

Abstract
Background and Purpose
Ischaemia compromises mitochondrial respiration. Consequently, the mitochondrial F1Fo-ATPsynthase reverses and acts as a proton-pumping ATPase, so maintaining the mitochondrial membrane potential (ΔΨm), while accelerating ATP depletion and cell death. Here we have looked for a molecule that can selectively inhibit this activity without affecting ATP synthesis, preserve ATP and delay ischaemic cell death.
Experimental Approach
We developed a chemoinformatic screen based on the structure of BMS199264, which is reported to selectively inhibit F1Fo-ATPase activity and which is cardioprotective. Results suggested the molecule BTB06584 (hereafter referred to as BTB). Fluorescence microscopy was used to study its effects on ΔΨm and on the rate of ATP consumption following inhibition of respiration in several cell types. The effect of BTB on oxygen (O2) consumption was explored and protective potential determined using ischaemia/reperfusion assays. We also investigated a potential mechanism of action through its interaction with inhibitor protein of F1 subunit (IF1), the endogenous inhibitor of the F1Fo-ATPase.
Key Results
BTB inhibited F1Fo-ATPase activity with no effect on ΔΨm or O2 consumption. ATP consumption was decreased following inhibition of respiration, and ischaemic cell death was reduced. BTB efficiency was increased by IF1 overexpression and reduced by silencing the protein. In addition, BTB rescued defective haemoglobin synthesis in zebrafish pinotage (pnt) mutants in which expression of the Atpif1a gene is lost.
Conclusions and Implications
BTB may represent a valuable tool to selectively inhibit mitochondrial F1Fo-ATPase activity without compromising ATP synthesis and to limit ischaemia-induced injury caused by reversal of the mitochondrial F1Fo-ATPsynthase.
Introduction
In eukaryotic cells, ATP is mainly produced through oxidative phosphorylation, which is dependent on the activity of the mitochondrial F1Fo-ATPsynthase. When the oxygen supply is compromised, for example, during ischaemia, the F1Fo-ATPsynthase runs in reverse, acting as an ATPase, hydrolysing ATP and maintaining the proton motive force and thus the mitochondrial membrane potential (ΔΨm) at the expense of the cellular supplies of ATP (Rouslin et al., 1990; Vinogradov, 2000). This ATP consumption, referred to as ATP wastage, makes a significant contribution to cell death (Harris and Das, 1991; Jennings et al., 1991).
The inhibitor protein of F1 subunit (IF1) is a small nuclear-encoded endogenous inhibitor of the F1 domain of the mitochondrial F1Fo-ATPsynthase. First discovered in bovine heart, it is ubiquitously expressed in the body albeit at varying levels, according to the species and the tissue considered (Rouslin, 1991; Green and Grover, 2000). In rare cases, its absence is responsible for a mitochondrial myopathy (Luft et al., 1962). This highly sequence-conserved protein is highly specific for the ATP hydrolase activity, acting as a reversible non-competitive inhibitor. Inhibition of the F1Fo-ATPase requires an acid pH (∼6.7), which promotes binding of the endogenous inhibitor to the enzyme, a condition encountered during ischaemia (Power et al., 1983; Rouslin et al., 1986; Cabezon et al., 2000). IF1 binding disrupts the connection between the central γ- and β-subunits, inhibiting ATP hydrolase activity of the F1 complex (Cabezon et al., 2001). Thus, IF1 may play a significantly protective role in the pathophysiology of ischaemia, as ATP wastage by the F1Fo-ATPase accounts for a large fraction of ATP consumption, in the rodent cardiomyocyte.
The binding of IF1 to the F1 subunit is capable of reducing this wastage by up to 80% (Rouslin et al., 1995). Indeed, overexpression of IF1 is significantly protective in a hypoxia re-oxygenation cell model (Campanella et al., 2008). A molecule that mimics or potentiates the action of IF1 could significantly improve the outcome in pathologies associated with impaired oxygen supply. Grover et al. (2004) developed such a compound, named BMS199264 (Atwal et al., 2004), which reduced infarct size in the isolated rat heart. However, as no further original publications have confirmed its positive effects, the question remains open: is it possible to find a molecule that can selectively inhibit mitochondrial ATPase activity without also compromising ATP synthesis? Could such a drug be clinically useful? And how could we explain its specificity towards the hydrolase activity of the F1Fo-ATPsynthase?
Following a chemoinformatic screen based on the molecular conformation of the BMS compound, we selected the molecule BTB06584 (hereafter referred as BTB), which we have tested for its biological efficacy in vitro and in vivo, characterizing a potential chemical lead to preserve cell integrity during acute pathological conditions (ischaemia) associated with reversal of the F1Fo-ATPsynthase and bio-energetic defects (haemoglobin synthesis) associated with reduction in endogenous IF1.
Methods
Virtual screening of compounds
We performed the screening for BTB according to the protocol published by Professor Grant Churchill and his collaborators (Naylor et al., 2009; Rosen et al., 2009). In detail, using ZINC, a free database of commercially available compounds for virtual screening, provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (Irwin and Shoichet, 2005), we ran a rapid overlay of chemical structures analysis based on shape and colour similarity with BMS scoring molecules between a range value of 0.1 and 1.0 (with the latter bearing greater similarity) and eliminated those characterized by CH3 groups making these biologically inert. The most robust hit was ZINC01044546, also known as Oprea1_170890, AC1ME324, MolPort-002–891-783, BTB06584, CCG-42771, 2-nitro-5-(phenylsulfonyl)phenyl 4-chlorobenzoate, SR-01000632744-1. Molecular formula: C19H12ClNO6S, molecular weight: 417.81968, InChIKey: WNDWKKPBLAKXMI-UHFFFAOYSA-N.
Following this, the program http://autodock.scripps.edu, a suite of automated docking tools, was used to predict how and where the chosen compound would bind to the F1Fo-ATPsynthase and then generate a three-dimensional structure of the complex.
Cell cultures
HL-1 cells, a cardiac cell line derived from a mouse atrial cardiomyocyte tumour lineage, were cultured according to the instructions from Claycomb et al. (1998). HeLa cells, derived from a human cervix cancer lesion, were grown in standard DMEM supplemented with FBS (10%), penicillin (100 U·mL−1), streptomycin (100 μg·mL−1) and L-glutamine (4 nM). The cells were cultured in DMEM-F12 (Gibco, Life Technologies, Paisley, UK) (10% inactivated FBS and penicillin/streptomycin) at 37°C, 5% CO2 and 95% air in an incubator for cell culture. FuGENE® 6 transfection reagents (Roche, Penzberg, Upper Bavaria, Germany) were used for the transfection experiments in HL-1 cells. Cells were co-transfected with either cyan fluorescent protein (CFP) or yellow fluorescent protein (YFP) in addition to IF1 construct (a gift from Professor Sir John Walker) or IF1 siRNA (Qiagen, Manchester, UK) to overexpress or silence IF1 respectively.
Fluorescent imaging plate reader (FLIPR) experiments
To assess the appropriate concentration of BTB required to inhibit the F1Fo-ATPase, cells were first exposed to rotenone (5 μM), which inhibits complex I of the electron transport chain (ETC), resulting in a reversal of the activity of the F1Fo-ATP synthase. Subsequently, they were challenged with BTB at several concentrations from 1 to 100 μM – effective inhibition of the ATPase will then cause a loss of ΔΨm. The potential was measured using rhodamine 123 (Rh123), a lipophilic cationic fluorescent dye used in ‘quench/dequench’ mode (loaded at 20 μM, for 10 min followed by a wash). At this concentration, when the ΔΨm falls, the dye, initially located in the mitochondria, redistributes to the cytosol and its mean fluorescence increases (Duchen et al., 2003).
Fluorescence imaging
All imaging experiments were performed with either a Zeiss 510 CLSM (Carl Zeiss Microscopy GmbH, Jena, Germany) or Leica SP5 confocal microscopes (Leica Microsystems, Wetzlar, Germany), using appropriate lasers and emission filters. ΔΨm was assessed using Rh123 or tetramethylrhodamine methyl ester (TMRM). HL-1 cells loaded with Rh123 were imaged intermittently during 18 min using a 488 nm laser with the emission optimally monitored at 530 nm, and the changes in fluorescence were recorded after addition of inhibitors of the ETC (rotenone 5 μM and antimycin A 1 mg·mL−1 for HL-1) followed by addition of either BTB or oligomycin B (3 μM) as a positive control. We found that the best and most sensitive way to explore the changes in ΔΨm using Rh123 in quench/dequench mode (10 μg·mL−1) was to calculate the SD of the mean fluorescence ratio. The SD of the signal directly reflects the redistribution of the dye: it is high when the mitochondria are polarized as the dye is located preferentially in the mitochondria. Following mitochondrial depolarization, the fluorescence in the cytosol increases, the fluorescence signal becomes more uniform and thus the SD goes down. It is necessary to correct changes in SD in relation to changes in the mean fluorescence, as the SD will vary with the mean signal. Thus, the SD/mean ratio provides the best sensitivity possible to detect significant changes in ΔΨm.
