

Abstract
Background and Purpose
Dysregulated tonic tension and calcium sensitization in blood vessels has frequently been observed in many cardiovascular diseases. Despite a huge therapeutic potential, little is known about natural products targeting tonic tension and calcium sensitization.
Experimental Approach
We screened natural products for inhibitory effects on vasoconstriction using the rat isolated thoracic aorta and found that an anthraquinone derivative, emodin, attenuated tonic tension. Organ bath system, primary vascular smooth muscle cells, confocal microscopy and Western blot analysis were employed to demonstrate the suppressive effects of emodin on PKCδ-mediated myosin phosphatase inhibition.
Key Results
Emodin, an active ingredient of Polygonum multiflorum extract, inhibited phenylephrine-induced vasoconstriction in rat isolated thoracic aorta, and inhibited vasoconstriction induced by 5-HT and endothelin-1. It also generally suppressed vasoconstrictions mediated by voltage-operated, store-operated calcium channels and intracellular calcium store. However, emodin did not affect agonist-induced calcium increases in primary smooth muscle cells. In contrast, post-treatment with emodin following phenylephrine stimulation potently suppressed tonic tension in rat aortic rings. Western blot analysis revealed that emodin inhibited phenylephrine-induced phospho-myosin light chain (pMLC) and the phosphorylation of myosin-targeting subunit and C-kinase-activated protein phosphatase-1 inhibitor (CPI-17). This was mediated by selective inhibition of PKCδ, whereas PKCα was not involved.
Conclusion and Implications
Emodin attenuates tonic tension through the blockade of PKCδ and CPI-17-mediated MLC-phosphatase inhibition. This new mode of action for the suppression of tonic tension and structural insights into PKCδ inhibition revealed by emodin may provide new information for the development of modulators of tonic tension and for the treatment of hypertension.
Table of Links
| TARGETS | LIGANDS |
|---|---|
| PKCδ | Phenylephrine |
| PKCα | 5-HT |
| VOCC | Endothelin-1 |
| MLCK | |
| ROCK |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a,b).
Introduction
Hypertension is a major risk factor for acute and chronic heart diseases. High arterial blood pressure can also increase the mortality and morbidity from many other life-threatening cardiovascular diseases such as stroke, renal failure and atherosclerosis (Roger et al., 2012). The prevalence of hypertension continually increases, reaching over 25–35% of the adult population, and 60–70% of the elderly population aged beyond 70 (Hajjar et al., 2006; CDC, 2011). To tackle this deadly disease, many molecular targets have been explored to develop effective anti-hypertensive agents that include diuretics, adrenoceptor agonists/antagonists, calcium channel blockers, angiotensin converting enzyme (ACE) inhibitors, vasodilators and renin inhibitors/receptor blockers. Indeed, these drugs have helped to save millions of lives. However, there are still a significant number of patients with hypertension, as many as 56% of all treated, whose blood pressure cannot be controlled with currently available drugs (Lloyd-Jones et al., 2010), underlining a urgent demand for novel approaches and more effective new anti-hypertensive drugs.
The vascular smooth muscle cell (VSMC) and its contractile machinery has been a subject of intense research for the discovery of novel anti-hypertensive drugs. VSMC contracts in response to various physiological contractile agonists and maintains vascular tension for a certain time to provide an adequate blood flow to tissues. Primary tension generation, that is phasic tension, is initiated by rapid and high cytosolic calcium increase that is provided from receptor-mediated, store-operated or voltage-gated calcium channels (Aksoy et al., 1983). Subsequently, calcium-calmodulin activated myosin light chain kinases phosphorylate myosin light chain (MLC20), ultimately generating contractile force. Concomitantly with phasic tension, receptor-mediated activation of PKC-CPI-17 (C-kinase-activated protein phosphatase-1 inhibitor) (Kitazawa et al., 2000) and RhoA–Rho-associated PK (ROCK) pathways inhibit MLC-phosphatase via the phosphorylation of a regulatory subunit, MYPT1, sustaining MLC phosphorylation for a certain time even after the initial calcium peak dissipates. This phenomenon is called calcium sensitization and the maintenance of the tension generated is described as tonic tension (de Godoy and Rattan, 2011). While most conventional anti-hypertensive drugs have focused on the control of phasic tension, recent interest is being transferred to the regulation of calcium sensitization and tonic tension (Christ and Wingard, 2005). Augmented calcium sensitization has been frequently observed in patients with cardiovascular diseases and in many animal models of hypertension (Satoh et al., 1994; Shimokawa et al., 1999; Seko et al., 2003; Chiba et al., 2005), indicating the importance of regulating calcium sensitization for the treatment of hypertension.
