

Abstract
This phase I, open-label, single-dose study evaluated the pharmacokinetics, safety, and tolerability of renally excreted drug dexpramipexole in subjects with normal and impaired renal function, i.e. mild, moderate, severe renal impairment, or end-stage renal disease (ESRD) requiring hemodialysis when matched by age and sex. Dexpramipexole area under the curves (AUCs), but not Cmax, were significantly increased with the severity of renal impairment after a single dose administration. The geometric mean ratio of dose-normalized AUC(0–72) was 1.4, 1.7, 2.7, and 4.5, respectively, in mild, moderate, severe renal impairment, and ESRD subjects when compared to healthy subjects. There was a strong association between renal function (eGFR) and dexpramipexole CLr. The slope (90% confidence interval(CI)) of eGFR and renal clearance (CLr) in the regression model was 3.1 (2.4, 3.7). Dexpramipexole elimination in ESRD subjects during both dialysis and non-dialysis (i.e., interval between dialysis) was insignificant. Single 75 mg and 150 mg doses of dexpramipexole were well tolerated, and the safety profile was comparable across renal function groups. Extensive drug accumulation may occur with repeated dosing in patients with significant renal impairment. It is recommended that dexpramipexole not to be given to patients with severe renal impairment or in those with ESRD.
Keywords: dexpramipexole, renal impairment, pharmacokinetics, eGFR, amyotrophic lateral sclerosis
Dexpramipexole is a novel synthetic amino-benzothiazole that has been shown in multiple in vitro and in vivo assays as neuroprotective.1 The drug substance is produced via a chiral synthetic route that yields the active pharmaceutical ingredient (API) in high chiral and chemical purity and is exclusively the (R)-(+) enantiomer of pramipexole (PPX, Mirapex®). PPX was discovered as neuroprotective through a mechanism independent of its dopaminergic pharmacology.2 Dexpramipexole is pharmacologically distinct from PPX with respect to dopamine receptor affinity, and thus was considered to possess a unique and potentially more useful profile for the treatment of neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS). In an ascending dose study in healthy volunteers, dexpramipexole was tolerated at doses up to 600 mg (single dose) and 300 mg twice daily for 3½ days. Dexpramipexole was also investigated for safety and efficacy in patients with ALS at 150 mg twice daily dose.3
Dexpramipexole appeared to be almost exclusively eliminated by renal excretion. In humans, approximately 80% of the dose is excreted in urine as unchanged parent compound at 72 hours post-dose. Estimates of renal clearance ranged from 3 to 5 times greater than the normal glomerular filtration rate (GFR) which is indicative of a substantial tubular secretion component. The in vitro transporter uptake assay in human embryonic kidney (HEK)-organic cation transport 2 (OCT2) cells suggested that the renal secretion of dexpramipexole was mediated by OCT-2. There was a minimal role of liver metabolism in the clearance of dexpramipexole as no metabolite was identified in the [14C] dexpramipexole study with human liver microsomes and hepatocytes. In humans, there was evidence of circulating dexpramipexole metabolites but they accounted for less than 5% of the dexpramipexole exposure.
In healthy subjects, dexpramipexole was rapidly absorbed with a mean time of maximum plasma concentration (Tmax) of approximately 2 hours; decline in plasma drug concentration was described by a mean terminal half-life (t½) that generally ranged from 6 to 8 hours. The plasma protein binding was low and appeared to be independent of dexpramipexole concentration with approximately 95% circulating as free drug. The drug exhibited linear pharmacokinetic (PK) up to the highest tested dose of 600 mg, and there were no dose-related trends in either clearance (CL/F) or volume of distribution uncorrected for bioavailability (Vz/F), indicating that saturation of excretion had not been reached at tested doses. The pharmacokinetic profiles in ALS patients were similar to those in healthy volunteers.
In the United States, more than 10% of people, or more than 20 million, aged 20 years or older have chronic kidney diseases (CKD) and the prevalence of CKD is more than 40% at age of 65 or higher.4 It can be anticipated that a significant percentage of patients with neurodegenerative disease are also likely to suffer from renal insufficiencies. Thus, the dominant role of renal elimination necessitates the evaluation of dexpramipexole pharmacokinetics in patients with various degrees of renal impairment, including mild, moderate, and severe GFR decrease, as well as end-stage renal disease (ESRD). The study was conducted to investigate if dose adjustments were needed in patients with renal insufficiency.
Materials and Methods
Methods
This phase 1, open-label, single-dose study was conducted at two sites, Orlando Clinical Research Center and Twin Cities Clinical Research Center. The protocol was approved by the institutional review boards of the study sites. The protocol was conducted in accordance with the guidelines on Good Clinical Practice and with ethical standards for human experimentation established by the Declaration of Helsinki Principles. All subjects gave written informed consent prior to the participation in the study.