Changes in cellular ATP content were measured using the fluorescent dye, magnesium green (MgG-AM, 5 μM) (Leyssens et al., 1996). MgG fluorescence is a measure of concentration of free [Mg2+], which has a strong affinity with ATP, whereas its affinity for ADP is 10-fold less. Therefore, ATP hydrolysis is associated with a rise in free Mg2+. All cells were imaged in a ‘normal recording saline’. For the experiments with MgG, glucose was replaced with 2-deoxyglucose to ensure inhibition of glycolytic production of ATP.
In ischaemia/reperfusion experiments, the cell death ratio was calculated after loading the cells with Hoechst (1 μg·mL−1) and propidium iodide (PI, 15 μg·mL−1) and corresponded to the PI-positive/Hoechst-positive ratio.
The mitochondrial network of HeLa cells was monitored via MitoTracker® Green FM (Invitrogen, Life Technologies, Paisley, UK). Briefly, cells were stained with 100 nM MitoTracker Green FM for 45 min at 37°C, 5% CO2. After staining, cells were washed twice with normal recording medium, then either directly imaged with a Leica SP5 confocal microscope or incubated with 100 μM BTB for 2 h at 37°C, 5% CO2 before imaging.
Ischaemia/reperfusion
Ischaemia/reperfusion experiments in HL-1 cells were performed using an airtight gas chamber (Wolf Laboratories, Pocklington, UK) filled with 95% N2/5% CO2 after replacing the Claycomb medium (Sigma Aldrich, St. Louis, MO, USA) with an ischaemic medium (in mM: NaCl 125, KCl 8, 2-deoxyglucose 20, Na lactate 0.5, MgSO4 1.25, CaCl2 1.25, KH2PO4 1.2, NaHCO3 6.25, HEPES 20; pH 7.4). The chamber was stored for 7 h in an incubator at 37°C. Reperfusion consisted of replacing the ischaemic medium with an oxygenated reperfusion medium (as described above but modified to: NaHCO3 25mM, CaCl2 1mM, HEPES 10mM; pH 7.4) and storing the culture plates for 1 h in a normoxic incubator. Five experimental groups were considered: sham, control (ischaemia/reperfusion without any intervention), cyclosporine A (CsA) 200 nM a potent inhibitor of the mitochondrial permeability transition pore (mPTP), diazoxide 10 μM, a mitochondrial K-ATP channel opener, and BTB 100 μM.
Cortical neuronal preparation, combined oxygen and glucose deprivation (OGD) plus reoxygenation (RX) and intravital staining
Procedures involving animals and their care were conducted in strict accordance with the Policy on Ethics approved by the Society for Neuroscience and with the European Communities Council Directive for Experimental Procedures. Every effort was made to minimize the number of animals used and any possible suffering caused to them. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 3 mice and 75 zebrafish embryos were used in the experiments described here.
Cortical neurons were isolated from 15-day-old C57BL/6N mouse embryos according to Dugan et al. (1995) and cultured as described in Carunchio et al. (2007). Cortical neurons were exposed to OGD for 3 h followed by 24 h RX (Goldberg and Choi, 1993). Briefly, the culture medium was replaced with a hypoxia medium previously saturated for 20 min with 95% N2 and 5% CO2 and containing NaCl 116 mM, KCl 5.4 mM, MgSO4 0.8 mM, NaHCO3 26.2 mM, NaH2PO4 1 mM, CaCl2 1.8 mM, glycine 0.01 mM and 0.001 w/v phenol red. Hypoxic conditions were maintained using a hypoxia chamber (temperature 37°C, atmosphere 95% N2 and 5% CO2). These experimental conditions induced 30% decrease of PO2 in the medium. Deprivation of oxygen and glucose was stopped by placing the cells in the usual culture medium, saturated with a mixture of 95% O2 and 5% CO2 for 10 min. Reoxygenation was achieved by returning neurons to normoxic conditions (37°C in a humidified 5% CO2 atmosphere) for 24 h (Savoia et al., 2011). BTB 100 μM was added during the 3 h of OGD.
After the experimental procedure, cortical neurons were washed with the standard medium and stained for 3 min at 22°C with a solution containing 36 μM fluorescein diacetate (FDA) (Sigma Aldrich) and 7 μM PI (Calbiochem, San Diego, CA, USA). The stained cells were examined immediately with a confocal laser-scanning microscope (Leica SP5). FDA, a non-polar ester, crosses the cell membrane and is hydrolysed by intracellular esterases to produce a green-yellow fluorescence. Cell injury curtails FDA staining and allows for cell permeation with PI, a polar compound that, by interacting with nuclear DNA, yields a bright red fluorescence (Manev et al., 1990).
Immunofluorescence assay
Successful overexpression or silencing of IF1 was demonstrated by immunofluorescence experiments. We used a rabbit polyclonal anti-mouse ATPIF1 antibody (ProteinTech Europe, Manchester, UK) (1/100) in conjunction with an anti-rabbit Cy3 antibody (1/50). Cells were fixed with paraformaldehyde 4% and stored at 4°C before imaging.
Gel electrophoresis and immunoblot analyses
Sample proteins were quantified using a BCA protein assay kit (Thermo Scientific, Loughborough, UK). Equal amounts of protein (20 μg) were resolved in kD TGX™ (Bio-Rad Laboratories, Hemel Hempstead, UK), 15% polyacrylamide gels and transferred to nitrocellulose membrane. The membrane was blocked in 2% non-fat dry milk in TBST [50 mM Tris, 150 mM NaCl, 0.05% Tween 20 (Sigma Aldrich), pH 7.5] for 1 h then incubated with the appropriate diluted primary antibody at 4°C overnight: mouse α-ATPIF1 (clone 5E2D7) (Abcam, Cambridge, UK) 1:1000 or α-GAPDH:Hrp conjugated (Abcam) 1:20 000. The membrane was washed in TBST (3 × 15 min at room temperature) and then incubated with corresponding peroxidase-conjugated secondary antibodies for 1 h at room temperature. After further washing in TBST, the blot was developed using an ECL Plus Western blotting detection kit (Amersham, GE Healthcare Life Sciences, Little Chalfont, UK). Immunoreactive bands were analysed by performing densitometry with ImageJ software (NIH, Bethesda, MD, USA).
Oxygen consumption experiments
HL-1 cells were trypsinized and suspended in normal recording medium at a concentration of 10 × 106·mL−1. Non-permeabilized cells (1 × 106·) were placed in a chamber in contact with a Clark electrode (Hansatech Instruments, Norfolk, UK), and oxygen tension was continuously monitored in basal conditions and after addition of either BTB, oligomycin B or carbonyl cyanide p-trifluoromethoxy-phenylhydrazone (FCCP) to determine a possible inhibiting effect on the ATP synthase or an inhibition of respiration that might limit the uncoupling induced maximal oxygen consumption.
Zebrafish experiments
Ethical approval for zebrafish experiments was obtained from the Royal Veterinary College and the Home Office (UK) under the Animal (Scientific Procedures) Act 1986. pnttq209 carrier zebrafish (Shah et al., 2012) were obtained from the Tübingen Stock Center. Zebrafish were housed in a multi-rack aquarium system at the Royal Veterinary College and kept on a constant 14/10 h light/dark cycle at 27–29°C, bred and staged as described (Westerfield, 2007). BTB (0.1 mM in DMSO) and DMSO alone as control, were diluted to a final concentration of 1 μM in aquarium water and added to embryos from a pnt heterozygous cross at 1 day post fertilization (dpf). This BTB concentration was chosen as it did not induce any toxicity on the animal, as was reported with greater concentrations. At 3 dpf, larvae were examined under a Nikon SMZ1500 microscope (Nikon, Kingston upon Thames, UK) and scored as having red or clear blood. The experiment was repeated four times. Images were taken using a Digital Sight DS-2 Mv camera (Nikon) and associated Digital Sight imaging software (Nikon).
