

Abstract
Range expansions can result in founder effects, increasing genetic differentiation between expanding populations and reducing genetic diversity along the expansion front. However, few studies have addressed these effects in long-distance migratory species, for which high dispersal ability might counter the effects of genetic drift. Monarchs (Danaus plexippus) are best known for undertaking a long-distance annual migration in North America, but have also dispersed around the world to form populations that do not migrate or travel only short distances. Here, we used microsatellite markers to assess genetic differentiation among 18 monarch populations and to determine worldwide colonization routes. Our results indicate that North American monarch populations connected by land show limited differentiation, probably because of the monarch's ability to migrate long distances. Conversely, we found high genetic differentiation between populations separated by large bodies of water. Moreover, we show evidence for serial founder effects across the Pacific, suggesting stepwise dispersal from a North American origin. These findings demonstrate that genetic drift played a major role in shaping allele frequencies and created genetic differentiation among newly formed populations. Thus, range expansion can give rise to genetic differentiation and declines in genetic diversity, even in highly mobile species.
Keywords: gene surfing, dispersal, cline, Columbus hypothesis, landscape genetics, ecological genetics
1. Introduction
Range expansions often result in decreased genetic diversity and increased between-population differentiation with increasing geographical distance from the source population [1–3]. Theoretical and empirical studies show that genetic drift and serial founder events are important mechanisms producing such patterns [4–7]. In Homo sapiens, migration out of Africa resulted in serial founder effects, including a decrease in heterozygosity with increasing distance from the African origin [8–10], as well as a loss of genetic diversity in their accompanying human malaria parasite, Plasmodium falciparum [11]. Importantly, genetic drift in edge populations can override selection during range expansion [12], sometimes causing fixation of harmful alleles and extinction of beneficial alleles [13].
Serial founder events can result in gene surfing: this spatial analogue of genetic drift occurs when neutral or even deleterious alleles reach higher than expected frequencies along the front of an expansion [4,14–17]. Gene surfing has been observed in multiple species, many with relatively low dispersal rates [18,19]. While these studies demonstrated the effects of genetic drift in shaping population genetics associated with range expansions, few studies have been carried out on organisms that undertake frequent and long-distance migrations. Simulations predicted that while serial founder events can cause genetic diversity loss and greater differentiation, these effects can be reduced by high dispersal [20–22]. Indeed, the disappearance or reduction of an isolation-by-distance pattern has been shown in a handful of species capable of long-range dispersal [23,24].
Here, we study how the range expansion of a migratory species has shaped its population genetics. We focus on the monarch butterfly, Danaus plexippus, which has expanded its ancestral range to colonize locations around the globe [25]. Monarchs are famous for their annual migration from the eastern parts of Canada and the United States to overwintering sites in central Mexico [26]; however, monarchs also occur in locations around the world, ranging from the New World tropics to the Pacific Islands [25,27] and southern Europe [28]. Most monarch populations outside of North America are, in fact, non-migratory [29,30], or locally travel only short distances in the form of modest range shifts [31]. Historical records and recent genomic analyses suggest that monarchs colonized other locations throughout the world from North American origins [32].
Records of monarchs outside of the New World are limited to the last 200 years, after suitable milkweed host plants (mostly in the genus Asclepias) were introduced [27]; however, recent genome-wide analyses suggest that these colonizations may have occurred earlier [32]. Monarchs likely colonized locations around the world through serial founder effects (stepwise dispersal) or multiple independent colonization events [27,33], and were aided in reaching new locations through human transportation of both milkweeds and monarchs [34], and extreme weather events [35]. For example, monarchs were recorded in Australia in 1870 and were most probably carried there on cyclonic winds from a source population in New Caledonia [35]. Regarding eastward dispersal across the Atlantic Ocean, monarchs have been spotted along the southern Iberian Peninsula and northern Africa, where their population sizes vary seasonally [36], pointing to a potential role for multiple introductions in facilitating population persistence [37].
We sampled monarchs from 18 locations worldwide (figure 1) and used microsatellite analyses to investigate patterns of isolation by distance and genetic diversity. We found that monarch populations connected with the North American source population by land maintained high levels of gene flow. We also detected strong genetic differentiation associated with range expansion out of North America. Across the Pacific Ocean, we found evidence of gene surfing and colonization through a pattern of serial stepwise dispersal. Conversely, across the Atlantic, monarchs showed evidence for multiple colonization events and dramatic founder effects within small, isolated populations.
Figure 1.

Map showing sampling locations of monarchs used for this study. Numbers represent sample sizes for each location. The inset shows a cluster of North American migratory monarchs at an overwintering site in central Mexico (left) and a female monarch laying eggs on the tropical milkweed Asclepias curassavica (right). (Photos by J.C.d.R.)