Subjects
Adult males and females aged 18–75 years inclusive and between 19 and 36 kg/m2 body mass index (BMI) were eligible to the study. Subjects with impaired renal function had to have stable renal disease (i.e., no change in disease status within the 28 days prior to dosing), and had to have two separate estimates of creatinine clearance that were within 25% of each other, obtained >5 days apart, but not >6 months apart (excluding ESRD subjects). Subjects with ESRD had to have dialysis ≤3 times a week. Healthy volunteers had to be matched individually to renally impaired subjects (four matched to mild subjects, four matched to moderate subjects and four matched to either severe or ESRD subjects) for age (± 10 years), sex, and, if possible, BMI (± 20%). Subjects had to have no clinically relevant abnormalities as determined by pre-study medical history, physical examination, and 12-lead ECG other than those consistent with renal impairment or related disease/disorder in the appropriate subject group. Subjects with renal insufficiency were not allowed to receive prescription medication within 14 days prior to dosing, except for birth control and medications taken at a stable dose for underlying conditions.
Subjects with an absolute neutrophil count (ANC) <1.96 × 103/µL at the screening visit or any documented history of neutropenia were excluded. Subjects were also excluded if they had received drugs that are known to inhibit renal tubular secretion of organic acids via the organic cationic transport system within 14 days prior to treatment and for the duration of the study. Other exclusion criteria included pregnancy; positive for HIV I or II, or positive for HCVAb, HBsAg and/or HBcAb at screening in healthy volunteers only; history of clinically relevant surgery, relevant hypersensitivity; history of alcoholism or drug abuse within 5 years; donation or loss of blood of greater than 400 mL 8 weeks prior to admission.
Thirty-six subjects were enrolled; all subjects received study treatment and completed the study between 06 July 2011 and 10 January 2012. Thirty-six subjects were assigned to dose groups based on renal function as assessed by eGFR using the Modification of Diet in Renal Disease (MDRD) equation, calculated within 30 days prior to the start of the study, of ≥80 (healthy volunteers), between 50 and 79 (mild decrease), between 30 and 49 (moderate decrease), or <30 (severe decrease), or end-stage renal disease undergoing hemodialysis ≤3 times a week ESRD. The MDRD equation is: eGFR (in mL/min/1.73 m2) = 175 × Serum Creatinine−1.154 × Age−0.203 × [1.212 if Black] × [0.742 if Female], where serum creatinine is expressed in mg/dL.
Six subjects each in the mild and moderate renal function groups (Cohorts 1 and 2, respectively) and eight healthy volunteers with normal renal function were dosed with 150 mg dexpramipexole. Six subjects each in the severe and ESRD cohorts (Cohorts 3 and 4, respectively) and four healthy volunteers with normal renal function were dosed with 75 mg dexpramipexole. Patient cohorts were dosed beginning with sentinel subjects who were assessed for safety and tolerability (two subjects each in mild and moderate renal function group, and one subject each in severe and ESRD group) before the remainder of the subjects in the renal function group were dosed.
A single oral dose of dexpramipexole was administered with 240 mL of water to all subjects, either as a round compressed oral tablet (75 mg), or a compressed film-coated tablet (150 mg). Subjects received a light meal prior to dosing. For subjects on hemodialysis, dexpramipexole was administered on the day of dialysis after dialysis was completed.
Blood samples were obtained at pre-dose and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, 24, 36, 48, and 72 hours after dosing in all subjects, and in addition at 96, 120, and 144 hours after dosing in subjects with severe renal impairment and ESRD. For ESRD subjects on dialysis, dosing occurred following completion of dialysis on Day 1 and additional blood samples for determination of dexpramipexole concentrations were obtained immediately before and after their first post-dose dialysis, as well as 12 hours post-dialysis. If timing overlapped with the pre-scheduled PK sampling time (± 2 hours) duplicate samples were not taken.
Urine was collected at pre-dose and at 0–2, 2–4, 4–8, 8–12, 12–24, 24–36, 36–48, and 48–72 hours in all subjects. Multiple voids within a collection interval were pooled. For ESRD subjects, urine samples were collected as available.
Dialysate sample was collected at every hour during the first post-dose dialysis in ESRD subjects. Time of dialysate collection and the total dialysate volume were recorded.
Bioanalytical Analysis Method
Blood samples (4 mL at each time point) were collected into sterile 4 mL lavender top K2EDTA tubes. After collection, each sample tube was gently inverted 8–10 times to ensure uniform mixing. The sample tubes were then kept on ice and centrifuged within 15 minutes of collection at 4°C and approximately 3000 rpm for 10 minutes to separate the plasma. Each plasma sample was then aliquoted (2.0 mL each) into vials and stored at −70°C.Urine samples were collected into 3-liter urine collection containers. A volume of approximately 30 mL was taken from each pooled collection interval and stored at −70°C. Hemodialysate samples were collected from subjects undergoing hemodialysis treatment using a 3-liter container. A volume of approximately 30 mL was taken from each pooled collection interval and stored frozen at −70°C.
Fully validated bioanalytical methods were used for the determination of dexpramipexole concentrations in plasma, urine, and hemodialysate samples. Calibration standard and quality control samples were prepared by spiking dexpramipexole neat solution into blank biological matrices. Calibration standards, quality controls, blanks, and study samples were spiked with deuterium labeled internal standards (pramipexole-d7) and extracted with a liquid–liquid extraction method using methyl tert–butyl ether. The extracted samples were analyzed using liquid chromatography coupled with tandem mass spectrometric detection (LC-MS/MS). The chromatographic separation was done with a Gemini C18 column (50 × 2 mm, 3 μm, Phenomenex, Torrance, CA). An Agilent 1100 Series LC pump (Agilent Technologies, Wilmington, DE), CTC autosampler (LEAP Technologies, Carrboro, NC) and a Sciex API4000 triple quadrupole mass spectrometer (MDS Sciex, Concord, Ontario) were used for analysis and quantification. The mass spectrometer was operated in positive ion electrospray mode (ESI+) using multiple reaction monitoring (MRM) for detection. The precursor/product ion transitions used for dexpramipexole and pramipexole-d7 were 212 →153 and 219 →153, respectively.