Natural resources have helped to enrich the pharmacotherapeutic arsenal for the treatment of hypertension by providing important insights into pharmacophore, molecular targets and structural properties for the development of new anti-hypertensive drugs. According to the analysis by Newman et al. (2003), 49 of 75 new chemical entities (NCEs) in the anti-hypertensive category have been derived from the knowledge gained from a study of natural products, highlighting the importance of natural products in development of new anti-hypertensive drugs. Recently, Seok et al. (2008) demonstrated that isoflavones can attenuate vascular contraction through inhibition of RhoA/ROCK-mediated calcium sensitization, providing an important line of evidence supporting the possibility that the anti-hypertensive effect of natural products may stem from the regulation of tonic tension. Incidentally, to the best of our knowledge there has been no report regarding natural products that target PKC-CPI-17-dependent tonic tension.
In the present study we demonstrated that emodin, an anthraquinone derivative widely present in herbal medicines (Srinivas et al., 2007), can modulate agonist-induced vasoconstriction. The decreased contraction induced by emodin resulted from the inhibition of tonic tension and calcium sensitization. Notably, this was induced by selective blockade of PKCδ, a novel calcium-independent PKC isoform in VSMCs (Salamanca and Khalil, 2005), and subsequent suppression of CPI-17-mediated MLC-phosphatase inhibition and contractile force generation.
Methods
Herb extracts
Herb extracts were provided by the National Centre for Standardization of Herbal Medicines in Korea. Dry powders of herbs (Achyranthes japonica, Atractylodes japonica, Aucklandia lappa, Foeniculum vulgare, Lycium chinense, Kalopanax pictus, Polygonatum stenophyllum, Polygonum multiforum, Rehmannia glutinosa) were extracted with 70% ethanol at 70–80°C for 3 h. The extraction was repeated three times. After filtration and concentration under reduced pressure, extracts were lyophilized and the resultant powder was stored at −20°C. For experiments extracts were dissolved in DMSO.
Animals
The entire animal protocol was approved by the Ethics Committee of the Animal Service Centre at Seoul National University. Male Sprague-Dawley rats (SamTako., Seoul, Korea) weighing 250–300 g were used (except for the primary smooth muscle cell culture assay which used animals weighing 150–170 g). Before the experiments, animals were acclimatized for 1 week in the laboratory animal facility maintained at constant temperature (22 ± 2°C) and humidity (55 ± 5%) with a 12 h light/dark cycle. Food and water were provided ad libitum. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Measurement of vasoconstriction in isolated aortic rings
After rats were humanely decapitated and exsanguinated, the thoracic aorta was carefully isolated and cut into ring segments in lengths of 3–4 mm on ice. Aortic rings without endothelium were prepared by gently rubbing the intimal surface of the aortic rings with a cotton swab. Complete removal of endothelium was checked by the absence of relaxation response to ACh challenge. The rings were then mounted in organ baths filled with Krebs-Ringer solution (KR solution; 115.5 mM NaCl, 4.6 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2, 25 mM NaHCO3 and 11.1 mM glucose, pH 7.4) continuously saturated with 95% O2/5% CO2 mixture gas and maintained at 37°C. The change in tension was measured with Grass FT03 force transducers (Grass Instrument Co., Quincy, MA, USA) and recorded using the AcqKnowledge III (BIOPAC Systems Inc., Goleta, CA, USA). To investigate the effect on vasoconstriction, the aortic rings were pretreated with emodin or vehicle (DMSO) and vasoconstriction was initiated by the cumulative addition of phenylephrine, 5-HT, ET-1 or PDBu. In order to examine whether emodin might affect the stage of tension maintenance, that is, the tonic tension, emodin was treated 10 min after induction of vasoconstriction by phenylephrine (10−5 M).
