

Abstract
Two previously conducted rivaroxaban studies showed that, separately, renal impairment (RI) and concomitant administration of erythromycin (P-glycoprotein and moderate cytochrome P450 3A4 [CYP3A4] inhibitor) can result in increases in rivaroxaban exposure. However, these studies did not assess the potential for combined drug–drug–disease interactions, which—in theory—could lead to additive or synergistic increases in exposure. This study investigated rivaroxaban pharmacokinetics and pharmacodynamics when co-administered with steady-state (SS) erythromycin in subjects with either mild or moderate RI. Similar to previous studies, rivaroxaban administered alone in RI subjects, or when co-administered with SS erythromycin in normal renal function (NRF) subjects, increased rivaroxaban exposure. When combined, the co-administration of rivaroxaban 10 mg with SS erythromycin in subjects with mild or moderate RI produced mean increases in rivaroxaban AUC∞ and Cmax of approximately 76% and 56%, and 99% and 64%, respectively, relative to NRF subjects, with PD changes displaying a similar trend. No serious adverse events occurred and no persistent adverse events were reported at the end of study. Although these increases were slightly more than additive, rivaroxaban should not be used in patients with RI receiving concomitant combined P-glycoprotein and moderate CYP3A4 inhibitors, unless the potential benefit justifies the potential risk.
Keywords: rivaroxaban, erythromycin, drug–drug–disease interaction, renal impairment, pharmacokinetics
Rivaroxaban is a potent, direct Factor Xa inhibitor with high oral bioavailability, predictable pharmacokinetics (PK), and a rapid onset and offset of action.1 Rivaroxaban has been shown to be an effective and well-tolerated alternative to traditional anticoagulants for the prevention and treatment of venous thromboembolism and for stroke prevention in patients with non-valvular atrial fibrillation.2–8
Rivaroxaban has a dual mode of elimination, in which approximately two-thirds of the absorbed dose is hepatically metabolized through oxidative and hydrolytic pathways via cytochrome P450 (CYP) enzymes (CYP3A4/3A5 and CYP2J2) and CYP-independent mechanisms (Figure S1 of Supporting information), then excreted as inactive metabolites in both the urine and the feces.9 The remaining third of the absorbed dose is eliminated as unchanged drug in the urine via P-gp-mediated and ABCG2 (also abbreviated as Bcrp for breast cancer resistance protein)-mediated secretion.2,10
Considering the percentage of the administered dose renally eliminated as unchanged drug and also metabolized via CYP3A4/3A5 enzymes, a renal impairment study and several drug–drug interaction studies, including an erythromycin drug–drug interaction study, were previously conducted to characterize their effect on the PK and pharmacodynamics (PD) of rivaroxaban.
Although the changes in rivaroxaban exposure observed in the renal impairment and erythromycin interaction studies were not considered clinically relevant when assessed independently from each other, the potential for a combined drug–drug–disease interaction potentially resulting in clinically relevant increases in rivaroxaban exposure could not be ruled out. This particular clinical scenario was assessed by the Food and Drug Administration (FDA) through the use of physiologically-based pharmacokinetic (PBPK) modeling, in which the authors concluded that a drug–drug–disease interaction, potentially leading to a synergistic increase in rivaroxaban exposure, may occur in these types of clinical scenarios.11
Therefore, this study was conducted to evaluate the actual extent of this type of interaction with the concomitant use of rivaroxaban and erythromycin (a moderate inhibitor of CYP3A isozymes and a reported inhibitor of P-gp-mediated secretion)11 in subjects with various degrees of renal impairment.
Methods
Subjects
Men or women aged 35–75 years were eligible for participation in this study if they: had a body mass index of 18–38 kg/m2; had a body weight of ≥50 kg; and had been characterized as having either normal renal function, mild renal impairment, or moderate renal impairment, but were otherwise healthy.
Subjects were excluded from the study if they had: a history of or current clinically significant medical illness or any other illness that could interfere with the interpretation of the study results; any condition that would preclude the use of erythromycin or rivaroxaban; clinically significant abnormal values for hematology, clinical chemistry, or urinalysis (other than CLCR 30–79 mL/min – see Study Design section); clinically significant abnormal physical examination, vital signs, or 12-lead electrocardiogram (ECG); presence or history of disorders known to be associated with increased risk of bleeding (e.g. acute gastritis, acute peptic ulcer, prior hemorrhage, coagulation disorders); concomitant use (≤2 weeks prior to the start of the study) of drugs that influence either the coagulation system or cytochrome P450 3A4 metabolism and P-gp transport systems; or a history of drug or alcohol abuse within the past 2 years. Subjects were also excluded if they were pregnant or breastfeeding or intended to become pregnant during the study. Renally impaired subjects taking medications for conditions associated with their underlying renal impairment, which were allowed as per protocol, were to maintain their original treatment schedule during the study, and these medications were to be documented. While the use of concomitant medications was allowed in the study, each medication was reviewed by the sponsor prior to subject enrollment to ensure they were not classified as inhibitors of the relevant CYP or P-gp pathways utilized by rivaroxaban or erythromycin.
All subjects gave written informed consent to participate in the study and the study protocol and amendments were reviewed by an Institutional Review Board (IRB). There were three separate IRBs in total: Independent Investigational IRB, Inc. (Plantation, FL); Aspire IRB, LLC (Santee, CA); and Crescent City IRB (New Orleans, LA). The study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice Guideline, and applicable regulatory requirements.
Study Design
This study had an open-label, sequential design and was conducted across four US study sites in Orlando, FL; St. Paul and Minneapolis, MN; and Knoxville, TN. Target enrollment was 24 subjects, with eight in each renal function group; however, subjects that did not complete the study were replaced by individuals with the same renal characteristics. To ensure demographic balance between the renal function groups, subjects with mild and moderate renal impairment were enrolled and completed the study first, followed by subjects with normal renal function who were matched for mean age (± 10 years), mean body mass (± 20%), and sex.
Renal function was determined by taking the mean value of two estimated CLCR measurements assessed using the Cockcroft–Gault equation.12 Measurements were taken approximately 1–2 weeks before the study, ≥7 days apart. Subjects were then assigned to one of three renal function groups: Group 1: CLCR ≥80 mL/min (normal renal function – healthy controls); Group 2: CLCR 50–79 mL/min (mild renal impairment); Group 3: CLCR 30–49 mL/min (moderate renal impairment). The Cockcroft–Gault method was selected based on current industry guidelines13 and in order to be consistent with previous rivaroxaban studies.
The study schedule began with a screening phase of approximately 20 days (in which informed consent, medical history, and other screening factors for eligibility were assessed), followed by an open-label treatment phase (Figure 1). This open-label phase consisted of two sequential treatment periods for subjects with normal renal function and three sequential treatment periods for subjects with mild or moderate renal impairment. Treatment phases were separated by a washout period of 7–14 days.
Figure 1.
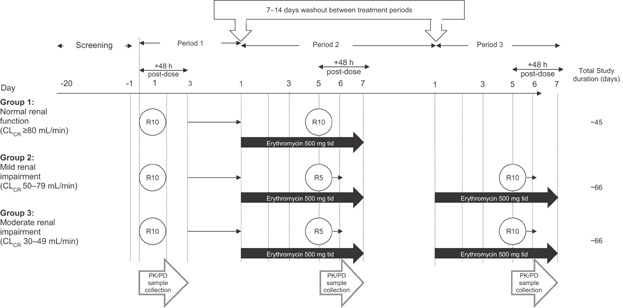
Trial design. CLCR, creatinine clearance; PK, pharmacokinetic; R, rivaroxaban; tid, three times daily.