The validated concentration ranges of the bioanalytical methods were: plasma (2.00–2000 ng/mL), urine (0.100–25.0 μg/mL), and hemodialysate (2.00–2000 ng/mL). The lower limits of quantitation (LLOQ) were: plasma (2.0 ng/mL), urine (0.100 μg/mL), and hemodialysate (2.0 ng/mL). The assay sample volume was 100 μL. The intra-assay and inter-assay accuracy of the method, determined by comparing the means of the measured concentrations with their nominal concentrations, was within the acceptable range of 85.0%–115.0% (80.0%–120.0% at the LLOQ). The precision of the assay, expressed as the relative standard deviation (RSD) for calibration standards (inter-assay) and QC samples (intra- and inter-assay), was <15.0% (<20.0% at LLOQ).
Pharmacokinetic And Statistical Analysis
Plasma pharmacokinetic parameters were estimated from total plasma dexpramipexole concentrations using non-compartmental methods and actual elapsed time from dosing. Urine pharmacokinetic parameters were calculated based on urinary drug concentrations. Dialysate pharmacokinetic parameters were calculated based on dialysate drug concentrations. All PK computations were performed using WinNonlin Professional 5.2 (Pharsight Corp., Mountain View, CA) and SAS® version 9.2 (SAS Institute, Inc., Cary, NC). Graphics were prepared with SAS Version 9.2 and SigmaPlot® 9.0 (Systat Software, Inc., San Jose, CA).
Treatments were defined with the dexpramipexole dose expressed as the anhydrous dihydrochloride salt. For calculation of apparent total body clearance, volume of distribution, and fraction of dose excreted into the urine or dialysate, the actual dexpramipexole dose expressed as free base was used. The free base dose was calculated as (Dose (salt)/284.25) × 211.33 (rounded to three significant figures) and used in the PK analysis. Molecular weights were 211.33 for free base dexpramipexole and 284.25 for the dihydrochloride salt, respectively. Therefore, actual free base dexpramipexole doses were 55.8 and 112 mg for the 75 and 150 mg treatments, respectively.
To allow pooling of the data for graphical comparison across treatments, plasma concentrations and select PK parameters (i.e., AUC(0–inf), AUC(0–72), AUC(0–144), Cmax, Ae(0–t), and Re(0–t)) were normalized to a dose of 150–mg by multiplying the data from the 75–mg treatment by two. Dexpramipexole exhibited linear PK between 75 mg and 150 mg doses, supported by the data from this study as well as from previous studies. The dose-normalized plasma concentration-time profiles were similar in healthy subjects between the 75 mg and 150 mg treatment groups, justifying pooling of PK data in all healthy subjects.
Dexpramipexole PK parameters were summarized across renal function groups using descriptive statistics. In the event that AUC(0–inf) could not be estimated in the majority of subjects in each renal function group, AUC(0–72) was used. Figures of dose-adjusted arithmetic mean plasma concentration versus time data were presented by renal function group on semi-log scales. Figures of PK parameters (AUC(0–72), Cmax, and CLr) were compared by renal function group. Regression models were used to assess and quantify the relationship between renal function, as measured by the eGFR, and dexpramipexole PK (Cmax, AUC(0–72), CLr). Note that data from ESRD subjects were not included in this analysis. Model parameter and 90% confidence interval (CI) were reported. The graph of each PK parameter versus the eGFR was provided with the regression line.
Safety and tolerability assessments included Clinical laboratory, electrocardiogram (ECG), vital and adverse events (AEs) coded using MedDRA 14.1.
Results
Subjects
The study was conducted at two sites. Thirty-six subjects were enrolled; all subjects received study treatment and completed the study. Six subjects each in the mild and moderate renal function groups (Cohorts 1 and 2, respectively) and eight healthy volunteers with normal renal function were dosed with 150 mg dexpramipexole. Six subjects each in the severe and ESRD cohorts (Cohorts 3 and 4, respectively) and four healthy volunteers with normal renal function were dosed with 75 mg dexpramipexole. All subjects were included in the PK Population and Safety Population.
Four healthy volunteers were matched individually to mild renally impaired subjects, four were matched to moderate subjects and four were matched to either severe or ESRD subjects based on age (± 10 years) and sex. By these criteria, most study subjects were matched by BMI (± 20%) as well. Demographic characteristics are summarized in Table1. The demographics were generally balanced across renal function groups. The mean age of subjects in the study was 59.9 years, ranging from 46 to 76 years. The study was balanced for sex: 50% were males. Most subjects (78%) were Caucasian. The mean weight was 78.5 kg (range: 50–120 kg), and the mean BMI was 27.5 kg/m2 (range: 20–35 kg/m2). Major known causes of renal impairment included those of diabetics and diseases of kidney origin. Seventy-five percent of subjects were reported to take concomitant medications, with aspirin and vitamins as the most frequent medications. Four subjects reported insulin use. None of the concomitant medications was considered to interact with dexpramipexole PK.