Measurement of lactate dehydrogenase leakage
To examine the non-specific cytotoxicity of emodin, the extent of LDH leakage from aortic rings was measured. After incubation with emodin for 2 h and lysophosphatidylcholine for 24 h, aliquots were collected. Fifty microlitres of aliquot was added to 1 mL of Tris-EDTA-NADH buffer (56 mM Tris(hydroxymethyl)aminomethane, 5.6 mM EDTA, 0.17 mM β-NADH, pH 7.4) and then incubated for 10 min at 37°C. After incubation, 100 μL of prewarmed 14 mM pyruvate solution 37°C was added. The reduction in absorbance at 340 nm by the conversion of NADH to NAD+ was measured for 1 min to evaluate LDH activity in the aliquots.
TUNEL staining of aortic rings and histopathological assessment
Aortic rings were placed in MEM containing 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin and emodin, monomethylarsonous acid or DMSO was added and the rings incubated in a 95% air/5% CO2 incubator for 24 h at 37°C. After this incubation, aortic rings were fixed in buffered formalin solution (10%) and embedded in paraffin. The TUNEL assay was performed using a commercial kit according to the manufacturer's instruction (Chemicon International, Temecula, CA, USA). The embedded tissue was cut into sections, 4 μm thick, and placed on an adhesive slide. The sections were deparaffinized by washing with xylene following serial dehydration with ethanol (100, 95, 80 and 70%). Dehydrated sections were treated with 0.3% H2O2 to quench endogenous peroxidase activity followed by 20 μg·mL−1 DNase-free proteinase K to retrieve antigenic epitopes. After that, the samples were treated with terminal deoxynucletidyl transferase enzyme reagent for 1 h at 37°C to label free 3′-OH termini with digoxigenin-dUTP. To detect incorporated digoxigenin-conjugated nucleotides, HRP-conjugated antidigoxigenin antibody and 3,3'-diaminobenzidine (DAB) was used. The samples were treated with anti-digoxigenin-peroxidase for 30 min at room temperature followed by DAB development. They were then counterstained with Mayer's haematoxylin. The dehydrated samples were cleared in xylene and mounted.
Intracellular and extracellular Ca2+ pathways
To examine the effect of emodin on the contraction mediated by release of the intracellular Ca2+ store, vasoconstriction was induced in Ca2+-free conditions. After the aortic rings without endothelium had been treated with emodin or vehicle for 2 h, the KR solution was replaced with Ca2+-free KR solution (120.0 mM NaCl, 5.9 mM KCl, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 25.0 mM NaHCO3, 2 mM EGTA and 11.5 mM glucose, pH 7.4) containing emodin, and phenylephrine (10−5 M) was added to initiate vasoconstriction. For selective activation of SOCC, the aortic rings without endothelium were pretreated with 1 μM thapsigargin (an inhibitor of sarcoplasmic reticulum Ca2+-ATPase) for 90 min in Ca2+-free KR solution to deplete the intracellular Ca2+ store. Aortic rings were then treated with emodin or vehicle for 2 h and the bath solution was exchanged for KR solution containing 2.5 mM Ca2+ to induce SOCC-mediated contraction. To investigate the effect of emodin on the contraction induced by an influx of extracellular Ca2+, Bay K8644, an L-type Ca2+ channel opener was used. Bay K8644 was added cumulatively in 15 mM K+ buffer solution (105 mM NaCl, 15 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2, 25 mM NaHCO3, 0.026 mM EDTA and 11.1 mM glucose, pH 7.4) to initiate vasoconstriction.
