet cell tumors. The present study suggests that acinar adenocarcinoma may also arise from the neurogenin 3 positive cells. Neurogenin 3 in the pancreatic progenitor cells is often considered as the master transcriptional factor required for the determination of the pancreatic endocrine destiny [29], [30], [31]. All pancreatic endocrine cells derive from neurogenin 3-positive cells, while deletion of neurogenin 3 eliminates the differentiation of pancreatic endocrine cells [32]. However, lineage tracing studies have revealed that neurogenin 3-expressing cells can eventually adopt acinar or ductal fates if its expression level is not sufficient to secure the islet destiny [33], [34], [35]. This observation is in line with our finding of acinar adenocarcinoma in Neuroge3-Tsc1−/− mice. Whether the competence of neurogenin 3 positive cells to give rise to acinar neoplasm is an intrinsic property of these progenitors or depends on the change of the pancreatic microenvironment in the Neuroge3-Tsc1−/− mice requires further investigation. Further experiments using other transcriptional factors such as Pdx-1 to drive the deletion of TSC1 or deletion of Pten are important to clarify whether the deletion of TSC1 gene driven by neurogenin3 specifically causes pancreatic acinar carcinoma. Since absence of Pten in pancreas driven by Pdx-1 has been reported to cause progressive premalignant lesions mainly in the ductal linage [36], it is unlikely that TSC1 gene deletion driven by Pten-cre will cause the development of acinar carcinoma. Whether deletion of TSC1 by p48 or elastase-cre will lead to the development of pancreatic acinar carcinoma is of interest.
The molecular events driving the development of pancreatic carcinoma remains poorly understood. Our study provides the first evidence demonstrating that activation of mTOR signaling in pancreas is sufficient to induce the development of pancreatic acinar carcinoma. This finding is consistent with previous studies showing that chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Evidences from the studies on hepatocellular carcinoma indicate that activation of mTOR may promote the carcinogenesis by a mechanism involving unresolved endoplasmic reticulum stress and defects in autophagy [37]. Previous study also demonstrates that inhibition of mTOR signaling by rapamycin significantly attenuates the growth of AR42J cells, a rat pancreatic exocrine tumor cell line. In addition, several evidences have suggested the relevance of mTOR signaling with human pancreatic acinar cell carcinoma. [1] Acinar cell carcinoma of the pancreas has been described in a patient with Peutz-Jeghers syndrome and biallelic inactivation of LKB1 allele demonstrated in this tumor, suggesting a causative role of LKB1 in the development of pancreatic acinar cell carcinoma [38], [39]. Of note, LKB1 activates AMPK which then inhibits mTOR signaling. [2] Treatment with mTOR inhibitor results in partial remission of the tumor [39]. [3] Somatic mutations of EIF4G2, the downstream effector of mTOR, have been identified in human pancreatic neoplasms with acinar differentiation [40]. As an important nutrient-sensitive protein kinase, mTOR may thus represent a key molecular link between pancreatic cancer risk and environmental factors such as high fat diet.
In conclusion, this study provides the first evidence that activation of mTOR signaling in the pancreatic progenitor cells may trigger the development of acinar carcinoma. These results imply that mTOR signaling in the pancreas may serve as a potential target for treatment of pancreatic acinar carcinoma.
Acknowledgements
The authors have no additional acknowledgements.
Footnotes
Conflict of interest: The authors do not have any disclosure to report.
Financial support: None of the above authors have been funded by any National Institutes of Health (NIH) or other applicable grants for this research. The research was funded independently by National Natural Science Foundation of China (81030012, 81330010, 81390354) and American Diabetes Association grant #1-13-BS-225.