Table 1.
Baseline Demographic Characteristics of the Study Population
| Parameters | Normal (150 mg) N = 8 | Mild (150 mg) N = 6 | Moderate (150 mg) N = 6 | Normal (75 mg) N = 4 | Severe (75 mg) N = 6 | ESRD (75 mg) N = 6 | Total N = 36 |
|---|---|---|---|---|---|---|---|
| Sex, n, M:F | 5:3 | 2:4 | 5:1 | 2:2 | 1:5 | 3:3 | 18:18 |
| Race, n, white:black:other | 7:1:0 | 6:0:0 | 5:1:0 | 2:2:0 | 5:1:0 | 3:2:1 | 28:7:1 |
| Age, mean (SD), y | 56.1 (7.1) | 67.5 (6.4) | 64.5 (9.3) | 52.3 (8.7) | 64.5 (4.8) | 53.5 (5.7) | 59.9 (8.8) |
| BMI, mean (SD), kg | 27.0 (3.5) | 24.5 (3.9) | 30.8 (4.4) | 28.3 (2.1) | 27.5 (6.2) | 27.0 (4.7) | 27.5 (4.5) |
| eGFR, mean (SD), mL/min/1.73 m^2 | 92.8 (8.8) | 61.7 (5.6) | 39.3 (3.1) | 102.5 (19.4) | 22.7 (3.7) | NA | NA |
Dexpramipexole PK
Mean plasma concentration-time profiles by renal function groups are shown in Figure 1. Individual and geometric means of dose adjusted Cmax and AUC(0–72) by renal function groups are shown in Figure 2. There was no difference in Cmax of dexpramipexole in plasma across subjects with various degrees of renal impairment after single dose administration (Figure 2A). The regression model also suggested that there was no association between eGFR and Cmax, with the slope (90% CI) of the regression line equal to 0.1 (−0.8, 1.0). In contrast to Cmax, the AUC values of dexpramipexole in plasma were significantly increased with the severity of renal impairment. The geometric mean ratio of dose-normalized AUC(0–inf) compared to healthy subjects was 1.4, 1.8, and 3.1, respectively, in subjects with mild, moderate, and severe renal impairment when compared to healthy subjects. Due to its prolonged elimination half-life, the AUC(0–inf) in most of the ESRD subjects could not be reliably estimated. The dose-adjusted AUC(0–72) is presented in Figure 2B, with the geometric mean ratios of 1.4, 1.7, 2.7, and 4.5, respectively, in mild, moderate, severe renal impairment, and ESRD subjects.
Figure 1.
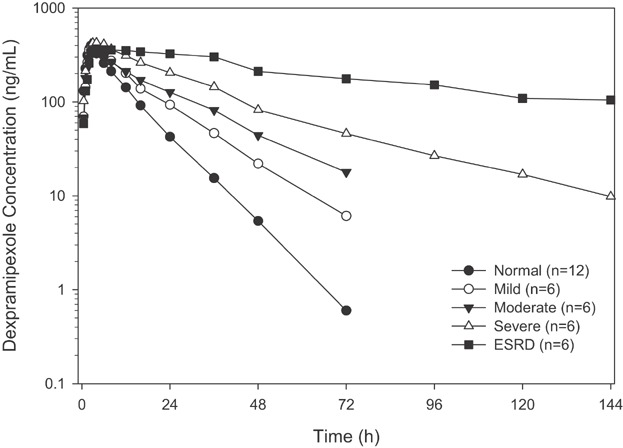
Dose adjusted mean plasma concentration of dexpramipexole versus time in subjects with normal renal function and in those with mild, moderate, severe renal impairment, and ESRD, after single dose administration of dexpramipexole at 150 mg or 75 mg. The dexpramipexole plasma concentration was adjusted to a dose of 150 mg based on PK linearity.
Figure 2.
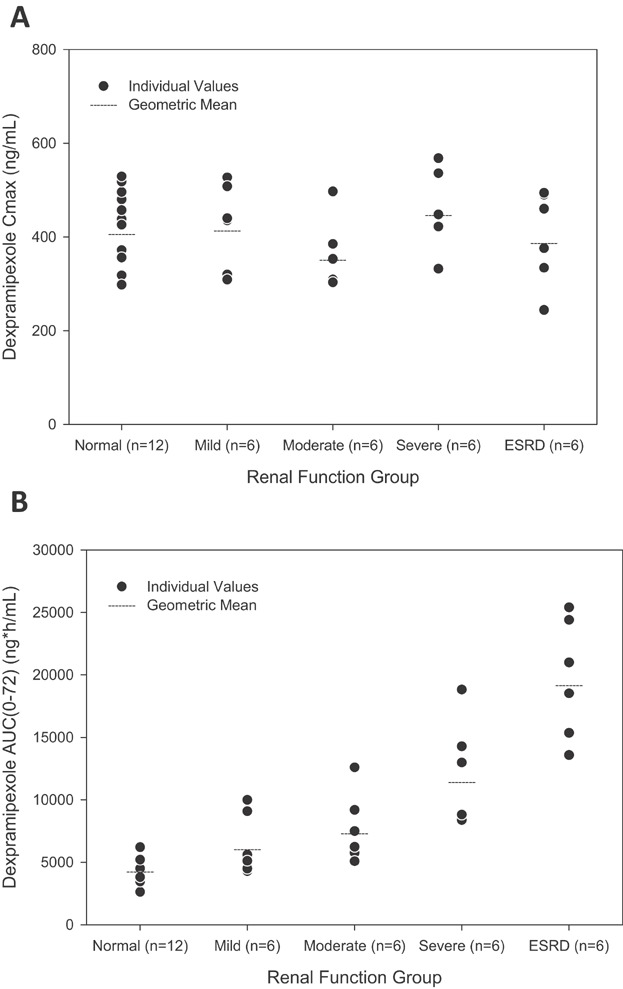
Cross comparison of dose adjusted Cmax (A) and AUC0–72 (B) of dexpramipexole in subjects with normal renal function and in those with mild, moderate, severe renal impairment, and ESRD, after single dose administration of dexpramipexole at 150 mg or 75 mg. The dexpramipexole Cmax and AUC0–72 were adjusted to a dose of 150 mg based on PK linearity.