2. Material and methods
(a). Monarch field collections
We obtained 746 monarchs from 18 locations around the world between 2007 and 2011 (figure 1 and table 1). From North American migratory monarchs, we included samples from one fall migratory stopover site in north Florida (St Marks: 100 monarchs), two California overwintering sites (Pismo Beach and Santa Barbara: 100 monarchs) and two Mexico overwintering sites (Cerro Pelon and Sierra Chincua: 27 monarchs). We refer to the monarchs sampled from the eastern and western United States as the USA sample from hereon because despite overwintering in different locations, eastern and western North American monarchs are genetically indistinguishable on the basis of microsatellites [38]. We also included samples from non-migratory New World monarchs inhabiting South Florida, southern North America (Costa Rica and Belize), Caribbean and Atlantic islands (Puerto Rico and Bermuda) and South America (Ecuador and Aruba). Additionally, we included non-migratory monarchs from islands across the Pacific Ocean (Hawaii, Samoa, Fiji, New Caledonia, New Zealand and Australia) and monarchs from the Iberian Peninsula and northern Africa (Spain, Portugal and Morocco).
Table 1.
Sampling sites and numbers of monarch butterflies.
| region | sampling location | number of monarchs | year collected |
|---|---|---|---|
| North America | United States | 200 | 2009, 2010 |
| Mexico | 27 | 2008 | |
| Belize | 31 | 2011 | |
| Costa Rica | 30 | 2009, 2010 | |
| South Florida | 36 | 2008, 2009 | |
| Bermuda | 13 | 2012 | |
| Puerto Rico | 29 | 2010 | |
| South America | Ecuador | 14 | 2008 |
| Aruba | 29 | 2012 | |
| Hawaiian Islands | Hawaii | 114 | 2007, 2009, 2010 |
| Pacific Islands | Samoa | 32 | 2006, 2007 |
| Fiji | 10 | 2009, 2010 | |
| New Caledonia | 40 | 1991, 2008, 2010 | |
| New Zealand | 22 | 2007, 2011 | |
| Australia | 27 | 2009 | |
| Iberian Peninsula | Spain | 31 | 2012 |
| Portugal | 32 | 2012 | |
| northern Africa | Morocco | 29 | 2012 |
(b). Genetic work
We used 16 microsatellite markers [39] to genotype each of our samples. For PCR, genomic DNA was extracted from a 0.5 mm section of butterfly thorax (females) or abdomen (males) using the Ultraclean DNA Isolation Kit from Mo-Bio (Carlsbad, CA, USA) and quantified using a Nanodrop 2000. We extracted DNA from females from the thorax rather than the abdomen to avoid possible contamination from male sperm. We followed PCR protocols as described [39]. Amplified DNA was genotyped on an ABI 3100 Genetic Analyzer (Perkin Elmer, Applied Biosystems, Foster City, CA, USA) at the Emory Integrated Genomics Core (EIGC; Atlanta, GA, USA) and alleles were scored using genemarker v. 4.0 (SoftGenetics LLC, State College, PA, USA).
(c). Microsatellite analyses
We used the software Arlequin v. 3.5.1.2 [40] to calculate observed and expected heterozygosity at each locus in each population and calculated deviations from Hardy–Weinberg equilibrium using sequential Bonferroni correction at α = 0.05 [41]. For subsequent analyses, we used 11 microsatellite loci that were in Hardy–Weinberg equilibrium in at least 11 of 18 sampling locations (electronic supplementary material, table S1).
(d). Worldwide population genetic analyses
To investigate worldwide population structure, we used the software structure v. 2.3.3 [42] and implemented 100 000 burn-ins and 200 000 Markov Chain Monte Carlo runs after the burn-in. We used an admixture model with uncorrelated allele frequencies to avoid the risk of overestimating the number of populations and used the locprior model to provide the software with location information for each butterfly to ensure the detection of subtle population structure. We started simulations with K = 18, to reflect the 18 sampling locations, and then ran simulations for K values of 18 through 1. For each K, we ran 10 simulations to check for consistency between runs, and used the log likelihood [42] and delta K method [43] to determine the most likely number of genetic populations.
We calculated Nei's standard genetic distance (DST) [44] using the GenAlEx Excel plugin [45]. With this measurement, we created a distance matrix and used it to build a neighbour-joining distance tree in mega5 [46] to visualize the genetic relationships between monarchs from our sampling sites. With the same distance matrix, we performed a Principal Coordinate Analysis using the cmdscale function in R (v. 3.0.1).
To confirm our results, we used FST and RST statistics [47,48] to measure genetic differentiation between monarch populations. Permutation tests (using 10 000 permutations), implemented in Arlequin v. 3.5.1.2 [40] were used to determine whether pairwise FST and RST values were significantly different from 0. To account for the potential occurrence of null alleles, we recalculated FST and RST values using corrected allele frequencies as determined by the software micro-checker, v. 2.2.3 [39]. Overall, statistics based on corrected and uncorrected allele frequencies were highly similar (electronic supplementary material, tables S2 and S3), and resulted in the same conclusions.
(e). Population genetic analyses of monarchs across the Pacific and Atlantic
We used structure (settings and determination of K as above) to investigate population structure at a smaller scale among locations across the Pacific versus the Atlantic. For six locations across the Pacific, we began with K = 6 and ran simulations for K values 6 through 1. For eight locations across the Atlantic, we started simulations with K = 8 and ran simulations for K values of 8 through 1. To analyse isolation by distance, we analysed the correlation between geographical distance and genetic differentiation (FST and RST) for locations across the Pacific and across the Atlantic using Mantel tests implemented in the vegan library v. 2.0-0 [49] in the statistical package R (v. 3.0.1).