The duration of treatment period 1 was 4 days, in which all subjects received a single 10 mg dose of rivaroxaban on the morning of day 1, followed by 48 hours of post-dose PK and PD blood sampling. Subjects were discharged from the study site on day 3, after the last blood sample collection and safety assessment. All subjects then proceeded through a washout period of 7–14 days and returned to their respective study site for treatment period 2. The duration of treatment period 2 was 8 days, in which all subjects received erythromycin 500 mg three times daily for 6 days (day 1 through day 6). On the morning of day 5, subjects with normal renal function received the concomitant administration of rivaroxaban 10 mg with the erythromycin dose and subjects with either mild or moderate renal impairment received the concomitant administration of rivaroxaban 5 mg with the erythromycin dose. This concomitant dose administration was followed by 48 hours of post-dose PK and PD blood sampling that ended on the morning of day 7. On completion of treatment period 2 and after an assessment of safety, subjects with normal renal function were discharged from the study and subjects with mild or moderate renal impairment proceeded through a second washout period of 7–14 days. On completion of this second washout period, subjects with renal impairment returned to their respective study site for treatment period 3. The design and duration of treatment period 3 mimicked that of treatment period 2, in which all subjects received the same erythromycin regimen for 6 days; however, on the morning of day 5, subjects now received the concomitant administration of rivaroxaban 10 mg with the erythromycin dose. Once again, PK and PD blood samples were collected for 48 hours post-dose and subjects were discharged from the study on the morning of day 7, after the final blood sample collection and safety assessments (Figure 1).
Pharmacokinetic and Pharmacodynamic Assessments
Bioanalytical Procedures
Blood samples for both the determination of rivaroxaban plasma concentrations (1 mL each) and PD markers – prothrombin time (PT); activated partial thromboplastin time (aPTT); HepTest®, and Factor Xa activity (total of 3 mL each) – were collected at the following time points: –1 (pre-dose), 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 36, and 48 hours (post-dose) for all treatment periods (on days 1–3 of treatment period 1 and days 5–7 for treatment periods 2 and 3). Blood samples (3 mL each) for the determination of erythromycin plasma concentrations during treatment periods 2 and 3 were collected prior to the morning administration of erythromycin on days 2–6 and again on the morning of day 7.
Urine samples for the determination of rivaroxaban concentrations were collected prior to rivaroxaban dosing (on day 1 of treatment period 1 and day 5 of treatment periods 2 and 3) and at intervals of 0–4, 4–8, 8–12, 12–24, and 24–48 hours post-dose on days 1–3 of treatment period 1 and days 5–7 for treatment periods 2 and 3. Subjects voided their bladders just before the administration of study drug (pre-dose) and just before starting each timed urine collection interval. The exact collection date and time of these samples, pH, and complete volume of each collection interval of these samples were recorded.
Plasma and urine samples for rivaroxaban concentrations were frozen and stored at approximately −20 °C until shipped and analyzed by the department of Drug Metabolism and Pharmacokinetics at Bayer Pharma AG (Wuppertal, Germany). Rivaroxaban concentration was determined in plasma after protein precipitation with acetonitrile (including an internal standard, [2H5·15N] rivaroxaban; MW 441.9 g/mol [WITEGA-Adlershof GmbH]) followed by separation employing high-performance liquid chromatography–tandem mass spectrometry; the specific assay has been described in detail elsewhere.14 The calibration range of the procedure was 0.50–500 µg/L; the inter-assay precision was ≤6.0% and inter-assay accuracy was between 98.2% and 101.2%. Rivaroxaban concentration was determined in urine after solid phase extraction (including an internal standard, BAY 60–4758; MW 463.9 g/mol [Bayer HealthCare AG]) followed by separation employing high-performance liquid chromatography with ultraviolet detection. The calibration range for this procedure was 0.1–20 mg/L; the inter-assay precision was ≤3.0% and inter-assay accuracy was between 98.5% and 101.6%.
Concentrations of erythromycin were analyzed from plasma samples using a validated specific and sensitive liquid chromatography coupled with tandem mass spectrometry (LC/MS/MS) method by PRA International Early Development Services (Assen, The Netherlands). The method was validated according to the FDA Guidance for Industry – Bioanalytical Method Validation.15 Briefly, frozen plasma samples were thawed and prepared for bioanalysis by protein precipitation in methanol followed by reverse phase liquid chromatography using a Waters XBridge Shield RP18 column (50 × 2.1 mm, 35 μm particle size) at a temperature of 40 °C coupled with a PE Sciex API 3000 triple quadrupole mass spectrometer. Monitored ions were 734.7 → 158.3 (erythromycin) and 738.7 → 162.4 (internal standard). The internal standard used was erythromycin-13C-d3; 737.9 g/mol (Sigma–Aldrich [Zwijndrecht, The Netherlands]). The blank matrix used was human K2-EDTA (Sera Laboratories International, Haywards Heath, UK). The quantification range for erythromycin in plasma was 20.0–5,000 ng/mL. The inter-assay precision was ≤8.0% and the inter-assay accuracy ranged from 95.6 to 104.5%. The intra-day precision (CV%) ranged from 2.2 to 5.6% and the inter-day precision (CV%) ranged from 1.7 to 8.0% (total CV%: 4.8 to 9.8%).
Plasma samples for the determination of PT, aPTT, HepTest®, and Factor Xa activity were frozen and stored at approximately −20 °C until shipped and analyzed by the departments of Toxicology Pathology/Clinical Pathology and Clinical Biomarkers at Bayer Pharma AG (Wuppertal, Germany). PT samples were automatically processed by an STA Compact Hemostasis workstation (Stago Asnieres, France) using STA Neoplastin as the reagent (Roche Diagnostics; Mannheim, Germany). The aPTT samples were automatically processed by an STA Compact Hemostasis workstation (Stago Asnieres, France) using STA aPTT Kaolin and calcium chloride 0.025 M as the reagents (Roche Diagnostics; Mannheim, Germany). HepTest® samples were semi-automatically processed using a Ball Coagulometer KC 10A (Amelung, Lemgo, Germany) and HepTest® Heparin Test as the reagent. Factor Xa samples were measured by a two-step photometric method. During the first step, the total Factor X is activated via Russell's Viper venom (Haemochrom, Germany) in the presence of calcium ions to Factor Xa. In the second step, Factor Xa hydrolyzes the chromogenic substrate Z-D-Arg-Gly-Arg-pNA (S-2765; Haemochrom, Germany), releasing the chromogenic group p-nitroanilin. The color intensity is determined with a photometer at 405 nm and the quantity of the released p-nitroanilin is proportional to the Factor Xa activity of the sample.
PK Assessments
The following key rivaroxaban plasma PK parameters were determined for each treatment by subject group via non-compartmental analysis methods utilizing Win Nonlin® software (version 5.2.1; Pharsight Corporation, California, USA): maximum plasma concentration (Cmax); time to maximum concentration (tmax); area under the concentration–time curve from time 0 to infinite time (AUC∞); elimination half-life (t1/2); total apparent oral clearance (CL/F); and apparent volume of distribution based on the terminal elimination phase (Vd/F).