Dexpramipexole was excreted in urine to a significant extent and the excretion rate reached its peak at approximately 4 hours after dosing. The urinary excretion rate during the first 4 hours post-dose decreased with the severity of renal impairment (Figure 3A). Urinary recovery of dexpramipexole within 72 hours post-dosing (Fe % dose) was approximately 80% in mild renal impairment, and was similar to that in subjects with normal renal function and was consistent with previous data. Urinary recovery of dexpramipexole was 70% in subjects with moderate, and 65% in subjects with severe renal impairment (Figure 3B). There was a strong association between renal function (eGFR) and dexpramipexole CLr (Figure 4). The slope (90% CI) of eGFR and CLr in the regression model was 3.1 (2.4, 3.7). In subjects with extremely low eGFR (e.g., less than 15 mL/min), an accurate measure of CLr could not be estimated due to insufficient urine production.
Figure 3.
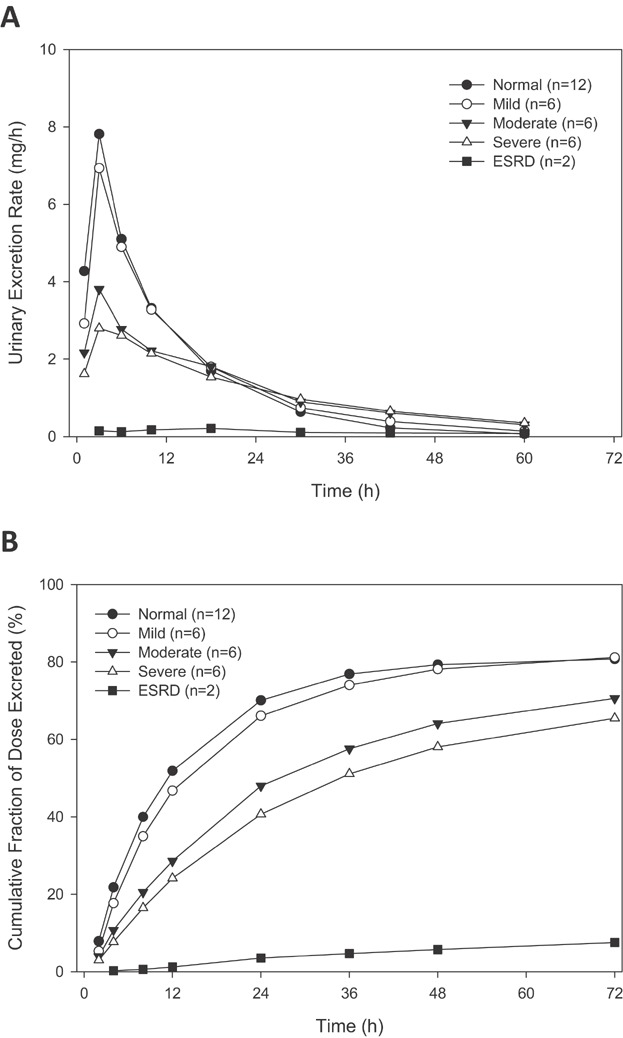
Mean urinary excretion rate (A) or cumulative fraction of dexpramipexole dose excreted unchanged in urine (B) versus time in subjects with normal renal function and in those with mild, moderate, severe renal impairment, and ESRD, after single dose administration of dexpramipexole at 150 mg or 75 mg.
Figure 4.
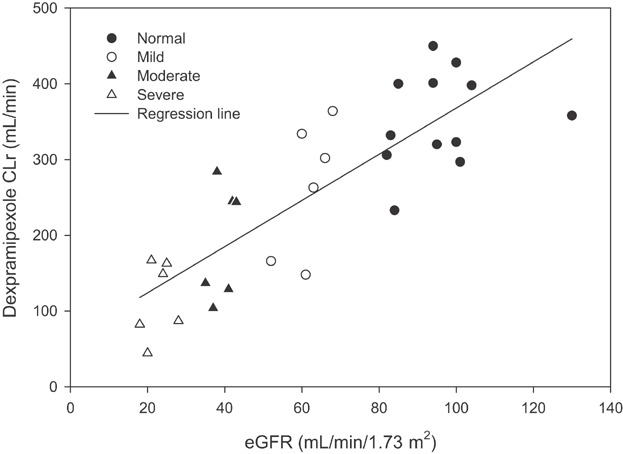
Dexpramipexole renal clearance (CLr) versus eGFR in subjects with normal renal function and in those with mild, moderate, severe renal impairment, after single dose administration of dexpramipexole at 150 mg or 75 mg. A regression line was applied. The slope (90% CI) of eGFR and CLr in the regression model was 3.1 (2.4, 3.7).