To determine likely dispersal routes, we resampled monarchs from each location with replacement using Poptools [50] to standardize sample size across sites (n = 22) prior to comparison of relative levels of genetic diversity (using the value 1-Qinter, the inter-individual diversity within populations), which were measured using genepop v. 4.1.0 [51]. We also calculated allelic richness, the number of alleles per locus, using adze-1.0 [52], which uses a rarefaction approach to account for differences in sample size. Locations with fewer than 20 samples were excluded from these analyses. We then grouped locations based on proximity to either the Pacific or Atlantic Ocean to investigate the possibility of allelic clinal patterns.
We calculated levels of gene flow, measured as the number of migrants reaching one population from another per generation, using likelihood ratio tests implemented in the coalescent-based software package migrate-n 3.2 [53]. We used a Brownian motion approximation to the ladder model and Bayesian inference with multiple heating chains to jointly estimate parameters [54,55]. We arbitrarily chose four locations for analysis of gene flow across the Pacific and four locations for determining gene flow across the Atlantic to maintain program accuracy and efficiency. Each analysis was run independently with five replicates.
3. Results
(a). Worldwide patterns
Monarchs showed substantial genetic differentiation among the sampled locations (figure 2). structure analyses supported a total of seven worldwide monarch populations composed of (i) North America (including USA, Mexico, Central America, and neighbouring Caribbean/Atlantic islands), (ii) South America (Ecuador), (iii) Aruba, (iv) Spain, (v) Portugal/Morocco, (vi) the Hawaiian Islands and, (vii) a series of Pacific Islands (including Australia and New Zealand; figure 2). We inferred similar relationships from a distance tree based on Nei's standard genetic distance (figure 3) with the locations grouping intuitively based on geographical proximity. In addition, the relatively long branch lengths between USA/Mexico monarchs and those from two neighbouring locations (Puerto Rico and South Florida/Bermuda) support the intermediate levels of differentiation identified by structure. The nested structure of the distance tree (figure 3) also offers insight into the colonization route. For example, the branch capturing the Pacific Islands shows Hawaii having intermediate differentiation, with other Pacific sites showing increased differentiation from the USA; this pattern supports the serial stepwise dispersal hypothesis. Similarly, the clustered nature of the Iberian Peninsula and northern Africa locations suggests common colonization events. Similar patterns were observed using Principal Coordinate Analysis (electronic supplementary material, figure S1). These relationships are further supported by FST and RST values, which are much lower between populations within the same geographical group and in closer geographical proximity than between populations in different geographical groups (electronic supplementary material, tables S2 and S3).
Figure 2.
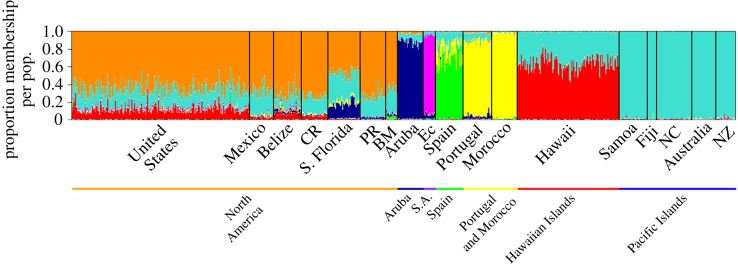
Worldwide structure plot showing that K (number of distinct populations) = 7. Microsatellite loci show that monarchs across the globe form seven genetic populations (orange bar, North America; dark blue bar, Aruba; purple bar, South America (S.A.); green bar, Spain; yellow bar, Portugal & Morocco; red bar, Hawaiian Islands; blue bar, Pacific Islands). The North American population is composed of the United States, Mexico, Belize and Costa Rica (CR) as well as slightly more genetically differentiated locations such as South Florida (S. Florida), Puerto Rico (PR) and Bermuda (BM). South America is composed of Ecuador (Ec) and the Pacific Islands are composed of Samoa, Fiji, New Caledonia (NC), Australia and New Zealand (NZ). Individual monarchs are indicated by vertical bars and colour denotes population membership.
Figure 3.

Distance tree based on Nei's standard genetic distance (DST). Sampling locations group intuitively based on geographical proximity and indicate groupings similar to the structure plot in figure 2. Branches are coloured to demonstrate group membership (orange, North America; dark blue, Aruba; purple, South America; green, Spain; yellow, Portugal and Morocco; red, Hawaiian Islands; blue, Pacific Islands).
We found evidence for a panmictic population spanning the continent of North America, composed of the USA, Mexico, Belize and Costa Rica. South Florida, Puerto Rico and Bermuda were clearly differentiated from these North American mainland populations, as indicated by FST and RST values (electronic supplementary material, tables S2 and S3) and structure analysis (figure 2).
(b). Monarchs across the Pacific
In our worldwide analyses, when compared with other populations, monarchs from the Pacific Islands appear very homogeneous. To examine more subtle patterns of variation, we ran structure for the Hawaiian and Pacific Islands only (figure 4e) and were able to detect more population differentiation among the Pacific Island locations. To further explore this pattern, we examined allele frequencies for each of the 11 loci and found a distinct clinal pattern (electronic supplementary material, figure S2), consistent with stepwise dispersal and gene surfing. Allelic richness was highest in the USA, with subsequent loss of some alleles and enrichment of others with increasing distance westward towards Australia and New Zealand. This pattern was observed across all 11 loci (electronic supplementary material, figure S2), which is highly indicative of a stepwise dispersal across the Pacific and the presence of gene surfing in these colonization events.