Measuring ΔΨm and haemoglobin in zebrafish larvae
WT and pnt embryos were separated at 3 dpf and either treated with a vehicle (DMSO) or 1 μM BTB diluted in PBS for 1.5 h for TMRM and 3 h for o-dianisidine at 28°C.
For ΔΨm, larvae were simultaneously exposed to the cell-permeant, cationic red fluorescent dye TMRM (300 nM) that is sequestered by polarized mitochondria. After incubation, embryos were washed twice in PBS before mounting in 2% low melting point agarose gel in PBS onto a glass-bottomed culture dish. Z-stack images were taken using a 40X objective with a Leica SP5 confocal microscope. Microscope parameters including gain, offset, z-stack slice number and laser power were kept constant between experiments. The olfactory bulb of each embryo was selected for imaging as this region exhibited consistent TMRM loading, permitting comparison between conditions. Ten mitochondrial regions of interest were demarcated in the olfactory bulb per embryo, and the mean maximum fluorescence intensity was calculated from this.
For o-dianisidine staining after BTB treatment, larvae were washed in PBS then stained for 15 min in the dark in o-dianisidine (0.6 mg·mL−1) (Paffett-Lugassy and Zon, 2005), 0.01 M sodium acetate (pH 4.5), 0.65% H2O2 and 40% (v/v) ethanol. The stained larvae were washed once in PBS prior to fixing in 4% PFA overnight at 4°C. After fixation, larvae were washed in PBS again before placing in 70% glycerol/PBS solution where they were equilibrated for at least 1 h before imaging on a Nikon SMZ1500 microscope using a Digital Sight DS-2 Mv camera and associated Digital Sight imaging software.
Measurement of mitochondrial matrix pH
Mitochondrial matrix pH was assessed using the cell-permeant pH indicator probe 5-(and-6)-carboxy SNARF®-1 AM acetate (Molecular Probes®, Invitrogen) as reported previously in Shah et al. (2012). HeLa cells were seeded on Ø22 mm borosilicate coverslips and, following overnight growth at 37°C, 5% CO2, co-loaded with 10 μM carboxy SNARF-1 AM acetate and 200 nM MitoTracker Green FM in recording medium (30 min incubation at 37°C, 5% CO2). After 30 min incubation, the dye was removed and cells were incubated in recording medium for 2.5 h to allow the hydrolysis of cytosolic carboxy SNARF-1 AM acetate and the preferential compartmentalization of the probe into the mitochondria. Cells were imaged using a ‘Fluar’ 40×/1.30 oil immersion objective; to avoid contamination between carboxy SNARF-1 AM acetate and MitoTracker Green FM emissions, a sequence of three consecutive scans was performed throughout, using a green helium–neon laser (543 nm line) for the former and an argon laser (488 nm line) for the latter. Fluorescence emission of carboxy SNARF-1 AM acetate at 590 and 680 nm was captured in two consecutive scans through the HTF 488/543 dichroic mirror with BP 565/615 band pass filter and LP 650 high pass filter, while MitoTracker Green FM, whose staining was used to select only mitochondrial pixels and derive the mitochondrial calibration curve avoiding contaminations from cytosolic signal, through the HTF 488/543 dichroic mirror with BP 510/520 band pass filter. In situ pH calibration of carboxy SNARF-1 AM acetate was performed using control DMSO-treated cells; cells were exposed to high-K+ buffer supplemented with 13 mM nigericin, 1 μM FCCP and 20 μg·mL−1 oligomycin to achieve equilibration of the external and internal pH and of cytosolic and mitochondrial matrix pH through permeabilization of plasma membrane and mitochondrial membrane in conjunction with suppression of the F1Fo-ATPsynthase activity. The pH of the solution was set to four different values (6.0, 7.0, 8.0 and 9.0), and the calibration was performed both from low to high pH and vice versa. Averaged SNARF-1 AM acetate ratios (680/590 nm) were plotted against pH values to build up the calibration curve and measure mitochondrial matrix pHm (maintaining the same settings, chosen to minimizing bleaching). Basal pHm measurements of treatment were performed in recording medium, and emission intensities were quantified with the LSM Image Browser software (Carl Zeiss Microscopy GmbH).
Data analysis
Confocal images were processed using the FIJI software (Schindelin et al., 2012). Data are expressed as means ± SEM. Statistical analysis was performed using the GraphPad Prism 5 software (GraphPad Software, La Jolla, CA, USA). We used one-way anova with either Dunnett's or Newman–Keuls's multiple comparison test as a post-test where necessary. For zebrafish data, the chi-square test was employed for prolonged treatments and Student's t-test for acute treatments. P < 0.05 was considered statistically significant.
Materials
All materials were purchased from Sigma Aldrich unless stated otherwise, and the fluorescent dyes from Molecular Probes (Life Technologies, Paisley, UK). The company Ambinter (Orléans, France) (http://www.ambinter.com/search-chemical-compounds) supplied the BTB 06584.
Results
Selection and characterization of BTB06584
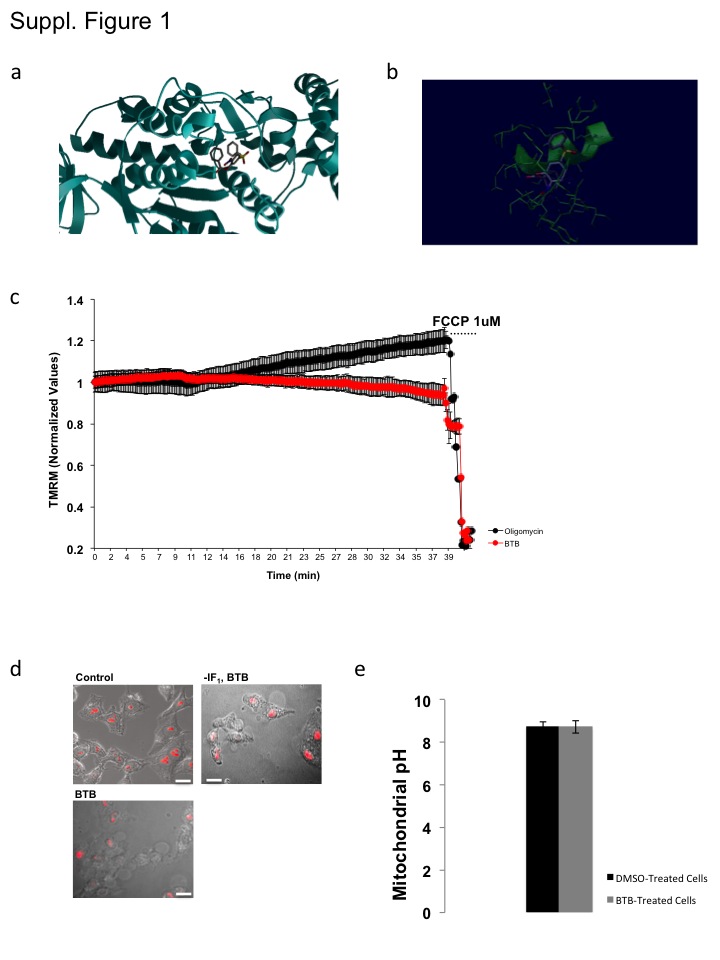
From the chemoinformatic screen of molecules potentially sharing the same structural characteristics as BMS199264 (and so with a similar biological activity) using the open database ZINC (http://zinc.docking.org), we obtained 10 initial hits without toxic chemical groups within their structure. Among these we then selected ZINC01044546, also known as BTB06584 as the one scoring higher among the others for similarity with the benchmark molecule BMS199264. Figure 1A shows the two-dimensional structure of the molecule. To corroborate this we performed an automated docking analysis to predict the binding sites for the BTB hit. This suggested that the selected molecule might have a significant interactivity with the soluble catalytic core, F1, of the F1Fo-ATPsynthase (Figure 1B). We have also generated a three-dimensional ribbon reconstruction (Papadimitriou et al., 2004) of the selected binder (BTB) in complex with the F1Fo-ATPsynthase of which we also present an animated version to better visualize the likely interactivity (Supporting Information Figure S1a,b).
Figure 1.