Dexpramipexole plasma t½ was prolonged corresponding to the severity of renal impairment. The geometric mean t½ was approximately 8 hours in healthy volunteers, and was 10.7 hours, 16.2 hours, 30.1 hours, and 80.2 hours in patients with mild, moderate, severe renal impairment, and ESRD subjects, respectively (Table2). This trend is consistent with reduced CLr since the kidneys serve as the primary route of elimination for dexpramipexole.
Table 2.
Summary Plasma, Urine and Dialysate PK Parameters of Dexpramipexole by Renal Function Groups
| Subjects by Category | ||||||
|---|---|---|---|---|---|---|
| Parametera | Normal (n = 8) | Mild (n = 6) | Moderate RI (n = 6) | Normal (n = 4) | Severe (n = 6) | ESRD (n = 6) |
| DEX dose, mg | 150 | 150 | 150 | 75 | 75 | 75 |
| AUC0-inf (h · ng/mL) | 4375.0 ± 1126.0 | 6558.3 ± 2643.7 | 8180.0 ± 3102.5 | 2212.5 ± 225.0 | 7156.7 ± 3126.2 | 22000 ± NAc |
| AUC0–72 (h · ng/mL) | 4360.0 ± 1123.4 | 6428.3 ± 2470.8 | 7728.3 ± 2792.9 | 2205.0 ± 225.8 | 6008.3 ± 2073.6 | 9855.0 ± 2387.0 |
| Cmax (ng/mL) | 406.5 ± 91.1 | 423.2 ± 91.7 | 358.2 ± 75.7 | 216.5 ± 40.2 | 227.2 ± 42.9 | 199.8 ± 49.9 |
| Median Tmax, h (min, max) | 2.5 (1.0, 3.0) | 2.5 (1.5, 4.0) | 3.0 (2.0, 4.0) | 2.8 (2.0, 4.0) | 3.0 (1.5, 6.0) | 5.0 (1.5, 16.0) |
| t1/2, h | 9.0 ± 2.7 | 11.0 ± 2.9 | 16.4 ± 2.5 | 7.2 ± 1.3 | 31.2 ± 8.8 | 82.1 ± 18.5 |
| CLr (mL/min) | 355.6 ± 75.1 | 262.8 ± 88.8 | 190.5 ± 75.8 | 350.3 ± 37.4 | 115.5 ± 51.0 | 5.5 ± 7.6b |
| CL/F(mL/min) | 454.4 ± 127.6 | 320.3 ± 107.3 | 253.0 ± 81.6 | 423.8 ± 43.3 | 149.1 ± 54.6 | 42.3 ± NAc |
| Vz/F (L) | 345.6 ± 114.8 | 287.7 ± 78.3 | 347.0 ± 82.0 | 264.3 ± 53.2 | 382.5 ± 140.3 | 204.0 ± NAc |
| fe (%) | 79.5 ± 7.7 | 81.2 ± 5.6 | 70.6 ± 10.5 | 83.3 ± 2.5 | 65.5 ± 12.3 | 7.5 ± 10.4b |
| CLd (mL/min) | NA | NA | NA | NA | NA | 138.7 ± 11.8 |
| fd (%) | NA | NA | NA | NA | NA | 6.8 ± 1.8 |
Data are mean ± SD unless otherwise specified.
n = 2
n = 1
AUC0–inf, plasma concentration curve from time zero extrapolated to infinity; AUC0–72, plasma concentration curve from time zero to 72 hours after oral administration; CL/F, apparent total body clearance (mL/min); Cmax, maximum plasma concentration; CLr, renal clearance; DEX, dexpramipexole; ESRD, end-stage renal disease; PK, pharmacokinetic; t1/2, terminal half-life; Tmax, time of maximum plasma concentration; Vz/F, apparent volume of distribution; fe %, Cumulative fraction of dose excreted unchanged into the urine from zero (pre-dose) to the last timepoint (%), calculated as Ae(0–last)/Dose; CLd, dialysis clearance; fd %, cumulative fraction of dose excreted unchanged into dialysate during the duration of dialysis (%), calculated as Ad/Dose.
A high-flux, high-efficiency dialyzer was used in each ESRD patient who underwent hemodialysis. Dexpramipexole elimination in ESRD subjects during both dialysis and non-dialysis (i.e., interval between dialysis) was very slow. During the non-dialysis period, the overall clearance (CL/F) was 42.3 mL/min (n = 1). Intermittent hemodialysis removed a small amount of dexpramipexole. The geometric mean dialysis clearance (CLd) in ESRD subjects was similar to CLr in severe renal impairment subjects, and was approximately one-third of CLr in subjects with normal renal function. Hemodialysis only decreased dexpramipexole plasma concentration by one-fourth, and the Fd over the first post-dose dialysis period was only 6.6% of the dexpramipexole dose. The insignificant elimination of dexpramipexole by hemodialysis is likely due to the wide tissue distribution of dexpramipexole and its intrinsic dependence on active renal secretion for elimination. Given the high-free fraction, the unbound dexpramipexole PK parameters approximated the parameters in its total form and were not calculated separately.