Figure 4.

Genetic trends across the Pacific. (a) Genetic diversity (using the value 1-Qinter, the inter-individual diversity within populations) was highest in the United States and tended to decrease with increasing distance from the source population. (b) Allelic richness was higher in the United States and significantly decreased across the Pacific. (c) A weak pattern of isolation by distance was found (r = 0.3833, p = 0.062). (d) Gene flow ranged from 2 to 15 individuals per generation and was highest from Hawaii to the United States. (e) Pacific structure plot indicating K = 3. Increased population differentiation and a clinal pattern appear when examined at a finer spatial scale.
In addition to this clinal pattern, we found a decrease in genetic diversity and allelic richness as monarchs moved farther west from North America (figure 4a,b), further supporting stepwise serial dispersal. Conversely, the observation of relatively high frequencies of rare alleles in more distant locations is also consistent with the possibility that some sites were founded by independent colonization events. Additionally, there was only a weak pattern of isolation by distance (figure 4c; p = 0.06), as has been seen in other highly mobile species, such as the European starling in South Africa [23], which might arise from ongoing gene flow or intermittent dispersal events among colonized locations. To determine levels of gene flow, we ran MIGRATE-N using samples across the Pacific. Estimated numbers of migrants per generation between locations ranged from only two individuals from New Zealand to Australia to 15 individuals from Hawaii to the USA (figure 4d). This confirms the presence of ongoing gene flow even among distantly separated populations.
(c). Monarchs across the Atlantic
We ran structure using samples from the USA and locations across the Atlantic Ocean, including South Florida, Puerto Rico, Bermuda, Spain, Portugal and Morocco. Results supported the same structure shown in figure 2, which is unsurprising as we were already able to detect ample population structure. We observed a decrease in allelic richness and genetic diversity among monarchs sampled in these Old World locations relative to the USA, although the pattern was not as strong as that observed across the Pacific (figure 5a,b). To investigate potential allelic clinal patterns, we examined allele frequencies for the USA, Spain, Portugal and Morocco. Many of the alleles found in the Iberian Peninsula and northern Africa represent a subset of alleles found in the USA (electronic supplementary material, figure S3), indicative of founder effects.
Figure 5.

Genetic trends across the Atlantic. (a) Genetic diversity (using the value 1-Qinter, the inter-individual diversity within populations) was highest in the United States and tended to decrease with increasing distance from the source population. (b) Allelic richness was higher in the United States and significantly decreased across the Atlantic. (c) A weak pattern of isolation by distance was found (r = 0.3181, p = 0.048), with increasing geographical distance correlating with increasing genetic distance. (d) Gene flow ranged from 2 to 6 individuals per generation.
We found a weak pattern of isolation by distance (figure 5c, p = 0.048), with increasing distance from the USA associated with increasing genetic differentiation. To determine gene flow across the Atlantic and among Old World locations, we ran MIGRATE-N and found relatively low levels of gene flow in comparison to the sites across the Pacific (figure 5d). This suggests that while there may have been multiple colonization events from North America, they have occurred infrequently or involved few individuals.
4. Discussion
A strong signal of worldwide genetic population structure was detected in monarch butterflies, despite their high propensity for flight and their long-distance migration. With increasing distance from the likely source population in North America, we found a trend of decreasing genetic diversity and clines in allelic richness, with some alleles disappearing and others becoming enriched. It thus appears that genetic drift has been the main driver in determining allele frequencies as monarchs colonized new areas. Importantly, spatial expansions can result in rare alleles reaching high frequencies through gene surfing, a process associated with genetic drift [4], and this process has likely been a strong force shaping allele frequencies of newly colonized monarch populations across both the Atlantic and the Pacific Oceans.
(a). Pacific versus Atlantic
Our results support a serial founder effect in monarchs from locations across the Pacific. The robust clinal pattern in allele frequencies (electronic supplementary material, figure S2) and decreased allelic richness and genetic diversity (figure 4) strongly suggest that stepwise dispersal was the primary mechanism of colonization (see also [33]), as has been seen in other range expansions, like that of the spur-thighed tortoise in Spain [19]. Conversely, across the Atlantic, we found relatively higher genetic diversity as well as evidence for multiple colonization events and dramatic founder effects with rare alleles in the donor population found at relatively high frequencies. This pattern is more similar to that found in the speckled wood butterfly, in which range expansion is characterized by multiple colonization events resulting from long-distance dispersal. There, it was found that while dispersal events resulted in reduced genetic diversity, this effect was lessened by subsequent gene flow [56]. Indeed, in monarchs, historical records provide evidence of infrequent ‘migrants’ or vagrants that strayed across the Atlantic on winds from North America with only a few individuals arriving in each dispersal event. In the Iberian Peninsula and northern Africa, monarchs are faced with hot and dry summers, resulting in host plants being scattered and restricted to wet soils (river beds, ponds, etc.) [28]. Although this fragmented distribution could contribute to reduced gene flow between different patches, the most likely explanation for rare alleles is the high fluctuations in butterfly density in those patches. It is common for Iberian monarchs to suffer periodic bottlenecks as a consequence of excessive larval density or the destruction of plants by flooding rivers [37].