The following key rivaroxaban PK parameters in urine were also calculated using non-compartmental methods: the cumulative amount excreted into the urine (Ae) over 48 hours; renal clearance (CLR) calculated as Ae/AUC0–48h; CLCR estimated using the Cockcroft–Gault equation;12 clearance by glomerular filtration (CLGFR) calculated as fraction unbound of rivaroxaban(fu)*CLCR; and active renal clearance of drug (CLact) calculated as CLR–CLGFR.
Based on pre-dose PK samples obtained, erythromycin plasma concentrations at trough were determined and attainment of steady-state conditions was assessed visually.
PD Assessments
PD parameters were calculated via non-compartmental analysis methods utilizing Win Nonlin® software (version 5.2.1; Pharsight Corporation, California, USA) and using change from baseline versus time profiles for each of the PD markers, in which baseline was defined as the pre-dose (−1 hour) PD sample collection per treatment period. However, for the sake of brevity, only Factor Xa inhibition and PT prolongation are described in detail within this manuscript. The PD parameters calculated were: area under the PD–time curve from time 0 to 48 hours (AUC0–48h) and the maximum effect (Emax).
Safety and Tolerability
Safety and tolerability were evaluated continuously throughout the study by monitoring the incidence of bleeding events and other treatment-emergent adverse events (TEAEs), as well as the severity and relationship of adverse events (AEs) to the study drugs. Safety was also evaluated by monitoring vital signs (heart rate, blood pressure), ECG variables, and changes in clinical laboratory test values (hematology, clinical chemistry, coagulation tests [PT and aPTT], and urinalysis) and by performing regular physical examinations. All subjects who received ≥1 dose of the study drug were included in the safety and tolerability analysis.
Statistical Methods
Statistical evaluations were performed using the SAS v9.1.3 software package (SAS Institute, Cary, NC, USA). The design of this study allowed for several different test and reference conditions. (1) When assessing the effects of renal impairment on the PK and PD of rivaroxaban, the test condition consisted of subjects with either mild or moderate renal impairment receiving rivaroxaban, with a reference condition consisting of subjects who had normal renal function and had received rivaroxaban. (2) When assessing the effects of concomitant erythromycin use, the test condition consisted of subjects with normal renal function receiving co-administered rivaroxaban and erythromycin, with a reference condition consisting of the same subjects, but receiving rivaroxaban alone. (3) When assessing the combined effects of renal impairment and concomitant erythromycin use on the PK and PD of rivaroxaban, the test condition consisted of subjects with either mild or moderate renal impairment receiving concomitantly administered rivaroxaban and erythromycin, with a reference condition consisting of subjects who had normal renal function and had received rivaroxaban alone. (4) Lastly, when the comparison was performed within the same renal function groups, the test condition consisted of subjects with either mild or moderate renal impairment receiving concomitantly administered rivaroxaban and erythromycin, with a corresponding reference condition consisting of the same subjects, but receiving rivaroxaban alone.
PK Assessments
Plasma concentrations of rivaroxaban were plotted versus time for each renal function group and PK parameters were summarized using descriptive statistics for each treatment period and renal function group. The primary parameters of interest for the statistical analysis were the log-transformed AUC∞ and Cmax values of rivaroxaban. When assessing the rivaroxaban 5 mg data, all PK parameters were dose-normalized to 10 mg. For each log-transformed, dose-normalized PK parameter, a mixed-effects ANOVA model was used to estimate the least squares means and intra- or inter-subject variance. The model included treatment, renal function group, and the interaction between treatment and renal function group as fixed effects, and subject as a random effect. Using the estimated least squares means and intra-/inter-subject variance, the point estimate and 90% confidence intervals (CIs) for the difference in means on a log-scale were constructed for the test to reference conditions. The limits of the CIs were retransformed using antilogarithms to obtain 90% CIs for the ratios of the mean area under the concentration–time curve (AUC) and Cmax of the test to reference condition. Attainment of steady-state for erythromycin concentrations was assessed through visual inspection of the concentration–time plots.
Only concentration data from subjects who completed the study were included in the statistical analysis. A subject or sample was excluded if there was non-compliance with study procedures, >10% of data were missing, or the pre-dose rivaroxaban concentration was >5% of the Cmax. Where data were below the limit of quantification, those data were treated as zero in the summary statistics and for the calculation of PK parameters.
PD Assessments
The PD parameters AUC0–48h and Emax (based on change from baseline values) for each of the PD markers were summarized by arithmetic mean, standard deviation, coefficient of variation (CV), median, minimum, and maximum at each post-dose time point. Median change from baseline versus time profiles for Factor Xa activity, PT, aPTT, and HepTest® were plotted by treatment and renal function group. The primary parameters of interest for the statistical analysis were the log-transformed AUC0–48h and Emax values. Only data from subjects who completed the study were included in the statistical analysis. If a subject had an inestimable PD parameter, then the data for that subject were not included in the statistical analysis of that parameter. Mixed-effects ANOVA models were used to generate point estimates and 90% CIs (for the ratios of the mean AUC0–48h and Emax of the test to reference condition) using the same methods as for the PK parameters above. The PD parameters (AUC0–48h and Emax) were not dose-normalized for the rivaroxaban 5 mg dose because not all of the parameters have a linear response with increasing rivaroxaban exposure, and thus, a true comparison could not be made and were not included in this manuscript.
Sample Size Determination
Based on prior study results for rivaroxaban in renally impaired subjects,16 the inter-subject CVs for PK parameters (AUC and Cmax) were estimated to be <35%, and the inter-subject CVs for PD parameters (AUC0–48h and Emax for PT) were estimated to be <45% for rivaroxaban in subjects with moderate renal impairment and in subjects with normal renal function. Using these cutoffs, a sample size of eight subjects completing the study in each renal function group was considered sufficient to determine point estimates of the ratio of mean PK (dose-normalized) and PD parameters of the renally impaired groups versus the normal renal function group subjects to fall within 73.5–136.1% and 67.3–148.6% of the true values with 90% confidence, respectively.
Based on the results of an earlier assessment of rivaroxaban/clarithromycin drug interaction [Bayer Pharma AG, data on file], the intrasubject CV for the PK parameters Cmax and AUC, was estimated to be <15% and the intrasubject CV for the PD parameters Emax and AUC for PT was estimated to be <12% for rivaroxaban in subjects with normal renal function. Using an estimated intrasubject CV of 15% for the PK parameters and 12% for the PD parameters for rivaroxaban, a sample size of eight subjects completing the study in each renal function group was considered sufficient to determine point estimates of the ratio of mean PK and PD parameters so as to fall within 86.8–115.3% and 89.3–112.0% of the true values with 90% confidence, respectively.
Results
Demographics
In total, 29 subjects were enrolled in the study (aged 46 − 75 years). There were 8 subjects in the normal renal function group, 12 subjects in the mild renal impairment group, and 9 subjects in the moderate renal impairment group. All subjects were correctly placed into their respective renal function groups through the calculation of their mean CLCR value during screening. Details of the baseline demographics according to renal function category are presented in Table 1. Of the 29 patients enrolled, 24 completed the study. One patient withdrew consent, two patients withdrew as a result of AEs, and two withdrew for other reasons (protocol violation & positive drug screen).
Table 1.