Dexpramipexole Safety and Tolerability
Single 75 mg and 150 mg doses of dexpramipexole were well tolerated by subjects with normal renal function, mild and moderate insufficiency (150 mg), and by subjects with severe insufficiency and ESRD (75 mg). The safety profile was comparable across renal function groups.
There were no SAEs, and no deaths reported in this study. All AEs were mild to moderate in severity. The most common AE was headache, assessed as mild to moderate in severity and considered related to study treatment by the Investigator. Analysis of hematology, blood chemistry, urinalysis, vital signs, and physical examination findings showed no abnormalities that were considered clinically significant by the Investigator or reported as AEs. Similarly, 12-lead ECG findings revealed no clinically significant abnormalities in the normal, mild, and moderate renal impairment groups; moderate prolongations in QTcB and QTcF were observed in severe and ESRD renal function groups, but these were also present at baseline and unchanged by exposure to dexpramipexole. The QT interval prolongation observed in this study was attributable to the underlying renal impairment of this subject population and not attributable to dexpramipexole.
Discussion
This study was conducted to evaluate the pharmacokinetics, safety, and tolerability of dexpramipexole in patients with various degrees of renal insufficiency.
Single 75 mg or 150 mg dexpramipexole doses were administered during this study. Based on prior knowledge of significant renal clearance, it was predicted that subjects with renal insufficiency may have increased exposure to dexpramipexole. Therefore it was considered prudent to dose healthy volunteers and patients with mild and moderate renal impairment with a single 150 mg dose, and subjects with severe renal impairment and ESRD with a 75 mg dose. A dose of 150 mg was half of the maximum tolerated single dose in healthy volunteers at the time of the study. Dexpramipexole exhibited linear PK. The formulation difference of round compressed oral tablet (75 mg) versus compressed film-coated tablet (150 mg) did not affect its PK. Therefore, the PK parameters for both treatments were dose normalized. On this basis, the single dose results from this trial could be extrapolated to understand drug exposure in the intended repeated dose regimen in the target patient population.
The study results illustrated a significant increase in dexpramipexole AUC with increasing severity of renal insufficiency. The increase in drug exposure in renally impaired subjects is likely due to the reduction in renal clearance as opposed to an increase in drug bioavailability because1 there was no difference in Cmax of dexpramipexole in plasma across subjects with various degrees of eGFR after single dose administration,2 dexpramipexole is primarily eliminated in kidney with minimal liver metabolism, and3 the renal clearance of dexpramipexole decreased with increasing severity of renal insufficiency. In fact, dexpramipexole is highly soluble in aqueous solutions (>300 mg/mL in water) and highly permeable (17 × 10−6 cm/sec in human intestinal Caco-2 cell monolayers in both directions); its bioavailability was close to 100% in rodent studies, and was predicted to be high in humans. In this study, approximately 80% of the administered dose was recovered in the urine of volunteers with normal renal function (Table2). Although comparable following a single dose, Cmax of dexpramipexole at steady state was expected to be different across renal function groups, after twice daily administration of dexpramipexole. In the current study, the dose normalized AUC(0–72) ratios were 1.4, 1.7, 2.7, and 4.5, respectively, in mild, moderate, severe renal impairment, and ESRD subjects to those with normal renal function. The highest dose of dexpramipexole tested in human was 300 mg twice daily, and given PK linearity, the exposure of dexpramipexole in subjects with severe renal impairment at 150 mg twice daily will exceed that in subjects with normal renal function at 300 mg twice daily. In ESRD patients, given its significant accumulation after a single dose and minimal elimination by hemodialysis, it was anticipated that the accumulation of dexpramipexole in a chronic use setting would be more significant. It is recommended that dexpramipexole not be given to patients with severe renal impairment or in those with ESRD.
The dose regimen recommendation in patients with mild or moderate renal impairment was less definitive. In a previous ascending dose study in healthy volunteers, heart rate transiently increased >20 bpm from baseline following single 450 mg and 600 mg doses and following 225 mg and 300 mg administered twice daily. The increase appeared to be dose and concentration dependent. This study population included only healthy volunteers and had a small sample size (n = 7 per dose), and the relationship of these findings to disease population was not definitive. In addition, the study only investigated short-term dosing (7 doses), and the relevance of these findings in the context of long-term dosing was unknown. Eventually, the safety data in larger patient population would reveal the significance of the heart rate elevation.
It is also worth mentioning that the contraindication of dexpramipexole in severe renal insufficiency or ESRD patients might not necessarily preclude the general ALS patient population. An analysis from the Northeast ALS Consortium (NEALS) database of more than 400 ALS patients (unpublished) indicated that most patients had normal renal function, and the percentage of patients with mild, moderate, severe, or ESRD at the entry of the trial was 34%, 2%, 0% and 0%, respectively. Therefore, dexpramipexole dose adjustment may only impact a very limited number of ALS patients.