Like many introduced or invasive species, monarch colonization across both the Pacific and the Atlantic was made possible by anthropogenic interactions, specifically human-driven colonization by milkweeds (which are non-native in most locations where monarchs are now found). Additionally, colonization was probably affected by between-location distances and wind patterns [27]. The distances between locations inhabited by monarchs in the Iberian Peninsula and northern Africa are much shorter (and often connected by land) than the distances between oceanic islands in the Pacific. Importantly, both normal wind patterns and severe weather events such as hurricanes or cyclones have been known to transport monarchs across oceans [35], thus facilitating colonization and gene flow. Winds in both directions moving east and west away from North America might have aided monarchs in trans-oceanic movements; additionally, the presence of the Gulf Stream and strong winds in middle latitudes from the USA to Europe may have resulted in more regular introductions across the Atlantic. Our analyses suggest that gene flow was generally higher among the Pacific locations than among those across the Atlantic, suggesting that monarchs move frequently enough between Pacific islands to limit population divergence.
(b). Implications
Our analyses show that monarch migration provides sufficient gene flow to create a panmictic population of butterflies that spans the mainland North American continent. These results indicate that the long-distance migration of monarchs from the USA and Canada to Mexico each year likely results in genetic mixing which affects monarchs in other North American locations. One hypothesis for how this occurs is that rather than solely re-migrating north each spring, a portion of monarchs radiate outwards from their overwintering sites in search of larval host plants [57,58]. Owing to the panmictic nature of the population in North America, our results provide a new outlook on monarch butterfly migration pathways.
Results here also indicate that different migration strategies, such as the use of different overwintering sites or the presence of non-migratory behaviour, can be maintained between populations despite high levels of gene flow [38], as also seen in the admixed populations of migratory and sedentary groups of broad-tailed hummingbirds in North America [59]. A similar lack of neutral genetic differentiation has been observed in other highly mobile, migrating species, such as hoverflies with different overwintering strategies in France [60] and blackcaps with different migratory behaviours and destinations in Europe [61].
To investigate evolutionary changes in monarchs and other species that might have arisen from the selective pressures of long-distance migration, different genetic approaches are required. For example, despite a lack of genetic differentiation based on selectively neutral markers, monarchs from populations with different migratory destinations or strategies could be differentiated at loci under differential selection in their respective geographical areas [32,62]. Thus, it is possible that locally adapted alleles are selected for during the breeding and migration seasons despite a high influx of neutral genes via gene flow. To better understand the role of genetics in migration as well as the potential role of local adaptation, comparative studies investigating non-neutral alleles and traits should be addressed in the future.
With respect to other systems, our work clearly demonstrates that range expansion crucially affects levels of genetic diversity and population differentiation. Although these results are generally expected for species with low levels of dispersal, theoretical models have suggested that high levels of dispersal could limit the effects of range expansion on reducing genetic diversity and creating genetic differentiation [20–22]. Some empirical studies have confirmed these predictions. In European starlings inhabiting South Africa, for example, and peat mosses in the Stockholm archipelago, long-distance dispersal events have maintained genetic diversity [23,24] and multiple colonization events are hypothesized to have maintained relatively high genetic diversity in expanding populations of the Mediterranean damselfly [63]. Additionally, when bats with different dispersal abilities suffering from habitat fragmentation were compared, highly mobile bats presented less genetic diversity loss [64]. In contrast to these findings, our results suggest that the genetic signature of range expansion can be maintained even in species with great capacity for dispersal. Beyond furthering our understanding of the population genetic effects of range expansion, these results are relevant for conservation biology by showing that even highly mobile species can suffer from reduced genetic diversity in widely separated geographical fragments.
In summary, our work demonstrates that genetic drift has played a key role in shaping allele frequencies in colonized monarch butterfly populations throughout the world. In addition to demonstrating the impact of migration on gene flow and its role in creating a panmictic population spanning the North American continent, this work is also relevant for conservation, as connectivity impacts population size and dynamics. Especially with global climate change becoming increasingly important and resulting in habitat and range shifts for numerous species [65], a deeper understanding of the mechanisms and effects of range expansions is critical in conservation efforts and the prevention and control of invasive species. Finally, our results confirm theoretical predictions that range expansions can result in allele clines [4,66] and gene surfing [14–17,67], and suggest that these patterns may occur even for species with great migratory ability.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
We thank J. Lyons, S. Barribeau, E. Sternberg and A. Mongue for discussion and technical support during the initial phases of this project; M. Maudsley, B. Ballister, D. Cook, R. Rarick, E. Osburn, R. Bartel, E. Rendon, D. Frey and R. Obregon for assistance with field collections; and the De Roode lab and L. Morran for helpful comments on a previous version of the manuscript.
Data accessibility
Microsatellite data uploaded to Dryad doi:10.5061/dryad.mb437.
Funding statement
A.A.P. was supported by NIH training grant no. 5T32AI055404-10 (L. Real, PI); J.C.d.R. was supported by NSF grants nos DEB-1019746 and DEB-1257160; J.F.H. was supported by Fundación Migres; M.R.K. was supported by NSF grant no. DEB-1316037 and S.A. was supported by NSF no. grant no. DEB-0643831.