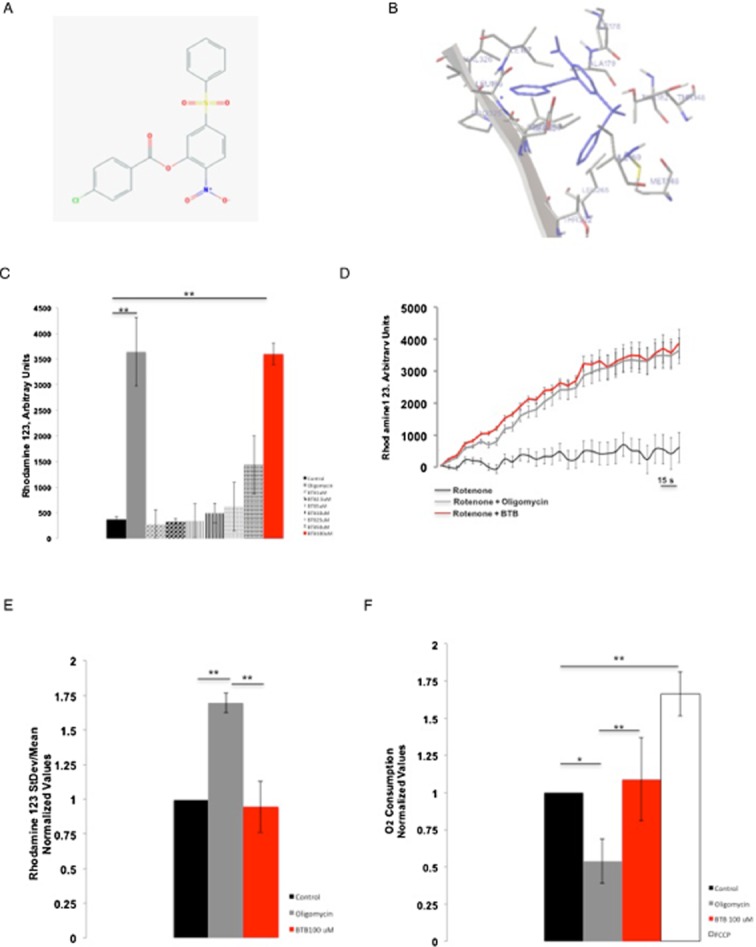
BTB chemical structure, ribbon diagram and effect on mitochondrial respiration. (A) Chemical structure of BTB. (B) BTB interaction conformer. (C) Determination of the optimal concentration of BTB in a FLIPR assay run in HL-1 cells by monitoring the ΔΨm after inhibition of the ETC by rotenone, which leads to the reversal of the ATP synthase. The Y axis shows the mean fluorescence of Rh123 in arbitrary units. In these experimental conditions, an increase in the mean fluorescence reports mitochondrial depolarization. Concentrations of BTB from 1 to 100 μM were tested; only 100 μM of BTB resulted in a significant increase in mean fluorescence in conditions when the ATP synthase is reversed, suggesting the inhibition of the ATP hydrolase activity. **P < 0.001, significantly different from control. (D) Snapshot of representative FLIPR recording of rotenone, rotenone + oligomycin and rotenone + BTB. (E) Effect of BTB on ΔΨm in HL-1 cells loaded with Rh123 assessed by the changes in SD/mean fluorescence ratio in fluorescence microscopy. The bars show the change (normalized) in ΔΨm 15 min after the addition of BTB or oligomycin. Control, n = 16; oligomycin B, n = 18; BTB (100 μM), n = 46. **P < 0.001, significantly different as shown. (F) Effect of BTB on oxygen consumption in HL-1 cells assessed using a Clark electrode. The bars show the change in oxygen consumption following addition of the drugs: BTB did not affect the rate of O2 consumption whereas oligomycin B decreased it (inhibition of synthesis of ATP) and FCCP increased it (uncoupling effect). Control, n = 6; oligomycin B, n = 8; BTB 100 μM, n = 10; FCCP 500 nM, n = 5. **P < 0.001, *P < 0.05, significantly different as shown.
Determination of the optimal working concentrations
A number of BTB concentrations were initially tested on cardiac muscle cell line HL-1, by assessing the effect on ΔΨm with a FLIPR, using Rh123 in ‘dequench mode’, whereby an increase in fluorescence reports mitochondrial depolarization. Rotenone (5 μM) was applied first to the cells to inhibit respiration. In these cells, ΔΨm is then maintained quite effectively by reversal of the F1Fo-ATPase, revealed with a mitochondrial depolarization in response to subsequent addition of oligomycin. Thus, BTB was tested using the same paradigm and caused a loss of ΔΨm at 100 μM (Figure 1C and D). The effect of BTB was not significantly different from that of oligomycin B (3 μM) (Matsuno-Yagi and Hatefi, 1993), the inhibitor of the F1Fo-ATPsynthase that was used as a positive control. This experiment was also confirmed by confocal microscopy with identical results (data not shown).
Effects of BTB on ‘resting’ mitochondrial function
To determine whether, at this concentration (100 μM), BTB had any direct effects on mitochondrial respiration or F1Fo-ATPsynthase activity, we measured its effect on ΔΨm, without prior addition of an inhibitor of the ETC. In HL-1 cells, BTB did not induce any significant change in ΔΨm (measured as a change in the ratio of the SD/mean of the fluorescence signal), supporting its selectivity towards the ATP hydrolase, compared with oligomycin B, which, in contrast, increased ΔΨm as expected, reflecting inhibition of the proton flux through the F1Fo-ATP synthase (Figure 1E).
Loading a different cell type, HeLa, with the fluorescent dye TMRM, we monitored the ΔΨm over time and no effect was recorded in the same resting respiratory conditions. In contrast, oligomycin B increased ΔΨm as expected following inhibition of the F1Fo-ATPsynthase (Supporting Information Figure S1c).
To further establish any direct effect on mitochondrial respiration, we measured O2 consumption in cell suspensions using a Clark-type electrode (Figure 1F). Addition of oligomycin B reduced O2 consumption as expected in response to inhibition of the F1Fo-ATPsynthase, whereas addition of the uncoupler FCCP (500 nM) maximized the rate of O2 consumption. BTB did not induce any significant change compared with basal conditions, nor did it alter maximal oxygen consumption in response to FCCP, supporting its selectivity on the F1Fo-ATPase.
BTB slows intracellular ATP consumption following inhibition of respiration
The next step was to determine the effects of BTB on rates of ATP depletion during inhibition of respiration. For this, we measured the level of free intracellular Mg2+ through the mean fluorescence of the dye MgG, using fluorescence microscopy. The level of free Mg2+ in a cell is inversely related to the availability of the unbound ATP, as its affinity to ATP is 10 times higher than its affinity for ADP. In response to inhibition of respiration using rotenone (5 μM) and antimycin A (1 μM), MgG fluorescence rose progressively. BTB (100 μM) significantly slowed the increase in MgGfluorescence under the same conditions. Similar results were obtained using oligomycin B (Figure 2A).
Figure 2.

BTB spares mitochondrial ATP consumption and protects from ischaemic cell death in an IF1-dependent fashion. (A) Indirect assessment of ATP intracellular levels through monitoring of MgG fluorescence in HL-1 cells in conditions when the F1Fo-ATPsynthase is reversed using fluorescence microscopy. The bars show the change of MgG mean fluorescence 12 min after subsequent addition of BTB or oligomycin B following reversal of the enzyme by rotenone and antimycin A. The inhibitors of the ETC caused an increase in the MgG fluorescence (control), suggesting a decrease in intracellular (ATP) and thus a reversal of the F1Fo-ATPsynthase. BTB and oligomycin significantly reduced this increase in fluorescence, suggesting preservation of ATP through inhibition of the reversed F1Fo-ATPsynthase. Control, n = 26; oligomycin B, n = 30; BTB 100 μM, n = 29. **P < 0.001, significantly different from control. (B) Time course of MgG fluorescence in HeLa cells with and without BTB following inhibition of respiration. At 80 min of NaCN and IAA treatment, n = 8. **P < 0.001, *P < 0.05, significant effects of BTB. (C) Kinetics of ΔΨm dissipation in HeLa cells. TMRM fluorescence values are normalized to the initial baseline; mean data are shown from 3 experiments. (D) Protective effect of BTB against ischaemia/reperfusion in HL-1 cells: compared with the ischaemic control, BTB significantly reduced the cell death ratio after simulated ischaemia/reperfusion, as assessed by the ratio of PI-positive cells over Hoechst-positive cells, similarly to the positive controls of cardioprotection using CsA and diazoxide. Sham control, n = 5; CsA 200 nM, n = 16; diazoxide 100 μM, n = 12; BTB 100 μM, n = 5. **P < 0.001, significantly different as shown. (E) Immunofluorescence assay confirming an efficient IF1 overexpression or silencing. (F) Quantification of the efficiency of modulation of IF1 in HL-1 cells. n = 5; **P < 0.001, significantly different from control. (G) Effect of the modulation of IF1 expression on the efficiency of BTB in conditions of reversion of the F1Fo-ATPsynthase in HL-1 cells loaded with Rh123 using fluorescence microscopy. The ΔΨm was monitored using the SD/mean fluorescence ratio. Similarly to Figure 1E, the bars show the change in ΔΨm 15 min after the addition of BTB when the F1Fo-ATPsynthase was reversed by rotenone and antimycin A. Addition of BTB in +IF1 cells resulted in a potentiation of its effect on ΔΨm (mitochondrial depolarization) compared with non-transfected HL-1 cells, suggesting that overexpression of IF1 potentiated the effect of BTB while silencing of IF1 (–IF1) reduced its effect. n = 112; **P < 0.001, significantly different from control. (H) Histogram of BTB-mediated protection in HeLa cells from ischaemic cell death, scored by PI staining. **P < 0.001, significantly different from control.