References
- 1.Parkin D.M., Bray F., Ferlay J., Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A., Tiwari R.C., Murray T., Ghafoor A., Samuels A., Ward E., Feuer E.J., Thun M.J., American Cancer S. Cancer statistics, 2004. CA Cancer J Clin. 2004;54(1):8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- 3.Warshaw A.L., Fernandez-del Castillo C. Pancreatic carcinoma. N Engl J Med. 1992;326(7):455–465. doi: 10.1056/NEJM199202133260706. [DOI] [PubMed] [Google Scholar]
- 4.Lowery M.A., Klimstra D.S., Shia J., Yu K.H., Allen P.J., Brennan M.F., O'Reilly E.M. Acinar cell carcinoma of the pancreas: new genetic and treatment insights into a rare malignancy. Oncologist. 2011;16(12):1714–1720. doi: 10.1634/theoncologist.2011-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abraham S.C., Wu T.T., Hruban R.H., Lee J.H., Yeo C.J., Conlon K., Brennan M., Cameron J.L., Klimstra D.S. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am J Pathol. 2002;160(3):953–962. doi: 10.1016/s0002-9440(10)64917-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones S., Zhang X., Parsons D.W., Lin J.C., Leary R.J., Angenendt P., Mankoo P., Carter H., Kamiyama H., Jimeno A. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez-Mancera P.A., Guerra C., Barbacid M., Tuveson D.A. What we have learned about pancreatic cancer from mouse models. Gastroenterology. 2012;142(5):1079–1092. doi: 10.1053/j.gastro.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Kataoka K., Fujimoto K., Ito D., Koizumi M., Toyoda E., Mori T., Kami K., Doi R. Expression and prognostic value of tuberous sclerosis complex 2 gene product tuberin in human pancreatic cancer. Surgery. 2005;138(3):450–455. doi: 10.1016/j.surg.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 9.Mak B.C., Yeung R.S. The tuberous sclerosis complex genes in tumor development. Cancer Invest. 2004;22(4):588–603. doi: 10.1081/cnv-200027144. [DOI] [PubMed] [Google Scholar]
- 10.Hay N., Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 11.Jiao Y., Shi C., Edil B.H., de Wilde R.F., Klimstra D.S., Maitra A., Schulick R.D., Tang L.H., Wolfgang C.L., Choti M.A. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331(6021):1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ying H., Elpek K.G., Vinjamoori A., Zimmerman S.M., Chu G.C., Yan H., Fletcher-Sananikone E., Zhang H., Liu Y., Wang W. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discov. 2011;1(2):158–169. doi: 10.1158/2159-8290.CD-11-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsubara S., Ding Q., Miyazaki Y., Kuwahata T., Tsukasa K., Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related function. Sci Rep. 2013;3:3230. doi: 10.1038/srep03230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khalaileh A., Dreazen A., Khatib A., Apel R., Swisa S., Kidess-Bassir N., Maitra A., Meyuhas O., Dor Y., Zamir G. Phosphorylation of ribosomal protein S6 attenuates DNA damage and tumor suppression during development of pancreatic cancer. Cancer Res. 2013;73(6):1811–1820. doi: 10.1158/0008-5472.CAN-12-2014. [DOI] [PubMed] [Google Scholar]
- 15.Ito D., Fujimoto K., Mori T., Kami K., Koizumi M., Toyoda E., Kawaguchi Y., Doi R. In vivo antitumor effect of the mTOR inhibitor CCI-779 and gemcitabine in xenograft models of human pancreatic cancer. Int J Cancer. 2006;118(9):2337–2343. doi: 10.1002/ijc.21532. [DOI] [PubMed] [Google Scholar]
- 16.Glienke W., Maute L., Wicht J., Bergmann L. The dual PI3K/mTOR inhibitor NVP-BGT226 induces cell cycle arrest and regulates Survivin gene expression in human pancreatic cancer cell lines. Tumour Biol. 2012;33(3):757–765. doi: 10.1007/s13277-011-0290-2. [DOI] [PubMed] [Google Scholar]
- 17.Okada T., Sawada T., Kubota K. Rapamycin enhances the anti-tumor effect of gemcitabine in pancreatic cancer cells. Hepatogastroenterology. 2007;54(79):2129–2133. [PubMed] [Google Scholar]
- 18.Stephan S., Datta K., Wang E., Li J., Brekken R.A., Parangi S., Thorpe P.E., Mukhopadhyay D. Effect of rapamycin alone and in combination with antiangiogenesis therapy in an orthotopic model of human pancreatic cancer. Clin Cancer Res. 2004;10(20):6993–7000. doi: 10.1158/1078-0432.CCR-04-0808. [DOI] [PubMed] [Google Scholar]
- 19.Wiedenmann B., Pavel M., Kos-Kudla B. From targets to treatments: a review of molecular targets in pancreatic neuroendocrine tumors. Neuroendocrinology. 2011;94(3):177–190. doi: 10.1159/000329386. [DOI] [PubMed] [Google Scholar]
- 20.Li Y., Jiang C., Xu G., Wang N., Zhu Y., Tang C., Wang X. Homocysteine upregulates resistin production from adipocytes in vivo and in vitro. Diabetes. 2008;57(4):817–827. doi: 10.2337/db07-0617. [DOI] [PubMed] [Google Scholar]
- 21.Xu G., Li Y., An W., Li S., Guan Y., Wang N., Tang C., Wang X., Zhu Y., Li X. Gastric mammalian target of rapamycin signaling regulates ghrelin production and food intake. Endocrinology. 2009;150(8) doi: 10.1210/en.2009-0372. [3637-3644.2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu K., Ding J., Chen C., Sun W., Ning B.F., Wen W., Huang L., Han T., Yang W., Wang C. Hepatic transforming growth factor beta gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology. 2012;56(6):2255–2267. doi: 10.1002/hep.26007. [DOI] [PubMed] [Google Scholar]