References
- 1.Eckert CG, Samis KE, Lougheed SC. 2008. Genetic variation across species’ geographical ranges: the central–marginal hypothesis and beyond. Mol. Ecol. 17, 1170–1188. ( 10.1111/j.1365-294X.2007.03659.x) [DOI] [PubMed] [Google Scholar]
- 2.Peter BM, Slatkin M. 2013. Detecting range expansions from genetic data. Evolution 67, 3274–3289. ( 10.1111/evo.12202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schulte U, Veith M, Mingo V, Modica C, Hochkirch A. 2013. Strong genetic differentiation due to multiple founder events during a recent range expansion of an introduced wall lizard population. Biol. Invasions 15, 2639–2649. ( 10.1007/s10530-013-0480-5) [DOI] [Google Scholar]
- 4.Slatkin M, Excoffier L. 2012. Serial founder effects during range expansion: a spatial analog of genetic drift. Genetics 191, 171–181. ( 10.1534/genetics.112.139022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF. 1998. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 7, 453–464. ( 10.1046/j.1365-294x.1998.00289.x) [DOI] [PubMed] [Google Scholar]
- 6.Francois O, Blum MGB, Jakobsson M, Rosenberg NA. 2008. Demographic history of European populations of Arabidopsis thaliana. PLoS Genet. 4, e1000075 ( 10.1371/journal.pgen.1000075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Austerlitz F, JungMuller B, Godelle B, Gouyon PH. 1997. Evolution of coalescence times, genetic diversity and structure during colonization. Theor. Popul. Biol. 51, 148–164. ( 10.1006/tpbi.1997.1302) [DOI] [Google Scholar]
- 8.Li JZ, et al. 2008. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319, 1100–1104. ( 10.1126/science.1153717) [DOI] [PubMed] [Google Scholar]
- 9.Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL. 2005. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc. Natl Acad. Sci. USA 102, 15 942–15 947. ( 10.1073/pnas.0507611102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deshpande O, Batzoglou S, Feldman MW, Cavalli-Sforza LL. 2009. A serial founder effect model for human settlement out of Africa. Proc. R. Soc. B 276, 291–300. ( 10.1098/rspb.2008.0750) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanabe K, et al. 2010. Plasmodium falciparum accompanied the human expansion out of Africa. Curr. Biol. 20, 1283–1289. ( 10.1016/j.cub.2010.05.053) [DOI] [PubMed] [Google Scholar]
- 12.Muller MJI, Neugeboren BI, Nelson DR, Murray AW. 2014. Genetic drift opposes mutualism during spatial population expansion. Proc. Natl Acad. Sci. USA 111, 1037–1042. ( 10.1073/pnas.1313285111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hallatschek O, Hersen P, Ramanathan S, Nelson DR. 2007. Genetic drift at expanding frontiers promotes gene segregation. Proc. Natl Acad. Sci. USA 104, 19 926–19 930. ( 10.1073/pnas.0710150104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flaxman SM. 2013. Surfing downhill: when should population range expansion be characterized by reductions in fitness? Mol. Ecol. 22, 5963–5965. ( 10.1111/mec.12564) [DOI] [PubMed] [Google Scholar]
- 15.Edmonds CA, Lillie AS, Cavalli-Sforza LL. 2004. Mutations arising in the wave front of an expanding population. Proc. Natl Acad. Sci. USA 101, 975–979. ( 10.1073/pnas.0308064100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klopfstein S, Currat M, Excoffier L. 2006. The fate of mutations surfing on the wave of a range expansion. Mol. Biol. Evol. 23, 482–490. ( 10.1093/molbev/msj057) [DOI] [PubMed] [Google Scholar]
- 17.Hallatschek O, Nelson DR. 2008. Gene surfing in expanding populations. Theor. Popul. Biol. 73, 158–170. ( 10.1016/j.tpb.2007.08.008) [DOI] [PubMed] [Google Scholar]
- 18.White TA, Perkins SE, Heckel G, Searle JB. 2013. Adaptive evolution during an ongoing range expansion: the invasive bank vole (Myodes glareolus) in Ireland. Mol. Ecol. 22, 2971–2985. ( 10.1111/mec.12343) [DOI] [PubMed] [Google Scholar]
- 19.Gracia E, Botella F, Anadon JD, Edelaar P, Harris DJ, Gimenez A. 2013. Surfing in tortoises? Empirical signs of genetic structuring owing to range expansion. Biol. Lett. 9, 20121091 ( 10.1098/rsbl.2012.1091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fayard J, Klein EK, Lefevre F. 2009. Long distance dispersal and the fate of a gene from the colonization front. J. Evol. Biol. 22, 2171–2182. ( 10.1111/j.1420-9101.2009.01832.x) [DOI] [PubMed] [Google Scholar]
- 21.Bialozyt R, Ziegenhagen B, Petit RJ. 2006. Contrasting effects of long distance seed dispersal on genetic diversity during range expansion. J. Evol. Biol. 19, 12–20. ( 10.1111/j.1420-9101.2005.00995.x) [DOI] [PubMed] [Google Scholar]
- 22.Ray N, Excoffier L. 2010. A first step towards inferring levels of long-distance dispersal during past expansions. Mol. Ecol. Resour. 10, 902–914. ( 10.1111/j.1755-0998.2010.02881.x) [DOI] [PubMed] [Google Scholar]
- 23.Berthouly-Salazar C, Hui C, Blackburn TM, Gaboriaud C, Van Rensburg BJ, Van Vuuren BJ, Le Roux JJ. 2013. Long-distance dispersal maximizes evolutionary potential during rapid geographic range expansion. Mol. Ecol. 22, 5793–5804. ( 10.1111/mec.12538) [DOI] [PubMed] [Google Scholar]