To further corroborate these data, these experiments were duplicated using HeLa cells. Cells were treated with NaCN and iodoacetic acid (IAA) (Yang, 1957; Chatham et al., 1988) to inhibit all ATP-generating pathways. Following BTB treatment, the level of ATP was significantly preserved compared with the untreated cells (Figure 2B; P < 0.001), consistent with the BTB-mediated inhibition of the F1Fo-ATPase. This evidence matches the data obtained from HL-1 cells and indicates a selectivity of BTB in conditions of impaired mitochondrial respiration.
Figure 2C shows an average trace for the ΔΨm dissipation after BTB addition to cells bathed in NaCN. The kinetics of ΔΨm dissipation were very similar to that recorded in HeLa cells overexpressing IF1 in the same conditions of respiratory inhibition (Campanella et al., 2008).
BTB protects against ischaemic cell death
HL-1 cells were pretreated with BTB prior to a period of ischaemia, and cell death was assayed using PI staining. This analysis (Figure 2D) showed that cell death was reduced substantially by BTB (100 μM) to a level equivalent to that seen with either CsA (Sharov et al., 2007) or diazoxide (Wang et al., 1999; Murata et al., 2001).
Interaction between BTB and IF1
To further understand the mechanism via which BTB achieves protection, we asked whether its action could involve a participation of IF1. HL-1 cells were transfected with either the IF1 construct to induce overexpression of IF1 or the IF1 siRNA to silence this protein, in conjunction with CFP or YFP. Immunofluorescence experiments revealed a significant overexpression or silencing of IF1 (Figure 2E and F).
After manipulating IF1 expression, HL-1 cells were therefore monitored for the changes in ΔΨm in conditions in which the F1Fo-ATPsynthase was forced to act in reverse (i.e. after inhibition of the ETC by rotenone and antimycin A). In these, when BTB was present, we observed a significant additional decrease in the SD/mean ratio in the +IF1 cells compared with the non-transfected cells and a significantly lower decrease in this ratio in cells devoid of IF1 (Figure 2G).
Such variations are therefore likely to depend on the level of IF1 expression, thus suggesting a role for IF1 in the action of BTB. Confirmation of this was given by the correlation with cell survival in HeLa cells (Figure 2H), as cell death following ischaemia was significantly higher in –IF1 cells and lower in +IF1 cells when treated with BTB as shown in Supporting Information Figure S1d.
The BTB-mediated protection was tested further in neurons. Primary cultured cortical neurons of mice were exposed to OGD, treated or not with BTB, followed by RX. The resulting cell death was significantly reduced by BTB, as scored by PI staining, compared with cells left untreated during OGD (Figure 3A and B)
Figure 3.
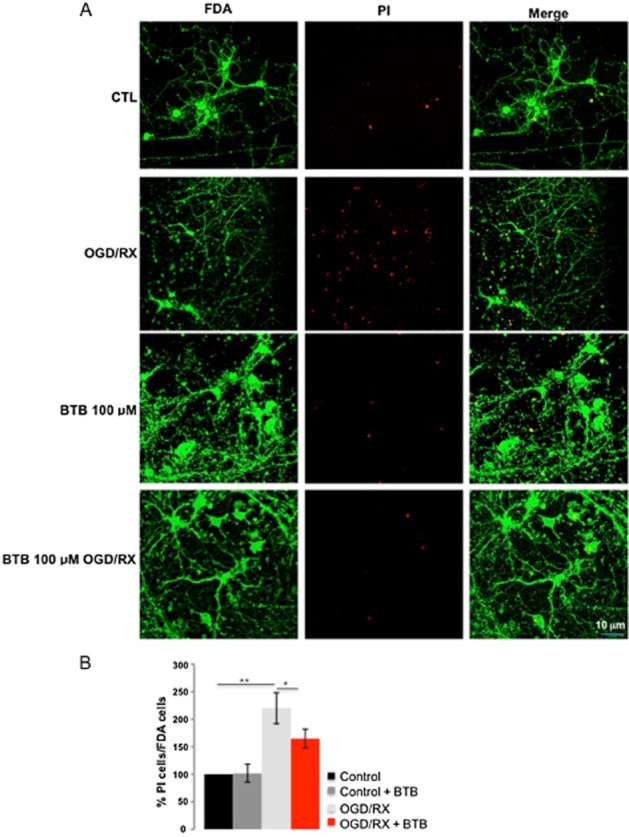
BTB prevention of neuronal cell death following treatment with OGD/RX. (A) Cortical neurons were exposed to OGD/RX (3 h OGD+24 h RX) with or without 100 μM BTB and stained for FDA and PI. (B) Summary data from PI-stained neurons, shown as % total number of neurons. n = 2; **P < 0.001, *P < 0.05, significantly different as shown.
These data imply a synergy between BTB and IF1 as (i) modulation of ΔΨm and (ii) protection from ischaemia are lost when IF1 is down-regulated. Ruling out that (i) BTB promotes any aberration of the mitochondrial network morphology (Figure 4A; n = 6) or (ii) matrix pH change (Supporting Information Figure S1e), which would predispose the activation of IF1, we investigated the ratio of expression between the dimeric and tetrameric conformations of the protein in order to gain further insight into the mechanism of action.
Figure 4.
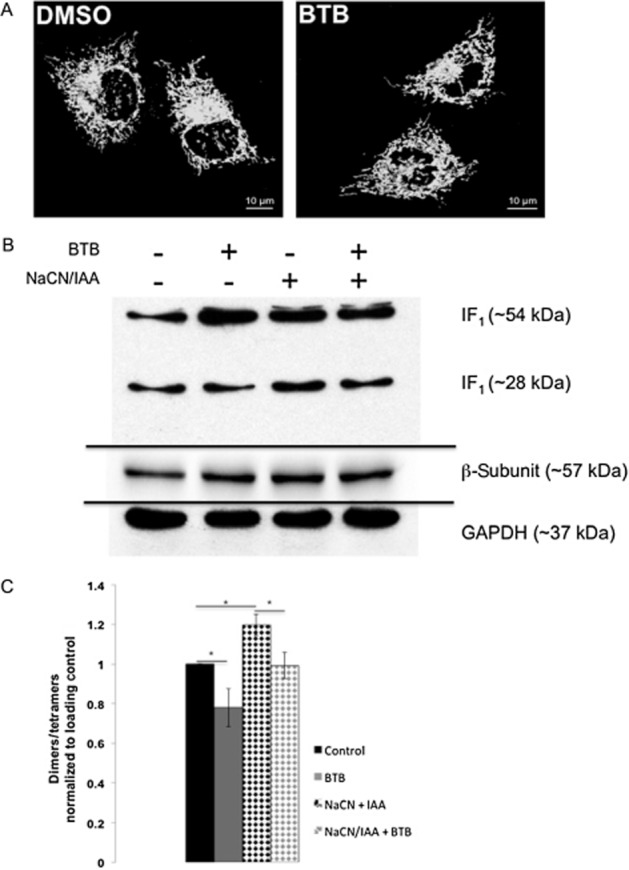
Mitochondrial network morphology in BTB-treated cells and IF1 dimers/tetramers ratio. (A) Mitochondrial network morphology in HeLa cells analysed via MitoTracker Green. (B) Immunoblotting of IF1 dimers and tetramers in control and ischaemic conditions with and without BTB treatment. (C) Quantification of IF1 dimers/tetramers ratio. n = 4; *P < 0.05, significantly different as shown.