- 23.Granville C.A., Warfel N., Tsurutani J., Hollander M.C., Robertson M., Fox S.D., Veenstra T.D., Issaq H.J., Linnoila R.I., Dennis P.A. Identification of a highly effective rapamycin schedule that markedly reduces the size, multiplicity, and phenotypic progression of tobacco carcinogen-induced murine lung tumors. Clin Cancer Res. 2007;13(7):2281–2289. doi: 10.1158/1078-0432.CCR-06-2570. [DOI] [PubMed] [Google Scholar]
- 24.Shapira M., Kakiashvili E., Rosenberg T., Hershko D.D. The mTOR inhibitor rapamycin down-regulates the expression of the ubiquitin ligase subunit Skp2 in breast cancer cells. Breast Cancer Res. 2006;8(4):R46. doi: 10.1186/bcr1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sastra S.A., Olive K.P. Quantification of murine pancreatic tumors by high-resolution ultrasound. Methods Mol Biol. 2013;980:249–266. doi: 10.1007/978-1-62703-287-2_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greten F.R., Wagner M., Weber C.K., Zechner U., Adler G., Schmid R.M. TGF alpha transgenic mice. A model of pancreatic cancer development. Pancreatology. 2001;1(4):363–368. doi: 10.1159/000055835. [DOI] [PubMed] [Google Scholar]
- 27.Grippo P.J., Nowlin P.S., Demeure M.J., Longnecker D.S., Sandgren E.P. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63(9):2016–2019. [PubMed] [Google Scholar]
- 28.Schaeffer B.K., Terhune P.G., Longnecker D.S. Pancreatic carcinomas of acinar and mixed acinar/ductal phenotypes in Ela-1-myc transgenic mice do not contain c-K-ras mutations. Am J Pathol. 1994;145(3):696–701. [PMC free article] [PubMed] [Google Scholar]
- 29.Apelqvist A., Li H., Sommer L., Beatus P., Anderson D.J., Honjo T., Hrabe de Angelis M., Lendahl U., Edlund H. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400(6747):877–881. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- 30.Jensen J., Heller R.S., Funder-Nielsen T., Pedersen E.E., Lindsell C., Weinmaster G., Madsen O.D., Serup P. Independent development of pancreatic alpha- and beta-cells from neurogenin3-expressing precursors: a role for the notch pathway in repression of premature differentiation. Diabetes. 2000;49(2):163–176. doi: 10.2337/diabetes.49.2.163. [DOI] [PubMed] [Google Scholar]
- 31.Gu G., Dubauskaite J., Melton D.A. Direct evidence for the pancreatic lineage: NGN3 + cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 32.Gradwohl G., Dierich A., LeMeur M., Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97(4):1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schonhoff S.E., Giel-Moloney M., Leiter A.B. Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev Biol. 2004;270(2):443–454. doi: 10.1016/j.ydbio.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 34.Wang S., Yan J., Anderson D.A., Xu Y., Kanal M.C., Cao Z., Wright C.V., Gu G. Neurog3 gene dosage regulates allocation of endocrine and exocrine cell fates in the developing mouse pancreas. Dev Biol. 2010;339(1):26–37. doi: 10.1016/j.ydbio.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beucher A., Martin M., Spenle C., Poulet M., Collin C., Gradwohl G. Competence of failed endocrine progenitors to give rise to acinar but not ductal cells is restricted to early pancreas development. Dev Biol. 2012;361(2):277–285. doi: 10.1016/j.ydbio.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu H.N., Nioka S., Li L.Z. Imaging heterogeneity in the mitochondrial redox state of premalignant pancreas in the pancreas-specific PTEN-null transgenic mouse model. Biomarker Res. 2013;1(1):6. doi: 10.1186/2050-7771-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menon S., Yecies J.L., Zhang H.H., Howell J.J., Nicholatos J., Harputlugil E., Bronson R.T., Kwiatkowski D.J., Manning B.D. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci Signal. 2012;5(217):ra24. doi: 10.1126/scisignal.2002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Wilde R.F., Ottenhof N.A., Jansen M., Morsink F.H., de Leng W.W., Offerhaus G.J., Brosens L.A. Analysis of LKB1 mutations and other molecular alterations in pancreatic acinar cell carcinoma. Mod Pathol. 2011;24(9):1229–1236. doi: 10.1038/modpathol.2011.83. [DOI] [PubMed] [Google Scholar]
- 39.Klumpen H.J., Queiroz K.C., Spek C.A., van Noesel C.J., Brink H.C., de Leng W.W., de Wilde R.F., Mathus-Vliegen E.M., Offerhaus G.J., Alleman M.A. mTOR inhibitor treatment of pancreatic cancer in a patient With Peutz-Jeghers syndrome. J Clin Oncol. 2011;29(6):e150–e153. doi: 10.1200/JCO.2010.32.7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiao Y., Yonescu R., Offerhaus G.J., Klimstra D.S., Maitra A., Eshleman J.R., Herman J.G., Poh W., Pelosof L., Wolfgang C.L. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol. 2014;232(4):428–435. doi: 10.1002/path.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]