Calcium measurement in live cells
The intracellular calcium level was measured using a fluorometric method, which employed fura-2 and digital imaging as described previously (Lee et al., 2006). After the endothelium and adventitia were removed, aortic rings were chopped finely and smooth muscle cells were liberated from the tissue using collagenase and elastase (Worthington Biochemical Corp., Lakewood, NJ, USA). Vascular smooth muscle cells grown on coverslips were treated with emodin for 2 h. To load fura-2, cells were incubated in physiological salt solution (PSS; 140 mM NaCl, 5 mM KCl, 5 mM NaHCO3, 1.8 mM CaCl2, 1.4 mM MgCl2, 1.2 mM NaH2PO4, 11.5 mM glucose and 10 mM HEPES, pH 7.4) containing 1 μM fura-2/AM and 1% BSA for 60 min. Coverslips were mounted in a superfusion chamber on the microscope stage and were superfused with PSS (2 mL·min−1). All experiments were performed at 33°C. Cells were imaged with a Nikon Eclipse Ti-U inverted microscope equipped with a S Fluor 40X (N.A. 1.30, oil) objective lens (Nikon, Melville, NY, USA) and an evolve EMCCD camera (Photometrics, Tucson, AZ, USA). Illumination was provided by a Sutter DG-4 filter changer (Sutter Instruments, Novato, CA, USA). Excitation and emission wavelengths used for fura-2 were 340/380 and 535 nm respectively. Images were acquired and analysed with a Meta Imaging System (Molecular Devices, West Chester, PA, USA).
Western blotting
After the aortic rings without endothelium had been treated with emodin or vehicle for 2 h, 10−5 M phenylephrine was added for 2 min and the reaction was terminated by addition of acetone containing 10% TCA and 10 mM DTT precooled to −80°C. The aortic rings were washed three times with ice-cold acetone containing 10 mM DTT and then lyophilized at −80°C overnight. Aortic rings were homogenized in RIPA buffer (25 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate and 0.1% SDS, pH 7.6) containing 1% Halt protease and a phosphatase inhibitor cocktail and lysed at 4°C for 1 h with frequent vortexing. The protein extracts (20 μg) were subjected to Western blot analysis using SDS-PAGE and antibodies specific against MLC, pMLC, MYPT1, pMYPT1 (for phosphorylation site at Thr835), CPI-17, pCPI-17, PKCα, pPKCα, PKCδ and pPKCδ. The bands were detected and analysed with ChemidocTM XRS system (Bio-Rad, Hercules, CA, USA).
Statistical analysis
All the data are shown as mean ± SEM and were subjected to one-way anova followed by Duncan's multiple-ranged tests to determine which means were significantly different from the control. Statistical analysis was performed using the Statistical Package for the Social Sciences (SPSS) software (SPSS, Inc., Chicago, IL, USA). In all cases, a P value <0.05 was taken to indicate significant difference.
Reagents
The following chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, USA): emodin, phenylephrine, endothelin-1 (ET-1), 5-HT creatinine sulfate, NADH, pyruvate, (-)-(S)-Bay K8644, thapsigargin, phorbol 12,13-dibutyrate (PDBu), DMSO, trichloroacetic acid (TCA), DTT. Fura-2/AM was obtained from Molecular Probes (Eugene, OR, USA), and reagents and media used in cell culture were purchased from Gibco Co. (Calsbad, CA, USA). MLC antibody, phospho-myosin light chain (pMLC) antibody, MYPT1 antibody, pMYPT1 antibody, PKCα antibody, pPKCδ antibody and HRP-conjugated anti-rabbit secondary antibody were from Cell Signaling Technology, Inc. (Danvers, MA, USA). pPKCα antibody and PKCδ antibody were from Abcam (Cambridge, UK). Antibody to CPI-17 was from Upstate Biotechnology, Inc. (Charlottesville, VA, USA) and pCPI-17 was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Halt protease and phosphatase inhibitor cocktail and RIPA buffer were from Thermo Scientific (Rockford, IL, USA). All other reagents used were of the highest purity available.
Results
To find natural compounds with anti-hypertensive activities, rat isolated aortic rings were pretreated with 70% ethanol extracts of eight herbs, that included Achyranthes japonica, Atractylodes japonica, Aucklandia lappa, Foeniculum vulgare, Lycium chinense, Kalopanax pictus, Polygonatum stenophyllum, Polygonum multiflorum, Rehmannia glutinosa, at 250 μg·mL−1 for 30 min and a contractile agonist, phenylephrine, was cumulatively added to induce contraction. As shown in Figure 1A, the extract of Polygonum multiflorum exhibited strong inhibitory activities on phenylephrine-induced vasoconstriction. To identify the active ingredient for this effect, known components of Polygonum multiflorum were tested and the vasoconstriction was examined. Among the six compounds, emodin, an anthraquinone derivative, was the most effective (Figure 1B); it completely inhibited phenylephrine-induced vasoconstriction at a relatively low concentration, 10 μM (Figure 1C). The phenylephrine-induced vasoconstriction was inhibited by emodin in concentration- and time-dependent manner (Figure 1C and D), while the vehicle had no effect up to 2 h. We examined the effects of emodin on the vasoconstriction induced by other contractile agonists. As shown in Figure 2A and B, 5-HT- and ET-1-induced vasoconstrictions were also suppressed by emodin suggesting that it affects common contractile pathways following the activation of adrenoceptors, 5-HT2A and ETA receptors. These effects were not from non-specific cytotoxic or apoptotic effectss (Figure 2C and D). In addition, a role for reactive oxygen species (ROS) could be excluded by the failure of ROS-removal, using catalase and superoxide dismutase, to reverse the effects of emodin (data not shown).