BTB modifies the ratio of IF1 dimers/tetramers
IF1 is found in two conformations: dimeric or tetrameric, corresponding respectively to the active and inactive forms of the protein (Cabezon et al., 2000). Dimers of IF1 are found when mitochondrial respiration is inhibited as the conformation that binds the F1 complex of the F1Fo-ATPsynthase. We therefore ran a Western blot analysis of HeLa cell protein extracts incubated with BTB both with and without inhibition of cellular respiration by addition of NaCN and IAA.
Exposure to BTB reduced the dimerization of IF1 both under resting conditions and following addition of NaCN/IAA, while it increased the relative amount of the tetrameric form (Figure 4B, quantified in 4C). These data suggest that BTB promotes a shift of IF1 conformation towards the tetrameric form, without altering the ability to dimerize during reversal of the F1Fo-ATPsynthase.
BTB rescues anaemia in pinotage zebrafish
A recent study surprisingly revealed that a zebrafish mutant, ‘pinotage’ (pnt) that is profoundly deficient in haemoglobin, is caused by a loss-of-function mutation in the Atpif1a gene, one of two Atpif1 homologues found in the zebrafish (Figure 5A) (Shah et al., 2012). The mechanism appears to involve mitochondrial iron overloading that impairs the enzyme ferrochelatase (Fech), with consequent inhibition of haemoglobin synthesis. We therefore explored whether BTB could rescue this phenotype by promoting the activity of the remaining IF1 isoform, Atpif1b. This was carried out by incubating pnt zebrafish and control siblings with BTB (1 μM) for 24 h. Remarkably, exposure to BTB rescued the anaemic phenotype of pnt (Figure 5B and C) with a significant increase in haemoglobin synthesis, as measured by positive staining with o-dianisidine.
Figure 5.
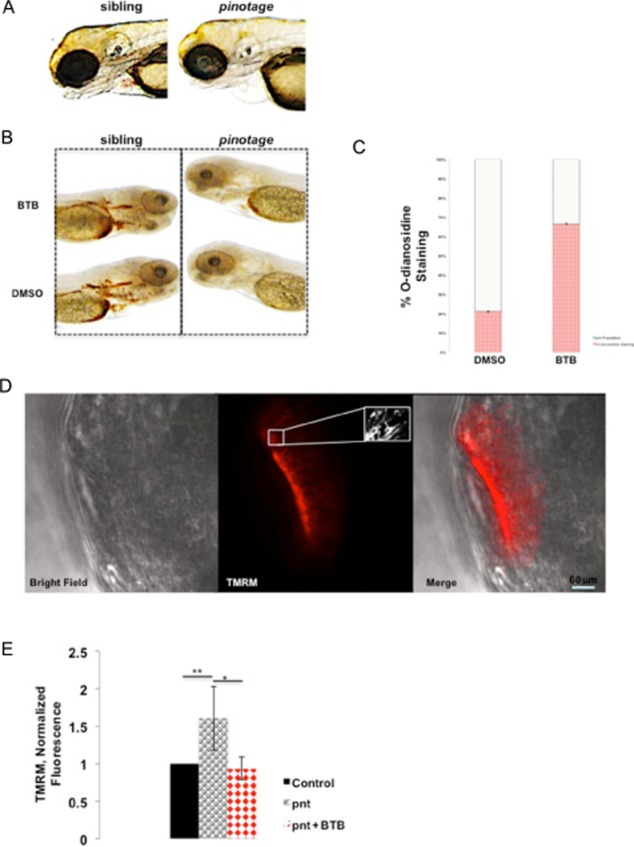
BTB-mediated rescue of haemoglobin synthesis in pnt zebrafish embryos. (A) Representative images of 3 dpf larvae from sibling and heterozygous pnt cross showing how anaemia is identified. (B) Detection of haemoglobin biosynthesis in pnt embryos, untreated and treated with BTB, using o-dianisidine staining. (C) Summary data of the pnt phenotype rescue by BTB, as illustrated in (B). Data are shown as percentage of total pnt population staining positively; n = 18; *P < 0.05. (D) Sample confocal images for the mitochondrial membrane potential (ΔΨm) measured with TMRM in the olfactory bulbs in living zebrafish embryos with detailed screenshot of mitochondria. (E) Summary data of TMRM values of ΔΨm in single mitochondria of wt and pnt embryos without and with BTB. Fluorescence intensity values are normalized to control; n = 4 days of analysis. **P < 0.001, *P < 0.05, significantly different as shown.
Using TMRM, we measured ΔΨm in pnt and control siblings to assess whether this response was related to altered mitochondrial bioenergetics. Measurement of TMRM accumulation in the olfactory bulbs (see images at different magnification in Figure 5D) showed that ΔΨm was significantly increased in the pnt fish compared with controls, and was reduced to control levels following short-term treatment with BTB (Figure 5E). Treatment with vehicle, DMSO, alone had no effect (normalized fluorescence values normalized to sibling + DMSO: 1.08 ± 0.11, pnt + DMSO: 1.46 ± 0.15, n = 4 days of analysis). These data indicate that concentrations of BTB that restore haemoglobin biosynthesis also alter mitochondrial bioenergetics in living fish (Shah et al., 2012).
Discussion and conclusions
The data presented here demonstrate that we have identified a drug candidate, BTB, which selectively inhibits the reversal of the F1Fo-ATPsynthase. This is a deleterious activity causing ATP wastage and represents a key factor contributing to ischaemic cell death, which is still the major cause of death in Western countries (McGovern et al., 1996; Dudas et al., 2011; Roger et al., 2012). BTB preserves ATP in ischaemia-like experimental protocols inhibiting the F1Fo-ATPase via a synergy with IF1 (Faccenda and Campanella, 2012).
A molecule consistent with previous findings
These results support the earlier work on BMS199264 (Grover et al., 2004), the compound on which we based our search for BTB. Grover et al. (2004) demonstrated, with sub-mitochondrial particles that BMS199264 was selective against the hydrolase activity of the F1Fo-ATPsynthase, as opposed to its synthase activity (Atwal et al., 2004). The question is how any low MW inhibitor might selectively act on only one direction of enzymic action. Our data suggest that this is feasible if the drug operates through potentiating the action of the endogenous inhibitor IF1. By showing changes in the efficacy of BTB in cells in which the expression of IF1 has been up- and down–regulated, we have shown that BTB efficacy depends on IF1 expression. It would be interesting to know whether this is also true of BMS199264, which we have been unable to obtain. Our findings also have the interesting corollary that the effects of BTB may vary in different tissues dependent on the expression level of IF1, which we have shown previously to vary significantly between different cell types (Campanella et al., 2008).
The physiological implications
If we consider myocardial ischaemia, the heart being the first tissue in which IF1 was discovered and probably one of the richest sources of this protein, our data suggests BTB is an interesting candidate to prevent cellular damage following myocardial ischaemia (Pullman and Monroy, 1963). The same considerations would apply to neurons in which the IF1 level is particularly high (Campanella et al., 2008), and also proximal tubules of the kidney (Hall et al., 2009). The data we present here in primary cultures of neurons sustain this hypothesis. On the same principles. the potential effects of BTB in neoplastic pathologies that show IF1 enrichment and resistance to chemotherapy-induced cell death (Luciakova and Kuzela, 1984; Yamada and Huzel, 1992; Capuano et al., 1997; Faccenda et al., 2013) could be explored.
The role of the F1Fo-ATPsynthase acting as an F1Fo-ATPase, hence consuming ATP, in ischaemia-induced cell death still remains controversial. The work of Chinopoulos et al. (2010; Chinopoulos, 2011) suggests that the ATP synthase and the adenine nucleotide translocase (ANT) do not share the same ‘reversal potential’, meaning that the mitochondrial F1Fo-ATPsynthase could work in reverse mode with the ANT working in the forward one. This would result in the F1Fo-ATPase hydrolysing the ATP stocks in the mitochondria but without any supply of cytosolic ATP through the ANT, and thus without any significant effect on the cytosolic ATP supplies which would be maintained by glycolytic activity. There is no doubt, however, that there is a decrease in total cellular ATP during ischaemia, which can be preserved both by oligomycin and by BTB, arguing strongly that ATP hydrolysis is indeed driven by the F1Fo-ATPsynthase in reverse.