- 24.Szovenyi P, Sundberg S, Shaw AJ. 2012. Long-distance dispersal and genetic structure of natural populations: an assessment of the inverse isolation hypothesis in peat mosses. Mol. Ecol. 21, 5461–5472. ( 10.1111/mec.12055) [DOI] [PubMed] [Google Scholar]
- 25.Ackery PR, Vane-Wright RI. 1984. Milkweed butterflies: their cladistics and biology. Ithaca, NY: Cornell University Press. [Google Scholar]
- 26.Urquhart FA, Urquhart NR. 1978. Autumnal migration routes of the eastern population of the monarch butterfly (Danaus p. plexippus L.; Danaidae; Lepidoptera) in North America to the overwintering site in the Neovolcanic Plateau of Mexico. Can. J. Zool. 56, 1759–1764. ( 10.1139/z78-240) [DOI] [Google Scholar]
- 27.Zalucki MP, Clarke AR. 2004. Monarchs across the Pacific: the Columbus hypothesis revisited. Biol. J. Linn. Soc. 82, 111–121. ( 10.1111/j.1095-8312.2004.00322.x) [DOI] [Google Scholar]
- 28.Fernandez Haeger J, Jordano Barbudo D. 2009. The monarch butterfly Danaus plexippus (Linnaeus, 1758) in the Strait of Gibraltar (Lepidoptera: Danaidae). Shilap Rev. Lepidopterol. 37, 421–438. [Google Scholar]
- 29.Altizer SM, Oberhauser KS, Brower LP. 2000. Associations between host migration and the prevalence of a protozoan parasite in natural populations of adult monarch butterflies. Ecol. Entomol. 25, 125–139. ( 10.1046/j.1365-2311.2000.00246.x) [DOI] [Google Scholar]
- 30.James DG. 1993. Migration biology of the monarch butterfly in Australia. In Biology and conservation of the monarch butterfly (eds Malcolm SB, Zalucki MP.), pp. 189–200. Los Angeles, CA: Natural History Museum of Los Angeles County. [Google Scholar]
- 31.Dingle H, Zalucki MP, Rochester WA. 1999. Season-specific directional movement in migratory Australian butterflies. Aust. J. Entomol. 38, 323–329. ( 10.1046/j.1440-6055.1999.00117.x) [DOI] [Google Scholar]
- 32.Zhan S, et al. 2014. The genetics of monarch butterfly migration and warning colouration. Nature 514, 317–321. ( 10.1038/nature13812) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shephard JM, Hughes JM, Zalucki MP. 2002. Genetic differentiation between Australian and North American populations of the monarch butterfly Danaus plexippus (L.) (Lepidoptera : Nymphalidae): an exploration using allozyme electrophoresis. Biol. J. Linn. Soc. 75, 437–452. ( 10.1046/j.1095-8312.2002.00034.x) [DOI] [Google Scholar]
- 34.Vane-Wright RI. 1993. The Columbus hypothesis: an explanation for the dramatic 19th century range expansion of the monarch butterfly. In Biology and conservation of the monarch butterfly (eds Malcolm SB, Zalucki MP.), pp. 179–187. Los Angeles, CA: Natural History Museum of Los Angeles County. [Google Scholar]
- 35.Clarke AR, Zalucki MP. 2004. Monarchs in Australia: on the winds of a storm? Biol. Invasions 6, 123–127. ( 10.1023/B:BINV.0000010120.29634.db) [DOI] [Google Scholar]
- 36.Fernandez Haeger J, Jordano D, Leon Melendez M. 2011. Status and conservation of Asclepiadaceae and Danaus in southern Spain. J. Insect Conserv. 15, 361–365. ( 10.1007/s10841-010-9354-7) [DOI] [Google Scholar]
- 37.Fernandez Haeger J, Jordano D, Zalucki MP. In press. Monarchs across the Atlantic Ocean: what's happening on the other shore? In Monarchs in a changing world: biology and conservation of an iconic insect (eds Oberhauser K, Altizer S, Nail KR.). Ithaca, NY: Cornell University. [Google Scholar]
- 38.Lyons JI, Pierce AA, Barribeau SM, Sternberg ED, Mongue AJ, de Roode JC. 2012. Lack of genetic differentiation between monarch butterflies with divergent migration destinations. Mol. Ecol. 21, 3433–3444. ( 10.1111/j.1365-294X.2012.05613.x) [DOI] [PubMed] [Google Scholar]
- 39.Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. 2004. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538. ( 10.1111/j.1471-8286.2004.00684.x) [DOI] [Google Scholar]
- 40.Excoffier L, Lischer HEL. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. ( 10.1111/j.1755-0998.2010.02847.x) [DOI] [PubMed] [Google Scholar]
- 41.Rice WR. 1989. Analyzing tables of statistical tests. Evolution 43, 223–225. ( 10.2307/2409177) [DOI] [PubMed] [Google Scholar]
- 42.Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. ( 10.1111/j.1365-294X.2005.02553.x) [DOI] [PubMed] [Google Scholar]
- 44.Nei M. 1973. Analysis of gene diversity in subdivided populations. Proc. Natl Acad. Sci. USA 70, 3321–3323. ( 10.1073/pnas.70.12.3321) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peakall R, Smouse PE. 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539. ( 10.1093/bioinformatics/bts460) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. ( 10.1093/molbev/msr121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holsinger KE, Weir BS. 2009. Genetics in geographically structured populations: defining, estimating and interpreting F(ST). Nat. Rev. Genet. 10, 639–650. ( 10.1038/nrg2611) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Slatkin M. 1995. A measure of population subdivision based on microsatellite allele frequencies. Genetics 139, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oksanen JF, et al. 2011. vegan: Community Ecology Package. R package version 2.0-0 See http://CRANR-projectorg/package=vegan.