Figure 1.
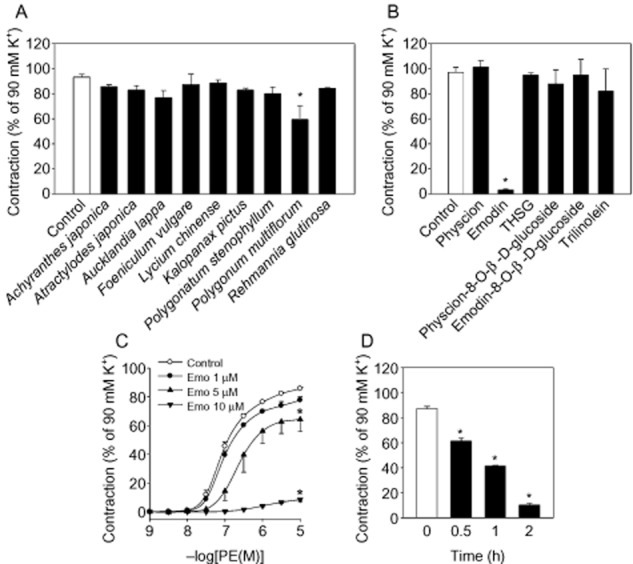
Effects of herbal extract on phenylephrine (PE)-induced vasoconstriction in aortic rings with intact endothelium. (A) After 250 μg·mL−1 of herbal extracts were treated to aortic rings with endothelium for 30 min, phenylephrine-induced contraction was measured. (B) Components of Polygonum multiforum (100 μM) were treat to aortic rings with endothelium for 30 min and phenylephrine-induced contraction were measured. THSG; 2,3,5,4-tetrahydroxystilbene-2-O-β-D-glucoside. *Represents significant difference from control (Student's t-test, P < 0.05). After emodin was treated to aortic rings with endothelium, (C) concentration- (with 2 h incubation) and (D) time-dependent effects (at 10 μM) on phenylephrine-induced vasoconstriction were obtained. Values are mean ± SEM of three to six independent experiments. *Represents significant differences from control (anova and Duncan's post hoc analysis, P < 0.05).
Figure 2.
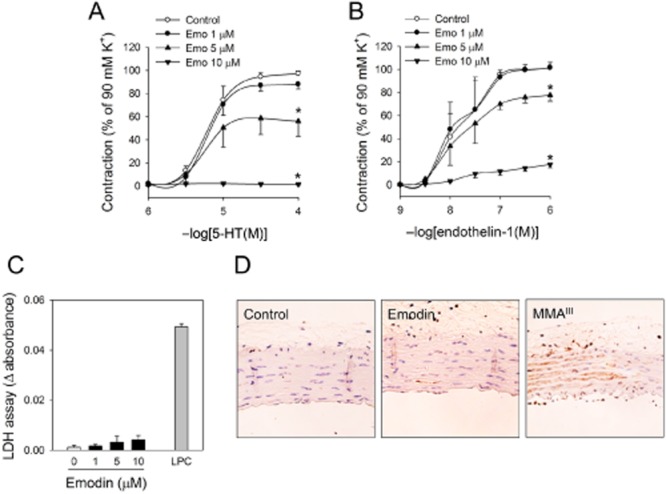
Effects of emodin on agonist-induced vasoconstriction and nonspecific cytotoxicities in aortic rings. Aortic rings with endothelium were pretreated with emodin for 2 h, then (A) 5-HT- and (B) ET-1-induced contractions were measured. (C) Cytotoxicity was determined by LDH leakage in emodin-treated aortic rings. Lysophosphatidylcholine (LPC; 100 μM) was used as positive control. (D) Aortic rings with endothelium were treated with emodin for 24 h, apoptosis was measured using TUNNEL assay. Monomethylarsonous acid (MMA, 5 μM) was used as positive control. Values are mean ± SEM of three to four independent experiments. *Represents significant differences from control (anova and Duncan's post hoc analysis, P < 0.05).