Demographics and Baseline Characteristics of the Participants with Normal and Impaired Renal Function
| Normal renal function (n = 8) | Mild renal impairment (n = 12) | Moderate renal impairment (n = 9) | |
|---|---|---|---|
| CLCR, mL/min | |||
| Mean (SD) | 99.5 (12.6) | 67.1 (9.4) | 42.5 (4.4) |
| Age, years | |||
| Mean (SD) | 61 (5) | 64 (10) | 63 (10) |
| Median (range) | 61 (55–69) | 67.5 (46–75) | 68 (46–71) |
| Sex, n (%) | |||
| Male | 2 (25) | 3 (25) | 3 (33) |
| Female | 6 (75) | 9 (75) | 6 (67) |
| Race, n (%) | |||
| White | 7 (88) | 9 (75) | 7 (78) |
| Black or African-American | 1 (13) | 3 (25) | 2 (22) |
| Weight, kg | |||
| Mean (SD) | 75.5 (10.6) | 76.7 (15.9) | 84.8 (19.8) |
| Height, cm | |||
| Mean (SD) | 164.5 (5.7) | 165.3 (8.3) | 163.1 (9.0) |
| Body mass index, kg/m2 | |||
| Mean (SD) | 27.9 (3.6) | 27.9 (4.0) | 31.7 (5.9) |
CLCR, creatinine clearance using the Cockcroft–Gault equation; SD, standard deviation.
Pharmacokinetics/Pharmacodynamics
Effect of Renal Function on Rivaroxaban PK and PD
When assessing the effects of renal function on rivaroxaban exposure, subjects with mild renal impairment receiving a single 10 mg dose of rivaroxaban had increases in AUC∞ and Cmax of approximately 15% and 23%, respectively, and subjects with moderate renal impairment had increases of approximately 17% and 36%, respectively, when compared with subjects with normal renal function (Figure 2a and Table S1 of Supporting information). Rivaroxaban CL/F and CLR values decreased with decreasing renal function. A similar trend was observed for CLGFR and CLact. Consequently, the total amount of drug secreted in the urine (Ae) also decreased with decreasing renal clearance parameters (Table 2). Additionally, subjects with renal impairment displayed a median tmax that occurred approximately 1 hour earlier and a mean elimination t1/2 that was prolonged by approximately 1–2 hours, although these values remained within the typical range of t1/2 previously observed (Table 2).
Figure 2.
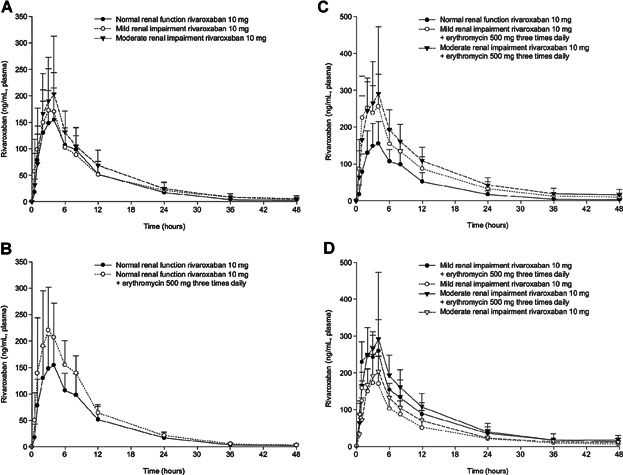
(a) Mean (+SD) plasma rivaroxaban concentrations following administration of rivaroxaban 10 mg in subjects with normal renal function and mild or moderate renal impairment. (b) Mean (+SD) plasma rivaroxaban concentrations following administration of rivaroxaban 10 mg with and without concomitant erythromycin 500 mg tid in subjects with normal renal function. (c) Mean (+SD) plasma rivaroxaban concentration following administration of rivaroxaban 10 mg with and without concomitant erythromycin 500 mg tid in subjects with normal renal function and mild or moderate renal impairment. (d) Mean (+SD) plasma rivaroxaban concentration following administration of rivaroxaban 10 mg with and without concomitant erythromycin 500 mg tid in subjects with mild or moderate renal impairment.
Table 2.
Arithmetic Mean (SD) Plasma and Urine Pharmacokinetic Parameters of Rivaroxaban with and without the Concomitant Administration of Erythromycin in Participants with Normal and Impaired Renal Function
| Rivaroxaban 10 mg | |||
|---|---|---|---|
| PK parameters | Normal renal function (n = 8) | Mild renal impairment (n = 12) | Moderate renal impairment (n = 9) |
| Cmax, ng/mL | 183 (44.6) | 210 (65.4) | 245 (96.6) |
| tmax, ha | 4.00 (2.00–8.00) | 3.00 (1.00–8.00) | 3.00 (1.00–4.00) |
| AUC∞, ng · h/mL | 1798 (459) | 2021 (835) | 2335 (738) |
| t½, h | 7.9 (2.5) | 9.9 (2.4) | 8.9 (1.7) |
| Vd/F, L | 67.7 (32.1) | 77.3 (26.9) | 58.5 (18.6) |
| CL/F, L/h | 5.91 (1.58) | 5.53 (1.74) | 4.68 (1.49) |
| Ae, mg | 3.51 (0.773) | 2.49 (0.697)a | 1.30 (0.654) |
| CLR, L/h | 2.12 (0.833) | 1.40 (0.517)a | 0.594 (0.278) |
| CLGFR, mL/min | 7.39 (2.51) | 6.40 (3.97) | 3.1 (1.15) |
| CLact, mL/min | 27.9 (13.8) | 16.8 (8.91)a | 6.80 (4.43) |
| Rivaroxaban 10 mg + erythromycin 500 mg tid | |||
|---|---|---|---|
| Normal renal function (n = 8) | Mild renal impairment (n = 8) | Moderate renal impairment (n = 8) | |
| Cmax, ng/mL | 256 (62.3) | 287 (77.3) | 323 (172) |
| tmax, ha | 2.50 (1.00–6.00) | 3.00 (1.00–4.00) | 3.00 (1.00–7.98) |
| AUC∞, ng · h/mL | 2,473 (468) | 3,228 (1194) | 3,584 (1005)b |
| t½, h | 7.4 (1.8) | 9.7 (2.7) | 9.8 (2.1)b |
| Vd/F, L | 44.2 (10.4) | 46 (14.7) | 40.9 (9.57)b |
| CL/F, L/h | 4.18 (0.804) | 3.38 (0.931) | 2.96 (0.75)b |
| Ae, mg | 4.80 (0.851) | 3.29 (0.893) | 2.74 (1.10) |
| CLR, L/h | 2.06 (0.650) | 1.15 (0.391) | 0.800 (0.383) |
| CLGFR, mL/min | 7.39 (2.51) | 6.40 (3.97)c | 3.10 (1.15)d |
| CLact, mL/min | 27.0 (11.6) | 12.2 (7.74) | 10.1 (7.01) |
| Rivaroxaban 5 mg + erythromycin 500 mg tid | |||
|---|---|---|---|
| Mild renal impairment (n = 10) | Moderate renal impairment (n = 9) | ||
| Cmax, ng/mL | – | 179 (54.5) | 164 (51.8) |
| DNCmax, ng/mL | – | 358 (109) | 329 (104) |
| tmax, ha | – | 3.00 (1.00–4.00) | 2.00 (1.00–4.00) |
| AUC∞, ng · h/mL | – | 1,967 (835) | 1,981 (510) |
| DNAUC∞, ng · h/mL | – | 3,935 (1,671) | 3,961 (1,020) |
| t½, h | – | 9.6 (3.3) | 10.0 (1.8) |
| Vd/F, L | – | 37.5 (11.7) | 38.1 (11) |
| CL/F, L/h | – | 2.89 (1.05) | 2.68 (0.739) |
| Ae, mg | – | 1.56 (0.692) | 1.04 (0.531) |
| CLR, L/h | – | 0.956 (0.475) | 0.555 (0.272) |
| CLGFR, mL/min | – | 6.40 (3.97)c | 3.10 (1.15) |
| CLact, mL/min | – | 9.39 (9.01) | 6.15 (4.41) |
Data presented as arithmetic mean (SD) unless otherwise noted. Ae, cumulative amount excreted into the urine over 48 hours; AUC∞, area under the plasma concentration–time curve from time 0 to infinity; CLact, active renal clearance; CL/F, total apparent oral clearance of drug; CLGFR, clearance by glomerular filtration; CLR, renal clearance; Cmax, maximum plasma concentration; DN, dose-normalized; h, hour; PK, pharmacokinetics; SD, standard deviation; t1/2, elimination half-life; tid, three times daily; tmax, time to reach the maximum plasma concentration; Vd/F, apparent volume of distribution.