Previous human studies indicated that dexpramipexole was renally filtered and secreted. In vitro substrate assessments indicated that secretion was potentially mediated by OCT-2. The current study results can be used to ascertain the impact of small changes in eGFR with changes in dexpramipexole renal clearance. Here, a single unit increase in eGFR (mL/min/1.73 m2) would be anticipated to produce a 3-unit increase in mean CLr (mL/min) of dexpramipexole. The slope of three also provided evidence that renal tubular secretion predominated excretion in comparison with passive glomerular filtration. Furthermore, the strong association between eGFR and CLr implied that in study subjects with various degrees of renal impairment, tubular secretion was reduced to a similar extent as glomerular filtration function, as estimated by eGFR.
The linear regression model appears to provide a good fit of the data. It should be noted that the linear regression model is limited with regard to extrapolation of CLr when eGFR is at or above the normal value for a young healthy subject (e.g., greater than 120 mL/min), or when eGFR is extremely low (e.g., less than 15 mL/min).
The insignificant effect of hemodialysis on the elimination of dexpramipexole may likely be due to the deep tissue distribution of dexpramipexole, which is plausible based on the compound's biopharmaceutic properties. The Vz/F of dexpramipexole was approximately 300 L, and therefore, after 4 hours of hemodialysis, only about 1/10th of Vz/F (the CLd of 138 mL/min × 4 hours of dialysis = 33 L) was exchanged. The majority of the dexpramipexole dose was not cleared and remained in the circulation and peripheral tissue.
There is one limitation of the study. Although the MDRD equation is considered superior to other methods of approximating renal function such as Cockcroft-Gault and to creatinine clearance measured from 24-hour urine collections,5 application of the equation to ALS patients with progressive muscle wasting may lead to large errors in GFR estimation due to accelerated muscle wasting. Therefore, the relationship of MDRD derived eGFR to the dexpramipexole PK might not be transferrable. Other renal function biomarkers were initially considered but were not studied. Inulin, an index for renal filtration function, is not readily available for intravenous use in humans and requires specialized assays for quantification. Cystatin C serum levels are virtually unaffected by age (>1 year), muscle mass, sex, and race, and therefore has recently been considered an alternative and more sensitive endogenous marker for the estimation of GFR than serum creatinine based GFR estimations in muscle wasting diseases.6 However, recent studies show correlations between CSF cystatin C levels to both ALS disease progression and patient survival.7
Dexpramipexole was well tolerated in all patients, including patients with various degrees of renal insufficiency. The safety profile was comparable across renal function groups, and the AEs noted in the study were consistent with what has been previously reported.
In conclusion, Dexpramipexole exposure was significantly increased with the severity of renal impairment, primarily due to the reduction in renal clearance. There was a positive linear association between renal function (eGFR) and dexpramipexole CLr. These results suggest that without dose regimen modification extensive drug accumulation may occur with repeated dosing in patients with significant renal impairment.
Acknowledgments
The authors gratefully acknowledge the contribution of Laurence Levasseur, Ph.D. of Quintiles, Inc for the pharmacokinetic analysis presented in the manuscript. The authors also thank Knopp Biosciences for its contribution.
Financial disclosure
This study was supported by Biogen Idec. PH was employee of Biogen Idec at the time of the study and has Biogen Idec stock ownership. The authors DK, WF, SS, YD, DW and MR are employees of Biogen Idec and are eligible for Biogen Idec stock options and have stock ownership. TM is employee of Orlando Clinical Research Center. DR is employee of Kidney Specialists of Minnesota. Neither TM nor DR has conflict to disclose.
References
- 1.Gribkoff VK, Bozik ME. KNS-760704 [(6R)-4,5,6,7-tetrahydro-N6-propyl-2, 6-benzothiazole-diamine dihydrochloride monohydrate] for the treatment of amyotrophic lateral sclerosis. CNS Neurosci Ther. 2008;14:215–226. doi: 10.1111/j.1755-5949.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schapira AH. Dopamine agonists and neuroprotection in Parkinson's disease. Eur J Neurol. 2002;9:7–14. doi: 10.1046/j.1468-1331.9.s3.9.x. [DOI] [PubMed] [Google Scholar]
- 3.Cudkowicz M, Bozik ME, Ingersoll EW, et al. The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nat Med. 2011;17:1652–1656. doi: 10.1038/nm.2579. [DOI] [PubMed] [Google Scholar]
- 4.National Chronic Kidney Disease Fact Sheet. Centers for Disease Control and Prevention Web site. 2014. http://www.cdc.gov/diabetes/pubs/factsheets/kidney.htm. Published 2014. Accessed May 6, 2014.
- 5.Shemesh O, Golbetz H, Kriss JP, Myers BD. Limitations of creatinine as a filtration marker in glomerulopathic patients. Kidney Int. 1985;28:830–838. doi: 10.1038/ki.1985.205. [DOI] [PubMed] [Google Scholar]
- 6.Pucci L, Triscornia S, Lucchesi D, et al. Cystatin C and estimates of renal function: searching for a better measure of kidney function in diabetic patients. Clin Chem. 2007;53:480–488. doi: 10.1373/clinchem.2006.076042. [DOI] [PubMed] [Google Scholar]
- 7.Wilson ME, Boumaza I, Lacomis D, Bowser R. Cystatin C: A candidate biomarker for amyotrophic lateral sclerosis. PLoS ONE. 2010;5:e15133. doi: 10.1371/journal.pone.0015133. [DOI] [PMC free article] [PubMed] [Google Scholar]