The molecular implications beyond the F1Fo-ATPsynthase
The BTB-mediated protection from ischaemia could imply an effect on the formation of mPTPs, which was very recently associated with the F1Fo-ATPsynthase (Giorgio et al., 2013). We have no data to suggest that BTB inhibits mPTP opening. We have also seen before that inhibition of the F1Fo-ATPase by oligomycin did not prevent opening of the mPTP in a cardiomyocyte cell model, although it did radically slow the rate of ATP depletion (Duchen, 2000). Notably, BTB has a chemical structure different from that of any known existing mPTP modulators, which would suggest that a direct regulatory effect could be excluded.
Rescuing haemoglobin synthesis in pinotage
The data from the pnt zebrafish imply that BTB is absorbed in the fish in vivo without causing significant toxicity and sustain the hypothesis that it might compensate pharmacologically for the deficiencies of IF1 expression. The defect in haemoglobin synthesis described in pnt is a consequence of impaired mitochondrial bioenergetics, resulting from a loss of Atpif1a that leads to mitochondrial iron (Fe2+) accumulation of ferrochelatase, the terminal enzyme in haem biosynthesis (Shah et al., 2012). Long-term treatment with BTB-restored haemoglobin synthesis in a large portion of the pnt zebrafish population, attributable to an action on mitochondrial bioenergetics as the ΔΨm, which was elevated in the untreated pnt mutant, was normalized following exposure to BTB.
In conclusion, our data suggest that BTB is an interesting potential candidate to reduce ischaemia-induced cellular injury, without impairing ATP generation under resting conditions and without evident in vivo toxicity, at least in the zebrafish. This compound specifically targets the reversal of the F1Fo-ATPsynthase, which, to the best of our knowledge, remains untargeted. The data help to establish an important proof of principle to suggest that the selective inhibition of the ATPase function of the F1Fo-ATPsynthase may represent a viable therapeutic approach to conditions in which mitochondrial respiration is impaired.
Acknowledgments
We are grateful to Professor G Churchill, Dr S Vasudevan and Professor A Ganesan for advising with the use of the chemoinformatic screening for BTB http://zinc.docking.org. A BBSRC Research Placement supported A. A. A. at the time of the experiments. A grant from the Federation Française de Cardiologie supported F. I. at the time of experiments. The research activities led by M. C. are supported by the BBSRC New Investigator Award Grant (BB/I013695/1), PPCT, Central Research Fund of the University of London, Local Funds of the Royal Veterinary College, EBRI-Rita Levi Montalcini Foundation Research Programme in Metabolism in Brain Diseases and LAM Research Grant on Brain Tumours. C. R. and C. H. K. C. (a BSc Project student) were supported by local funds of the Royal Veterinary College. E. A. was supported by University College London as an Intercalated BSc Project student located at the Royal Veterinary College supervised by M. C. and C. R.
Glossary
- ANT
adenine nucleotide translocase
- BTB06584
2-nitro-5-(phenylsulfonyl)phenyl 4-chlorobenzoate
- CFP
cyan fluorescent protein
- CsA
cyclosporine A
- ΔΨm
mitochondrial membrane potential
- ETC
electron transport chain
- FCCP
carbonyl cyanide p-trifluoromethoxy-phenylhydrazone
- FDA
fluorescein diacetate
- FLIPR
fluorescent imaging plate reader
- IAA
iodo acetic acid
- IF1
inhibitor protein of F1 subunit
- MgG
magnesium green
- mPTP
mitochondrial permeability transition pore
- OGD
oxygen-glucose deprivation
- pnt
pinotage zebrafish
- Rh123
rhodamine 123
- RX
reoxygenation
- TMRM
tetramethylrhodamine methyl ester
- YFP
yellow fluorescent protein
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12638
Figure S1 Ribbon modelling of the BTB binding to the F1Fo-ATPsynthase mitochondrial membrane potential (ΔΨm) and cell death images. (a) Ribbon model and (b) link to animated ribbon modelling of the BTB binding to the β-subunit of the F1Fo-ATPsynthase. (c) Kinetics of ΔΨm modulation in HeLa during exposure, respectively, to BTB and oligomycin. (d) Images of HeLa cells positive and not, to PI undergoing ischaemic treatment in the presence/absence of BTB with and without IF1 expression. (e) Mitochondrial pH data in DMSO and BTB-treated HeLa Cells.
{kind=link}
References
- Atwal KS, Wang P, Rogers WL, Sleph P, Monshizadegan H, Ferrara FN, et al. Small molecule mitochondrial F1F0 ATPase hydrolase inhibitors as cardioprotective agents. Identification of 4-(N-arylimidazole)-substituted benzopyran derivatives as selective hydrolase inhibitors. J Med Chem. 2004;47:1081–1084. doi: 10.1021/jm030291x. [DOI] [PubMed] [Google Scholar]
- Cabezon E, Butler PJ, Runswick MJ, Walker JE. Modulation of the oligomerization state of the bovine F1-ATPase inhibitor protein, IF1, by pH. J Biol Chem. 2000;275:25460–25464. doi: 10.1074/jbc.M003859200. [DOI] [PubMed] [Google Scholar]
- Cabezon E, Runswick MJ, Leslie AG, Walker JE. The structure of bovine IF(1), the regulatory subunit of mitochondrial F-ATPase. EMBO J. 2001;20:6990–6996. doi: 10.1093/emboj/20.24.6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, et al. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008;8:13–25. doi: 10.1016/j.cmet.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Capuano F, Guerrieri F, Papa S. Oxidative phosphorylation enzymes in normal and neoplastic cell growth. J Bioenerg Biomembr. 1997;29:379–384. doi: 10.1023/a:1022402915431. [DOI] [PubMed] [Google Scholar]
- Carunchio I, Pieri M, Ciotti MT, Albo F, Zona C. Modulation of AMPA receptors in cultured cortical neurons induced by the antiepileptic drug levetiracetam. Epilepsia. 2007;48:654–662. doi: 10.1111/j.1528-1167.2006.00973.x. [DOI] [PubMed] [Google Scholar]
- Chatham J, Gilbert HF, Radda GK. Inhibition of glucose phosphorylation by fatty acids in the perfused rat heart. FEBS Lett. 1988;238:445–449. doi: 10.1016/0014-5793(88)80529-5. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C. Mitochondrial consumption of cytosolic ATP: not so fast. FEBS Lett. 2011;585:1255–1259. doi: 10.1016/j.febslet.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Gerencser AA, Mandi M, Mathe K, Torocsik B, Doczi J, et al. Forward operation of adenine nucleotide translocase during F0F1-ATPase reversal: critical role of matrix substrate-level phosphorylation. FASEB J. 2010;24:2405–2416. doi: 10.1096/fj.09-149898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 2000;28:339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- Duchen MR, Surin A, Jacobson J. Imaging mitochondrial function in intact cells. Methods Enzymol. 2003;361:353–389. doi: 10.1016/s0076-6879(03)61019-0. [DOI] [PubMed] [Google Scholar]
- Dudas K, Lappas G, Stewart S, Rosengren A. Trends in out-of-hospital deaths due to coronary heart disease in Sweden (1991 to 2006) Circulation. 2011;123:46–52. doi: 10.1161/CIRCULATIONAHA.110.964999. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Bruno VM, Amagasu SM, Giffard RG. Glia modulate the response of murine cortical neurons to excitotoxicity: glia exacerbate AMPA neurotoxicity. J Neurosci. 1995;15:4545–4555. doi: 10.1523/JNEUROSCI.15-06-04545.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccenda D, Campanella M. Molecular regulation of the mitochondrial F(1)F(o)-ATPsynthase: physiological and pathological significance of the inhibitory factor 1 (IF(1)) Int J Cell Biol. 2012;2012:367934. doi: 10.1155/2012/367934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccenda D, Tan CH, Seraphim A, Duchen MR, Campanella M. IF1 limits the apoptotic-signalling cascade by preventing mitochondrial remodelling. Cell Death Differ. 2013;20:686–697. doi: 10.1038/cdd.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DW, Grover GJ. The IF(1) inhibitor protein of the mitochondrial F(1)F(0)-ATPase. Biochim Biophys Acta. 2000;1458:343–355. doi: 10.1016/s0005-2728(00)00085-2. [DOI] [PubMed] [Google Scholar]