Data presented as median (minimum–maximum).
n = 11.
n = 7.
n = 12.
n = 9.
In general, changes in the PD parameters displayed a similar trend for increased values; however, these changes were not as consistent for each of the PD parameters (Table S2 of Supporting information). Subjects with mild renal impairment displayed increases in Factor Xa inhibition AUC0–48h and Emax values by approximately 16% and 17%, respectively, and increases in PT AUC0–48h and Emax values by approximately 47% and 5%, respectively. Subjects with moderate renal impairment had very slight increases in Factor Xa inhibition, with AUC0–48h and Emax values increasing by approximately 3% and 17%, respectively, whereas AUC0–48h and Emax values for PT did not seem to change significantly (Table S3 and Figures S2a and S3a of Supporting information). Additionally, changes in aPTT and HepTest® were inconsistent and did not show any particular trend (data not shown).
Effect of the Co-administration of Erythromycin on Rivaroxaban PK and PD
When assessing the effects of erythromycin on rivaroxaban exposure, subjects with normal renal function receiving the concomitant administration of a single 10 mg dose of rivaroxaban with steady-state erythromycin had increases in AUC∞ and Cmax values by approximately 39% and 40%, respectively (Figure 2b and Table S1 of Supporting information). The concomitant administration of erythromycin decreased CL/F values with no apparent changes observed for either CLGFR or CLact (Table 2). The co-administration of erythromycin led to a mean tmax value that occurred approximately 1.5 hours earlier while producing a similar mean elimination t1/2 (Table 2). Additionally, steady-state erythromycin concentrations seem to have been obtained by the morning of day 5, prior to when the co-administration of erythromycin and rivaroxaban occurred.
Changes in the PD parameters displayed a similar trend for increased values; however, these changes were not as consistent for each of the PD parameters. The concomitant administration of rivaroxaban and erythromycin produced slightly lower Factor Xa inhibition AUC0–48h values by approximately 8% whereas Emax values increased by approximately 18%, and PT AUC0–48h and Emax values increased by approximately 22% and 10%, respectively (Table S3 and Figures S2b and S3b of Supporting information). Similar trends as above were also observed for aPTT and HepTest® (data not shown).
Effect of Renal Function and the Co-administration of Erythromycin on Rivaroxaban PK and PD (Comparisons across Renal Function Groups)
When assessing the combined effects of renal function and concomitant erythromycin use on rivaroxaban exposure across renal function groups, subjects with mild renal impairment receiving the concomitant administration of rivaroxaban 10 mg with steady-state erythromycin had increases in AUC∞ and Cmax values by approximately 76% and 56%, respectively, when compared with subjects with normal renal function receiving rivaroxaban 10 mg alone. For subjects with moderate renal impairment receiving the same regimen, AUC∞ and Cmax values increased by approximately 99% and 64%, respectively, when compared with subjects with normal renal function receiving rivaroxaban 10 mg alone (Table 3 and Figure 2c). In both above-mentioned scenarios, similar increases in the dose-normalized PK parameters were also observed with the administration of a single 5 mg dose of rivaroxaban with steady-state erythromycin, although with slightly higher values (Table S4 of Supporting information).
Table 3.
Ratio of Geometric Mean AUC∞ and Cmax Data and Associated 90% Confidence Interval After the Administration of Rivaroxaban 10 mg with and without Erythromycin 500 mg tid in Participants with Normal and Impaired Renal Function (n = 8 per Test and Reference Groups)
| Assessing the effects of renal function and concomitant erythromycin on rivaroxaban PK (comparisons across renal function groups) | ||||
|---|---|---|---|---|
| Test | Reference | |||
| Mild RI | ||||
| Rivaroxaban 10 mg + erythromycin 500 mg tid | Normal RF Rivaroxaban 10 mg | Geometric mean ratio (%) with 90% CI | Inter-subject CV (%) | |
| AUC∞, ng · h/mL | 3,078.7 | 1,745.5 | 176.4 (136.59–227.77) | 27 |
| Cmax, ng/mL | 277.2 | 178.17 | 155.6 (118.03–205.07) | 29 |
| Moderate RI | ||||
| Rivaroxaban 10 mg + erythromycin 500 mg tid | Normal RF Rivaroxaban 10 mg | Geometric mean ratio (%) with 90% CI | Inter-subject CV (%) | |
| AUC∞, ng · h/mL | 3,475.5a | 1,745.5 | 199.1 (152.81–259.44) | 27 |
| Cmax, ng/mL | 292.42 | 178.17 | 164.1 (124.51–216.33) | 29 |
| Assessing the effects of concomitant erythromycin on rivaroxaban PK (comparisons within renal function groups) | ||||
|---|---|---|---|---|
| Test | Reference | |||
| Mild RI | Mild RI | |||
| Rivaroxaban 10 mg + erythromycin 500 mg tid | Rivaroxaban 10 mg | Geometric mean ratio (%) with 90% CI | Intra-subject CV (%) | |
| AUC∞, ng · h/mL | 3,078.7 | 2,001.11 | 153.9 (137.27–172.43) | 14 |
| Cmax, ng/mL | 277.2 | 219.27 | 126.4 (110.59–144.51) | 16 |
| Moderate RI Rivaroxaban 10 mg + erythromycin 500 mg tid | Moderate RI Rivaroxaban 10 mg | Geometric mean ratio (%) with 90% CI | Intra-subject CV (%) | |
| AUC∞, ng · h/mL | 3,475.5a | 2,035.57 | 170.7 (151.14–192.88) | 14 |
| Cmax, ng/mL | 292.42 | 242.26 | 120.7 (105.59–137.98) | 16 |
AUC∞, area under the plasma concentration–time curve from time 0 to infinity; CI, confidence interval; Cmax, maximum plasma concentration; CV, coefficient of variation; h, hour; PK, pharmacokinetics; RF, renal function; RI, renal impairment; tid, three times daily.
n = 7.
As previously mentioned, both rivaroxaban CL/F and CLR values decreased with decreasing renal function, and although CL/F values were further reduced with the co-administration of erythromycin in these renally impaired subjects, a consistent trend was not observed for CLR values. Ae values also decreased with decreasing renal function, and this same decreasing trend was maintained with the addition of steady-state erythromycin, although with ultimately higher total amounts of unchanged drug recovered (Table 2).