- 50.Hood GM. 2010. PopTools version 3.2.5 See http://wwwpoptoolsorg.
- 51.Rousset F. 2008. GENEPOP ‘007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106. ( 10.1111/j.1471-8286.2007.01931.x) [DOI] [PubMed] [Google Scholar]
- 52.Szpiech Z, Jakobsson M, Rosenberg N. 2008. ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24, 1367–4811. ( 10.1093/bioinformatics/btn478) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beerli P. 2009. How to use MIGRATE or why are Markov chain Monte Carlo programs difficult to use? In Population genetics for animals conservation (eds Bertorelle G, Bruford MW, Hauffe HC, Rizzoli A, Vernesi C.), pp. 42–79. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 54.Beerli P. 2006. Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 22, 341–345. ( 10.1093/bioinformatics/bti803) [DOI] [PubMed] [Google Scholar]
- 55.Beerli P, Felsenstein J. 2001. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl Acad. Sci. USA 98, 4563–4568. ( 10.1073/pnas.081068098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vandewoestijne S, Van Dyck H. 2010. Population genetic differences along a latitudinal cline between original and recently colonized habitat in a butterfly. PLoS ONE 5, e13810 ( 10.1371/journal.pone.0013810) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pierce AA, Altizer S, Chamberlain NL, Kronforst MR, De Roode JC. In press Unraveling the mysteries of monarch migration and global dispersal through molecular genetic techniques. In Monarchs in a changing world: Biology and conservation of an iconic insect (eds Oberhauser K, Altizer S, Nail KR.). Ithaca, NY: Cornell University Press. [Google Scholar]
- 58.Wenner AM, Harris AM. 1993. Do California monarchs undergo long-distance directed migration? In Biology and conservation of the monarch butterfly (eds Macolm SB, Zalucki MP.), pp. 209–218. Los Angeles, CA: Natural History Museum of Los Angeles County. [Google Scholar]
- 59.Malpica A, Ornelas JF. 2014. Postglacial northward expansion and genetic differentiation between migratory and sedentary populations of the broad-tailed hummingbird (Selasphorus platycercus). Mol. Ecol. 23, 435–452. ( 10.1111/mec.12614) [DOI] [PubMed] [Google Scholar]
- 60.Raymond L, Plantegenest M, Gauffre B, Sarthou JP, Vialatte A. 2013. Lack of genetic differentiation between contrasted overwintering strategies of a major pest predator Episyrphus balteatus (Diptera: Syrphidae): implications for biocontrol. PLoS ONE 8, e72997 ( 10.1371/journal.pone.0072997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mettler R, et al. 2013. Contrasting patterns of genetic differentiation among blackcaps (Sylvia atricapilla) with divergent migratory orientations in Europe. PLoS ONE 8, e81365 ( 10.1371/journal.pone.0081365) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Altizer S, Davis AK. 2010. Populations of monarch butterflies with different migratory behaviors show divergence in wing morphology. Evolution 64, 1018–1028. ( 10.1111/j.1558-5646.2010.00946.x) [DOI] [PubMed] [Google Scholar]
- 63.Swaegers J, Mergeay J, Therry L, Larmuseau MHD, Bonte D, Stoks R. 2013. Rapid range expansion increases genetic differentiation while causing limited reduction in genetic diversity in a damselfly. Heredity 111, 422–429. ( 10.1038/hdy.2013.64) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meyer CFJ, Kalko EKV, Kerth G. 2009. Small-scale fragmentation effects on local genetic diversity in two phyllostomid bats with different dispersal abilities in Panama. Biotropica 41, 95–102. ( 10.1111/j.1744-7429.2008.00443.x) [DOI] [Google Scholar]
- 65.Thomas CD, Bodsworth EJ, Wilson RJ, Simmons AD, Davies ZG, Musche M, Conradt L. 2001. Ecological and evolutionary processes at expanding range margins. Nature 411, 577–581. ( 10.1038/35079066) [DOI] [PubMed] [Google Scholar]
- 66.Excoffier L, Foll M, Petit RJ. 2009. Genetic consequences of range expansions. A. Rev. Ecol. Evol. System 40, 481–501. ( 10.1146/annurev.ecolsys.39.110707.173414) [DOI] [Google Scholar]
- 67.Excoffier L, Ray N. 2008. Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol. Evol. 23, 347–351. ( 10.1016/j.tree.2008.04.004) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microsatellite data uploaded to Dryad doi:10.5061/dryad.mb437.