Vasoconstriction is achieved by the balanced sum of the vasodilator and contractile responses of endothelial cells and smooth muscle cells constituting the blood vessel. Inhibition of vasoconstriction by emodin was retained in endothelium-denuded aortic rings suggesting that it mainly affects the contractile function of vascular smooth muscle cells (Figure 3A). Contraction of vascular smooth muscle is accomplished in two distinct phases: phasic tension and tonic tension. Phasic tension, the generation of contractile force, is initiated by a rapid increase in cytosolic calcium, which is supplied from an intracellular calcium store (ICS), voltage-operated (VOCC) or store-operated calcium channels (SOCC). Emodin inhibited phasic tension without affecting ICS, VOCC and SOCC activation (Figure 3B), suggesting that emodin affected the contractile events downstream of calcium increases. Further supporting this, emodin did not affect agonist-induced increases in intracellular calcium in primary vascular smooth muscle cells (Figure 3C).
Figure 3.
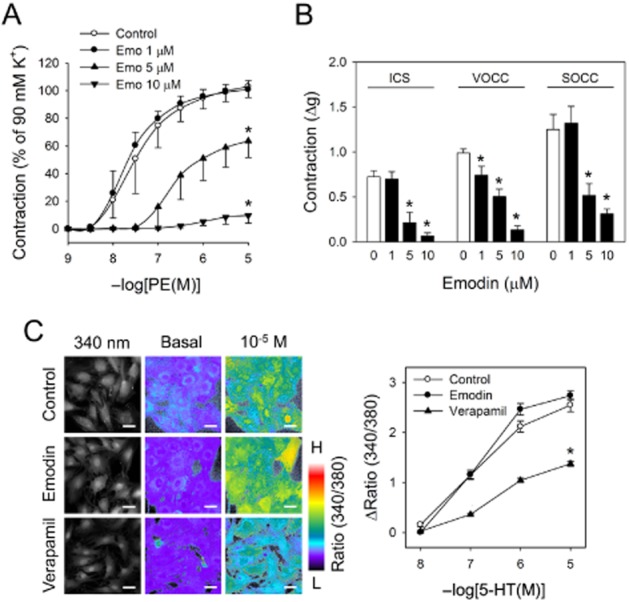
Effects of emodin on calcium mobilization and vasoconstriction. (A) Aortic rings without endothelium were pretreated with emodin for 2 h, phenylephrine-induced contraction was measured. (B) Effects of emodin on ICS-, VOCC- and SOCC-mediated vasoconstriction were measured in aortic rings without endothelium. (C) Effects of emodin (10 μM, 2 h) on calcium were observed in primary smooth muscle cells with a fluorometric method employing fura-2/AM. Verapamil was used as positive control. Values are mean ± SEM of three to four independent experiments. *Represents significant difference from control (anova and Duncan's post hoc analysis, P < 0.05).
We examined if emodin could affect tonic tension. After vasoconstriction was induced with phenylephrine, emodin or vehicle was added. As shown in Figure 4A, phenylephrine induced a long-lasting contraction of aortic rings, but the treatment with emodin shortened it significantly, suggesting that emodin may affect tonic tension. To confirm this, pMLC was measured in the presence and absence of emodin (10 μM). Emodin treatment significantly reduced the level of pMLC (Figure 4B). In the tonic tension, the pMLC level is regulated by the phosphorylation of MYPT1, which is the inhibitory regulatory subunit for MLC-phosphatase. A decreased pMYPT1 results in insufficient inhibition of MLC-phosphatase and an increase in pMLC. We observed that in the presence of emodin, phosphorylation of MYPT1 was almost completely prevented (Figure 4C), suggesting that emodin affects MYPT1 phosphorylation. MYPT1 phosphorylation is regulated by PKC-dependent CPI-17. Next, we examined the effects of emodin on CPI-17. Phenylephrine induced a robust activation of CPI-17, which was significantly attenuated by emodin, suggesting that emodin affects the PKC-CPI-17 pathways (Figure 4D).