In general, a similar trend was observed for the PD parameters. Subjects with mild renal impairment receiving the concomitant administration of rivaroxaban 10 mg with steady-state erythromycin exhibited increases in Factor Xa inhibition AUC0–48h and Emax values of approximately 19% and increases in PT AUC0–48h and Emax values of approximately 75% and 20%, respectively, when compared with subjects with normal renal function receiving rivaroxaban 10 mg alone (Table 4 and Figures S2c and S3c of Supporting information).
Table 4.
Ratio of Geometric Mean AUC0–48h and Emax Data and Associated 90% Confidence Interval After the Administration of Rivaroxaban 10 mg with and without Erythromycin 500 mg tid in Participants with Normal and Impaired Renal Function (n = 8 per Test and Reference Groups)
| Assessing the effects of renal function and concomitant erythromycin on rivaroxaban PD (comparisons across renal function groups) | ||||
|---|---|---|---|---|
| Factor Xa inhibition | ||||
| Test | Reference | |||
| Mild RI | ||||
| Rivaroxaban 10 mg + erythromycin 500 mg tid | Normal RF Rivaroxaban 10 mg | Geometric mean ratio (%) with 90% CI | Inter-subject CV (%) | |
| AUC0–48h | 708.5a | 593.43 | 119.4 (75.39–189.07) | 27 |
| Emax | 46.74a | 39.38 | 118.7 (97.71–144.16) | 15 |
| Moderate RI | ||||
| Rivaroxaban 10 mg + erythromycin 500 mg tid | Normal RF Rivaroxaban 10 mg | Geometric mean ratio (%) with 90% CI | Inter-subject CV (%) | |
| AUC0–48h | 842.3 | 593.43 | 141.9 (97.52–206.59) | 27 |
| Emax | 53.61 | 39.38 | 136.1 (116.13–159.54) | 15 |
| Prothrombin Time | ||||
|---|---|---|---|---|
| Test | Reference | |||
| Mild RI | ||||
| Rivaroxaban 10 mg + erythromycin 500 mg tid | Normal RF Rivaroxaban 10 mg | Geometric mean ratio (%) with 90% CI | Inter-subject CV (%) | |
| AUC0–48h | 118.7 | 67.82 | 175 (115.32–265.42) | 31 |
| Emax | 9.85 | 8.24 | 119.5 (79.73–179.18) | 41 |
| Moderate RI | ||||
| Rivaroxaban 10 mg + erythromycin 500 mg tid | Normal RF Rivaroxaban 10 mg | Geometric mean ratio (%) with 90% CI | Inter-subject CV (%) | |
| AUC0–48h | 115.1 | 67.82 | 169.8 (111.9–257.55) | 31 |
| Emax | 10.02 | 8.24 | 121.6 (81.11–182.28) | 41 |
AUC0–48h, area under the pharmacodynamic–time curve from time 0 to 48 hours; CI, confidence interval; CV, coefficient of variation; Emax, maximum pharmacodynamic change; PD, pharmacodynamics; RF, renal function; RI, renal impairment; tid, three times daily.
n = 4.
For subjects with moderate renal impairment receiving the same regimen, Factor Xa AUC0–48h and Emax values increased by approximately 42% and 36%, respectively, and PT AUC0–48h and Emax values increased by approximately 70% and 22%, respectively, when compared with subjects with normal renal function receiving rivaroxaban 10 mg alone (Table 4 and Figures S2c and S3c of Supporting information). Similar trends were also observed for aPTT and HepTest® (data not shown).
Effect of Renal Function and the Co-administration of Erythromycin on Rivaroxaban PK and PD (Comparisons within Renal Function Groups)
When assessing the combined effects of renal function and concomitant erythromycin use on rivaroxaban exposure within the same renal function group, subjects with mild renal impairment receiving the concomitant administration of rivaroxaban 10 mg with steady-state erythromycin had increases in AUC∞ and Cmax values by approximately 54% and 26%, respectively, compared with when the same subjects received rivaroxaban 10 mg alone. For subjects with moderate renal impairment receiving the same regimen, AUC∞ and Cmax values increased by approximately 71% and 21%, respectively, compared with when the same subjects received rivaroxaban 10 mg alone (Table 3 and Figure 2d). In both aforementioned scenarios, similar increases in the dose-normalized PK parameters were also observed with the administration of a single 5 mg dose of rivaroxaban with steady-state erythromycin, although with slightly higher values (Table S4 of Supporting information).
When assessing the changes in PD parameters, concomitant administration of rivaroxaban 10 mg with steady-state erythromycin in subjects with mild renal impairment resulted in very slight increases in Factor Xa AUC0–48h and Emax values by approximately 3% and 1%, respectively, and the AUC0–48h and Emax values for PT increased by approximately 19% and 14%, respectively, compared with when the same subjects received rivaroxaban 10 mg alone. For subjects with moderate renal impairment, the co-administration of rivaroxaban 10 mg with steady-state erythromycin resulted in increases in Factor Xa inhibition AUC0–48h and Emax values by approximately 38% and 17%, respectively, and increases in PT AUC0–48h and Emax values by approximately 79% and 20%, respectively, compared with when the same subjects received rivaroxaban 10 mg alone (Table S5 and Figures S2d and S3d of Supporting information). Similar trends were also observed for aPTT and HepTest® (data not shown).
Safety and Tolerability
The safety analysis included all randomized subjects who had received at least one dose of study drug. No clinically significant safety issues were noted during the study and no new safety signals were identified. The overall incidence of TEAEs was higher in the mild and moderate renal impairment groups compared with the normal renal function group. In total, 21 out of the 29 subjects enrolled (72.4%) reported one or more TEAEs; the most common of which (across all renal groups) were abdominal pain, diarrhea, and dizziness, which occurred mainly with erythromycin administration. Nausea and headache were other TEAEs commonly reported in the mild and moderate renal impairment groups that also occurred mainly with the administration of erythromycin.
There were no serious AEs reported during the study. Two subjects in the mild renal impairment group were discontinued from the study owing to AEs of nausea and vomiting that occurred during erythromycin monotherapy and were subsequently replaced. Four subjects (1 NRF; 3 moderate RI) reported bleeding events, which included AEs of hematoma, hemoptysis, skin lesion, vessel puncture site hemorrhage, and gingival bleeding, all of which were considered mild in intensity and none of which required any action to be taken. The hematoma and hemoptysis events in question occurred in two separate patients 4 days and 1 day, respectively, after receiving rivaroxaban in treatment period 1. The skin lesion occurred in one patient 18 days after receiving the last dose of erythromycin on Day 6 of treatment period 2. For the patient who experienced vessel puncture site hemorrhage (2 events) and gingival bleeding (1 event), the first vessel puncture site hemorrhage occurred 1 day before rivaroxaban administration in treatment period 1, while the second occurred nearly 3 hours after a rivaroxaban dose on Day 2 of treatment period 2. The gingival bleeding event occurred around 5 hours after administration of erythromycin 500 mg tid on Day 6 of treatment period 2. This same patient also experienced an infusion site extravasation 7.5 hours after erythromycin 500 mg tid on Day 4 of treatment period 3. All of the above-mentioned events were considered by the investigators as not related to rivaroxaban and erythromycin, except for skin lesion (doubtfully related to rivaroxaban and erythromycin) and gingival bleeding (probably related to rivaroxaban). No subjects reported persistent AEs at the end of the study or at the time of the last follow-up visit. Lastly, there were no clinically meaningful changes in clinical laboratory parameters, body weight, body mass index, vital signs, physical examinations, or 12-lead ECGs.