- Grover GJ, Atwal KS, Sleph PG, Wang FL, Monshizadegan H, Monticello T, et al. Excessive ATP hydrolysis in ischemic myocardium by mitochondrial F1F0-ATPase: effect of selective pharmacological inhibition of mitochondrial ATPase hydrolase activity. Am J Physiol Heart Circ Physiol. 2004;287:H1747–H1755. doi: 10.1152/ajpheart.01019.2003. [DOI] [PubMed] [Google Scholar]
- Hall AM, Unwin RJ, Parker N, Duchen MR. Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J Am Soc Nephrol. 2009;20:1293–1302. doi: 10.1681/ASN.2008070759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DA, Das AM. Control of mitochondrial ATP synthesis in the heart. Biochem J. 1991;280(Pt 3):561–573. doi: 10.1042/bj2800561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Shoichet BK. ZINC – a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings RB, Reimer KA, Steenbergen C. Effect of inhibition of the mitochondrial ATPase on net myocardial ATP in total ischemia. J Mol Cell Cardiol. 1991;23:1383–1395. doi: 10.1016/0022-2828(91)90185-o. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyssens A, Nowicky AV, Patterson L, Crompton M, Duchen MR. The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]i studied in isolated rat cardiomyocytes. J Physiol. 1996;496(Pt 1):111–128. doi: 10.1113/jphysiol.1996.sp021669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciakova K, Kuzela S. Increased content of natural ATPase inhibitor in tumor mitochondria. FEBS Lett. 1984;177:85–88. doi: 10.1016/0014-5793(84)80986-2. [DOI] [PubMed] [Google Scholar]
- Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B. A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J Clin Invest. 1962;41:1776–1804. doi: 10.1172/JCI104637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manev H, Favaron M, Vicini S, Guidotti A, Costa E. Glutamate-induced neuronal death in primary cultures of cerebellar granule cells: protection by synthetic derivatives of endogenous sphingolipids. J Pharmacol Exp Ther. 1990;252:419–427. [PubMed] [Google Scholar]
- Matsuno-Yagi A, Hatefi Y. Studies on the mechanism of oxidative phosphorylation. Different effects of F0 inhibitors on unisite and multisite ATP hydrolysis by bovine submitochondrial particles. J Biol Chem. 1993;268:1539–1545. [PubMed] [Google Scholar]
- McGovern PG, Pankow JS, Shahar E, Doliszny KM, Folsom AR, Blackburn H, et al. Recent trends in acute coronary heart disease – mortality, morbidity, medical care, and risk factors. The Minnesota Heart Survey Investigators. N Engl J Med. 1996;334:884–890. doi: 10.1056/NEJM199604043341403. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M, Akao M, O'Rourke B, Marban E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: possible mechanism of cardioprotection. Circ Res. 2001;89:891–898. doi: 10.1161/hh2201.100205. [DOI] [PubMed] [Google Scholar]
- Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, et al. Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol. 2009;5:220–226. doi: 10.1038/nchembio.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paffett-Lugassy NN, Zon LI. Analysis of hematopoietic development in the zebrafish. Methods Mol Med. 2005;105:171–198. doi: 10.1385/1-59259-826-9:171. [DOI] [PubMed] [Google Scholar]
- Papadimitriou C, Yapijakis C, Davaki P. Use of truncated pyramid representation methodology in three-dimensional reconstruction: an example. J Microsc. 2004;214(Pt 1):70–75. doi: 10.1111/j.0022-2720.2004.01298.x. [DOI] [PubMed] [Google Scholar]
- Power J, Cross RL, Harris DA. Interaction of F1-ATPase, from ox heart mitochondria with its naturally occurring inhibitor protein. Studies using radio-iodinated inhibitor protein. Biochim Biophys Acta. 1983;724:128–141. doi: 10.1016/0005-2728(83)90034-8. [DOI] [PubMed] [Google Scholar]
- Pullman ME, Monroy GC. A naturally occurring inhibitor of mitochondrial adenosine triphosphatase. J Biol Chem. 1963;238:3762–3769. [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics – 2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen D, Lewis AM, Mizote A, Thomas JM, Aley PK, Vasudevan SR, et al. Analogues of the nicotinic acid adenine dinucleotide phosphate (NAADP) antagonist Ned-19 indicate two binding sites on the NAADP receptor. J Biol Chem. 2009;284:34930–34934. doi: 10.1074/jbc.M109.016519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouslin W. Regulation of the mitochondrial ATPase in situ in cardiac muscle: role of the inhibitor subunit. J Bioenerg Biomembr. 1991;23:873–888. doi: 10.1007/BF00786006. [DOI] [PubMed] [Google Scholar]
- Rouslin W, Erickson JL, Solaro RJ. Effects of oligomycin and acidosis on rates of ATP depletion in ischemic heart muscle. Am J Physiol. 1986;250(3 Pt 2):H503–H508. doi: 10.1152/ajpheart.1986.250.3.H503. [DOI] [PubMed] [Google Scholar]
- Rouslin W, Broge CW, Grupp IL. ATP depletion and mitochondrial functional loss during ischemia in slow and fast heart-rate hearts. Am J Physiol. 1990;259(6 Pt 2):H1759–H1766. doi: 10.1152/ajpheart.1990.259.6.H1759. [DOI] [PubMed] [Google Scholar]
- Rouslin W, Frank GD, Broge CW. Content and binding characteristics of the mitochondrial ATPase inhibitor, IF1, in the tissues of several slow and fast heart-rate homeothermic species and in two poikilotherms. J Bioenerg Biomembr. 1995;27:117–125. doi: 10.1007/BF02110339. [DOI] [PubMed] [Google Scholar]
- Savoia C, Sisalli MJ, Di Renzo G, Annunziato L, Scorziello A. Rosuvastatin-induced neuroprotection in cortical neurons exposed to OGD/reoxygenation is due to nitric oxide inhibition and ERK1/2 pathway activation. Int J Physiol Pathophysiol Pharmacol. 2011;3:57–64. [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah DI, Takahashi-Makise N, Cooney JD, Li L, Schultz IJ, Pierce EL, et al. Mitochondrial Atpif1 regulates haem synthesis in developing erythroblasts. Nature. 2012;491(7425):608–612. doi: 10.1038/nature11536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov VG, Todor A, Khanal S, Imai M, Sabbah HN. Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial respiratory function in cardiomyocytes isolated from dogs with heart failure. J Mol Cell Cardiol. 2007;42:150–158. doi: 10.1016/j.yjmcc.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov AD. Steady-state and pre-steady-state kinetics of the mitochondrial F(1)F(o) ATPase: is ATP synthase a reversible molecular machine? J Exp Biol. 2000;203(Pt 1):41–49. doi: 10.1242/jeb.203.1.41. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hirai K, Ashraf M. Activation of mitochondrial ATP-sensitive K(+) channel for cardiac protection against ischemic injury is dependent on protein kinase C activity. Circ Res. 1999;85:731–741. doi: 10.1161/01.res.85.8.731. [DOI] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio. 5th edn. Eugene: Univ. of Oregon Press; 2007. [Google Scholar]
- Yamada EW, Huzel NJ. Distribution of the ATPase inhibitor proteins of mitochondria in mammalian tissues including fibroblasts from a patient with Luft's disease. Biochim Biophys Acta. 1992;1139:143–147. doi: 10.1016/0925-4439(92)90093-3. [DOI] [PubMed] [Google Scholar]
- Yang WC. Effect of iodoacetate and iodoacetamide on oxygen uptake of heart mitochondria. Science. 1957;125(3257):1087. doi: 10.1126/science.125.3257.1087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Ribbon modelling of the BTB binding to the F1Fo-ATPsynthase mitochondrial membrane potential (ΔΨm) and cell death images. (a) Ribbon model and (b) link to animated ribbon modelling of the BTB binding to the β-subunit of the F1Fo-ATPsynthase. (c) Kinetics of ΔΨm modulation in HeLa during exposure, respectively, to BTB and oligomycin. (d) Images of HeLa cells positive and not, to PI undergoing ischaemic treatment in the presence/absence of BTB with and without IF1 expression. (e) Mitochondrial pH data in DMSO and BTB-treated HeLa Cells.