Figure 4.
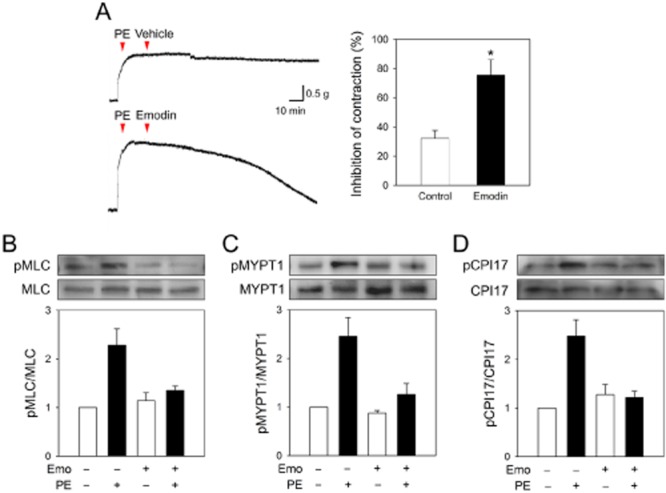
Effects of emodin on tonic tension, MLC phosphorylation, MYPT1 phosphorylation and CPI-17 phosphorylation in aortic rings without endothelium. (A) After aortic rings without endothelium were precontracted with phenylephrine (10−5 M, 15 min), they were treated with emodin (10 μM) and the change in contractility was measured after 2 h. After aortic rings without endothelium were treated with emodin (10 μM) for 2 h, aortic rings were stimulated with phenylephrine (10−5 M) for 2 min, and protein was extracted from aortic tissues. (B) MLC phosphorylation, (C) MYPT1 phosphorylation and (D) CPI-17 phosphorylation were determined by Western blotting. Values are mean ± SEM of three to four independent experiments. *Represents significant difference from control (Student's t-test, P < 0.05).
To confirm that emodin affects PKC-CPI-17 pathways PDBu, a PKC activator, was introduced. Emodin significantly inhibited the vasoconstriction and CPI-17 phosphorylation induced by PDBu (Figure 5A and B). To identify the PKC isoform affected by emodin, we examined the activation of PKC isoforms. Conspicuously, while a calcium-dependent typical PKC isoform, PKCα was not affected, emodin selectively suppressed the activation of a novel calcium-independent PKC isoform, PKCδ; this represents a potential new mode of action for the regulation of tonic tension and anti-hypertensive agents (Figure 5C and D, Supporting Information Figure S1).
Figure 5.
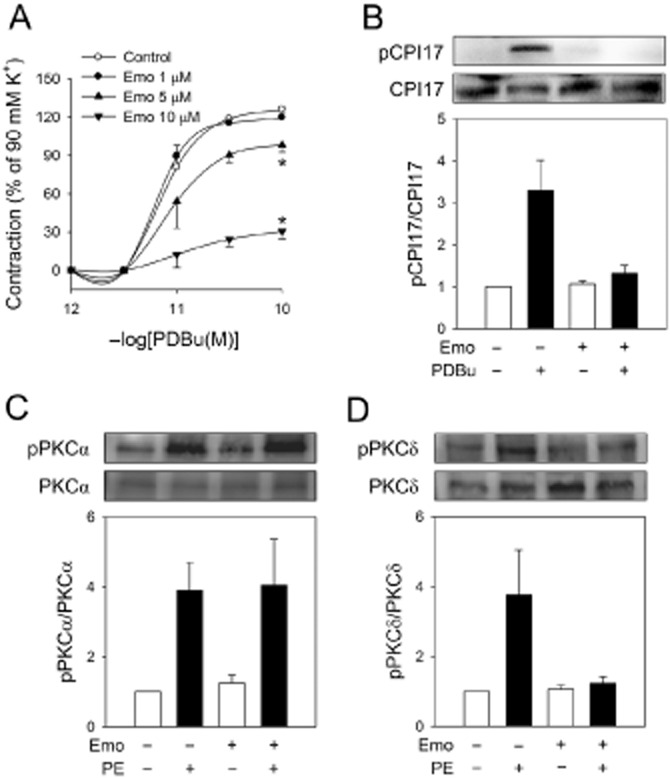
Effects of emodin on PKCδ activation in aortic rings without endothelium. (A) Aortic rings without endothelium were pretreated with emodin for for 2 h, PDBu-induced contraction was then measured. (B) Aortic rings without endothelium were pretreated with emodin (10 μM) for 2 h, aortic rings were stimulated with PDBu (10−10 M) for 2 min and protein was extracted from the aortic tissues. CPI-17 phosphorylation was determined by western blotting. (C, D) Aortic rings without endothelium were treated with emodin (10 μM) for 2 h, they were then stimulated with phenylephrine (10−5 M) for 2 min, and protein was extracted from aortic tissues. (C) PKCα and (D) PKCδ activation was determined by PKC phosphorylation through Western blotting. Values are mean ± SEM of three to four independent experiments. *Represents significant differences from control (anova and Duncan's post hoc analysis, P < 0.05).