Discussion
The clinical pharmacology of rivaroxaban has been extensively studied. The drug has multiple elimination pathways, with approximately 36% of the drug excreted unchanged by the kidneys, mainly by active renal secretion involving both the transporter proteins P-gp and ABCG2 (also abbreviated to Bcrp) and also by glomerular filtration. The remaining drug is metabolized by the liver, of which approximately 18% of the dose is metabolized via CYP3A4/3A5, 14% via CYP2J2, and 14% via CYP-independent hydrolysis.17,18 Considering the percentage of the administered dose renally eliminated as unchanged drug and also the percentage of drug metabolized via these pathways, a renal impairment study16 and several drug–drug interaction studies10 have been previously conducted.
Two of these previous studies, in which the effects of renal impairment16 and concomitant erythromycin use10 were assessed, showed that separately, subjects with mild or moderate renal impairment and subjects receiving the concomitant administration of erythromycin displayed increases in rivaroxaban exposure. However, these previous studies assessed the effects of renal impairment and inhibition of hepatic metabolism separately and did not account for a potential combined drug–drug and disease interaction that could occur. Such an interaction was previously explored by the FDA through the use of PBPK modeling, which simulated the potential effects in such a clinical scenario (e.g. the co-administration of rivaroxaban and erythromycin therapy in patients with renal impairment), which predicted a synergistic increase in rivaroxaban exposure11 (i.e. increases that are far greater than the sum of the increases observed with each factor alone). Therefore, for these reasons, an evaluation of this treatment scenario in a human clinical trial was conducted.
This study evaluated rivaroxaban PK and PD parameters across three renal function groups (normal renal function, and both mild and moderate renal impairment), with two doses of rivaroxaban (5 mg [subjects with mild/moderate renal impairment] and 10 mg [all renal function groups]) with and without the concomitant use of steady-state erythromycin. Accordingly, the following comparisons could be made: the effect of mild and moderate renal impairment on the PK and PD of rivaroxaban relative to subjects with normal renal function; the effect of erythromycin on the PK and PD of a 10 mg dose of rivaroxaban in patients with normal renal function; and the combined effects of erythromycin on the PK and PD of rivaroxaban (at 5 and 10 mg doses) in patients with mild or moderate renal impairment.
The first of these comparisons, the effect of renal impairment on the PK and PD of rivaroxaban, was originally demonstrated by Kubitza et al,16 in which the renal clearance of rivaroxaban was shown to decrease with decreasing renal function, thereby leading to an increase in rivaroxaban AUC∞ values of approximately 44% and 52% for subjects with mild and moderate renal impairment, respectively. Notably, the increases in rivaroxaban exposure in patients with renal impairment in this present study were lower than those observed in the previous study, with increase in AUC∞ values of approximately 15% and 17% for subjects with mild and moderate renal impairment, respectively. A potential reason for these differences in exposure may be that this current study enrolled a control group of healthy subjects with normal renal function that were older and in turn had a lower mean total clearance (CL/F) value, which was not substantially different from the value calculated for the subjects with renal impairment. The mean age of the control group in this study was 61 years, which was approximately 10 years older than the mean age of those enrolled in the previous renal impairment study. This outcome suggests that age may have an independent role from that of just renal impairment on drug exposure, perhaps via age-related alterations in the hepatic metabolism of rivaroxaban.
The second of these comparisons, the effect of concomitant erythromycin use on the PK and PD of rivaroxaban, was previously reported by Mueck et al.10 In this previous Phase I drug–drug interaction study, the administration of steady-state erythromycin led to increases in rivaroxaban AUC∞ and Cmax values by approximately 34%. When assessing the effects of co-administered erythromycin with rivaroxaban in this present study, similar increases in AUC∞ and Cmax of approximately 40% were observed.19,20
Lastly, when assessing the combined effects of renal impairment and the concomitant administration of erythromycin on rivaroxaban exposures in this study, an expected drug–drug and disease effect was observed. Compared with subjects with normal renal function who received 10 mg of rivaroxaban alone, those with mild renal impairment given rivaroxaban 10 mg with steady-state erythromycin exhibited mean increases in AUC∞ and Cmax of approximately 76% and 56%, respectively, and those with moderate renal impairment given rivaroxaban 10 mg with steady-state erythromycin exhibited mean increases in AUC∞ and Cmax of approximately 99% and 64%, respectively. Despite these changes in exposure being slightly more than additive, they were not to the degree previously predicted through the use of PBPK modeling.11 Grillo et al have identified some limitations of the PBPK model within their publication that may help to account for some of the differences between the exposure increases obtained through modeling and simulation and those observed in the present study.11 Notably, because active renal clearance did not seem to change with the addition of erythromycin in this study, the effects of erythromycin as an inhibitor of the P-gp pathway could not be confirmed. It would be informative to update the PBPK model with results from the present study.
The administration of a rivaroxaban 5 mg dose in the above scenarios provided similar increases in dose-normalized AUC∞ and Cmax parameters to that of the 10 mg dose. The 5 mg AUC and Cmax parameters were dose-normalized, because previous Phase I studies indicated rivaroxaban to be dose-proportional (based on AUC) through the 2.5–10 mg dose range under fasting conditions and through the 10–20 mg dose range under fed conditions. The slightly higher dose-normalized AUC∞ and Cmax values for the 5 mg dose relative to 10 mg observed in this study may reflect some slight solubility-limited absorption of the 10 mg dose.
The PD changes observed when administering a 10 mg dose of rivaroxaban in the aforementioned scenarios followed a similar trend to those observed with the PK parameters, but were generally smaller in magnitude. A discrepancy between the magnitude of increases in rivaroxaban exposures and inhibition of Factor Xa and PT has been observed on occasion in other studies in the rivaroxaban Phase I program (data on file). The small sample size included in the present study may have contributed to the inconsistencies reported in the magnitude of changes in the PK and PD parameters. Due to the crossover design of the study and the length of time it took to enroll and treat the subjects, not all PD samples were analyzed from the same subject in the same bioanalytical run, and this may have contributed to some of the variability observed. Additionally, the inter-assay variability for Factor Xa and PT is greater than the intra-day variability. These factors are potential limitations of this study.
Unlike the PK parameters, in which previous clinical pharmacology studies supported the use of dose-normalized comparisons, not all of the PD parameters assessed in this study have a linear response with increasing rivaroxaban exposure. Consequently, a decision was made not to dose-normalize the PD parameters for the rivaroxaban 5 mg dose because a true comparison between renal function groups could not be made.
Conclusion
The combination of renal impairment and the use of the P-gp and moderate CYP3A4/3A5 inhibitor erythromycin resulted in increases in rivaroxaban exposure that were slightly more than additive, although not to the extent of those previously predicted through published PBPK modeling. In general, similar results should be expected with other P-gp and moderate CYP3A4/3A5 inhibitors. Although data from the Phase III ROCKET AF trial, where the concomitant use of rivaroxaban with combined P-gp and weak or moderate CYP3A4 inhibitors was permitted, did not show an increase in bleeding in patients with a CLCR 30–49 mL/min,9,21 patients with advanced renal dysfunction are susceptible to an increased risk of bleeding due to their underlying disease.22–24 In light of these considerations, the US prescribing information for rivaroxaban states that rivaroxaban “should not be used in patients with creatinine clearance 15–80 mL/min who are receiving concomitant combined P-gp and moderate CYP3A4 inhibitors unless the potential benefit justifies the potential risk”.9 The European prescribing information states that “in patients with moderate renal impairment (creatinine clearance 30–49 mL/min) concomitantly receiving other medicinal products which increase rivaroxaban plasma concentrations, Xarelto [rivaroxaban] is to be used with caution”.2
To conclude, while this was a small clinical pharmacology study, a single dose of rivaroxaban, either 5 mg or 10 mg, appeared well tolerated, regardless of renal function and erythromycin co-administration, with no new safety signals being identified.
Acknowledgments
The authors acknowledge the assistance of Dagmar Kubitza, MD and Wolfgang Mueck, PhD, Bayer Pharma AG, for their support of the protocol design; Stephan Schwers, PhD; Alexius Freyberger, PhD, and Gabriele Rohde, PhD, Bayer Pharma AG, for the bioanalysis of the pharmacodynamic and pharmacokinetic samples; and Kevin Shalayda, Janssen Research & Development, LLC, for his support in the pharmacokinetic parameter analysis.
The authors would like to acknowledge Claudia Wiedemann, who provided medical writing services with funding from Bayer HealthCare Pharmaceuticals and Janssen Scientific Affairs, LLC.
Declaration of Conflicting Interests
This trial was sponsored by Janssen Research & Development, LLC. All authors are under the employment of Janssen Research & Development, LLC.
Funding
This trial was sponsored by Janssen Research & Development, LLC.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
Supporting Information.
References
- 1.Perzborn E, Roehrig S, Straub A, Kubitza D, Misselwitz F. The discovery and development of rivaroxaban, an oral, direct Factor Xa inhibitor. Nat Rev Drug Discov. 2011;10:61–75. doi: 10.1038/nrd3185. [DOI] [PubMed] [Google Scholar]
- 2.Bayer Pharma AG. Xarelto® (rivaroxaban) Summary of Product Characteristics. 2013. Available at: http://www.xarelto.com/en/information-on-xarelto/summary-of-product-characteristics/ Accessed May 1, 2014.
- 3.Bauer KA, Homering M, Berkowitz SD. Effects of age, weight, gender and renal function in a pooled analysis of four phase III studies of rivaroxaban for prevention of venous thromboembolism after major orthopedic surgery. Blood (ASH Annual Meeting Abstracts) 2008;112:166–167. Abstract 436. [Google Scholar]
- 4.Eriksson BI, Kakkar AK, Turpie AGG, et al. Oral rivaroxaban for the prevention of symptomatic venous thromboembolism after elective hip and knee replacement. J Bone Joint Surg Br. 2009;91:636–644. doi: 10.1302/0301-620X.91B5.21691. [DOI] [PubMed] [Google Scholar]
- 5.Eriksson BI, Borris LC, Friedman RJ, et al. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med. 2008;358:2765–2775. doi: 10.1056/NEJMoa0800374. [DOI] [PubMed] [Google Scholar]
- 6.Kakkar AK, Brenner B, Dahl OE, et al. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet. 2008;372:31–39. doi: 10.1016/S0140-6736(08)60880-6. [DOI] [PubMed] [Google Scholar]
- 7.Lassen MR, Ageno W, Borris LC, et al. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med. 2008;358:2776–2786. doi: 10.1056/NEJMoa076016. [DOI] [PubMed] [Google Scholar]
- 8.Turpie AGG, Lassen MR, Davidson BL, et al. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty (RECORD4): a randomised trial. Lancet. 2009;373:1673–1680. doi: 10.1016/S0140-6736(09)60734-0. [DOI] [PubMed] [Google Scholar]
- 9.Janssen Pharmaceuticals Inc. Xarelto® (rivaroxaban) Prescribing Information. 2014. Available at: http://www.xareltohcp.com/sites/default/files/pdf/xarelto_0.pdf Accessed May 1, 2014.
- 10.Mueck W, Kubitza D, Becka M. Co-administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br J Clin Pharmacol. 2013;76:455–466. doi: 10.1111/bcp.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grillo JA, Zhao P, Bullock J, et al. Utility of a physiologically-based pharmacokinetic (PBPK) modeling approach to quantitatively predict a complex drug-drug-disease interaction scenario with rivaroxaban during the drug review process: implications for clinical practice. Biopharm Drug Dispos. 2012;33:99–110. doi: 10.1002/bdd.1771. [DOI] [PubMed] [Google Scholar]
- 12.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 13.Food and Drug Administration Guidance for industry: Pharmacokinetics in patients with impaired renal function – study design, data analysis, and impact on dosing and labeling. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM204959.pdf Accessed May 1, 2014.
- 14.Rohde G. Determination of rivaroxaban – a novel, oral, direct Factor Xa inhibitor – in human plasma by high-performance liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;872:43–50. doi: 10.1016/j.jchromb.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 15.US Department of Health and Human Services. Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM). Guidance for Industry. Bioanalytical Method Validation. 2001. http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf Accessed May 1, 2014.
- 16.Kubitza D, Becka M, Mueck W, et al. Effects of renal impairment on the pharmacokinetics, pharmacodynamics and safety of rivaroxaban, an oral, direct Factor Xa inhibitor. Br J Clin Pharmacol. 2010;70:703–712. doi: 10.1111/j.1365-2125.2010.03753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cardiovascular and Renal Drugs Advisory Committee FDA Advisory Committee Briefing Document. 2014. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/UCM181524.pdf. Accessed May 1,
- 18.European Medicines Agency. CHMP assessment report for Xarelto. 2008. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000944/WC500057122.pdf Accessed May 1, 2014.
- 19.Mueck W, Eriksson BI, Bauer KA, et al. Population pharmacokinetics and pharmacodynamics of rivaroxaban – an oral, direct Factor Xa inhibitor – in patients undergoing major orthopaedic surgery. Clin Pharmacokinet. 2008;47:203–216. doi: 10.2165/00003088-200847030-00006. [DOI] [PubMed] [Google Scholar]
- 20.Mueck W, Lensing AWA, Agnelli G, Décousus H, Prandoni P, Misselwitz F. Rivaroxaban: population pharmacokinetic analyses in patients treated for acute deep-vein thrombosis and exposure simulations in patients with atrial fibrillation treated for stroke prevention. Clin Pharmacokinet. 2011;50:675–686. doi: 10.2165/11595320-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 21.Fox KAA, Piccini JP, Wojdyla D, et al. Prevention of stroke and systemic embolism with rivaroxaban compared with warfarin in patients with non-valvular atrial fibrillation and moderate renal impairment. Eur Heart J. 2011;32:2387–2394. doi: 10.1093/eurheartj/ehr342. [DOI] [PubMed] [Google Scholar]
- 22.Boccardo P, Remuzzi G, Galbusera M. Platelet dysfunction in renal failure. Semin Thromb Hemost. 2004;30:579–589. doi: 10.1055/s-2004-835678. [DOI] [PubMed] [Google Scholar]
- 23.Falga C, Capdevila JA, Soler S, et al. Clinical outcome of patients with venous thromboembolism and renal insufficiency. Findings from the RIETE registry. Thromb Haemost. 2007;98:771–776. [PubMed] [Google Scholar]
- 24.Holden RM, Harman GJ, Wang M, Holland D, Day AG. Major bleeding in hemodialysis patients. Clin J Am Soc Nephrol. 2008;3:105–110. doi: 10.2215/CJN.01810407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.