Discussion
Here we demonstrated that a natural anthraquinone derivative, emodin attenuates tonic tension and suppresses calcium sensitization through the blockade of PKCδ- mediated MLC-phosphatase inhibition. Importantly, emodin did not affect agonist-stimulated cytosolic calcium increase or phasic tension, but selectively inhibited the phosphorylation of CPI-17 and MYPT1, so preventing MLC-phosphatase activities, which resulted in a decreased level of pMLC. With this distinct mode of action, emodin inhibited phenylephrine-, ET-1- and 5-HT-induced vasoconstriction; thus revealing a new therapeutic target for the development of anti-hypertensive agents.
Conventional anti-hypertensive agents have focused on the modulation of phasic tension, but many cardiovascular diseases stem from the dysregulation of tonic tension and calcium sensitization (de Godoy and Rattan, 2011). Abnormally prolonged vasoconstriction or excessively sensitive vasoconstrictor responses are frequently observed in many cardiovascular diseases, like pulmonary artery hypertension, vasospasm and right ventricular hypertrophy, suggesting that the modulation of calcium sensitization may lower high blood pressure in these diseases. Indeed, modulators of calcium sensitization like the ROCK inhibitor, fasudil, are successfully going through clinical trials for the treatment of pulmonary hypertension (Fukumoto et al., 2005) and cerebral vasospasm (Zhao et al., 2006), supporting the fact that modulation of tonic tension may provide new therapeutic treatments for refractory hypertensive diseases.
At a similar intracellular calcium level, agonists induce a higher contraction than that achieved by simple membrane depolarization (DeFeo and Morgan, 1985). This phenomenon, agonist-induced force enhancement, has been coined as calcium sensitization (Kitazawa et al., 1991; Uehata et al., 1997). Importantly, there is increasing evidence suggesting that calcium sensitization has broad pathological implications in muscle-related gastrointestinal, respiratory and cardiovascular diseases. Dysregulation of calcium sensitization is involved in the pathogenesis of rectoanal incontinence, certain forms of constipation, recurrent anal fissures, haemorrhoids (de Godoy and Rattan, 2011), erectile dysfunction, asthma, vasospasm and congestive heart failure (Brozovich, 2002), underlining the therapeutic importance of modulators of calcium sensitization. We believe that it would be interesting to examine the effects of emodin in these diseases in the future.
Calcium sensitization is mainly accomplished by the inhibition of MLC-phosphatase. MLC-phosphatase modulates the level of pMLC and tension generation. The inhibition of MLC-phosphatase activity generates a stronger contractile force, while its stimulation attenuates the vasoconstriction. MLC-phosphatase is composed of three subunits; a small ∼20-kDa subunit with unidentified function, a ∼38-kDa catalytic subunit (protein phosphatase 1c), and a large regulatory subunit, myosin-targeting subunit (MYPT1) of 110–133 kDa (Brozovich, 2002). Phosphorylation of MYPT1 inhibits the catalytic activities of MLC-phosphatase (Ichikawa et al., 1996). Phosphorylation of MYPT1 and resultant inhibition of MLC-phosphatase occurs through two pathways, Rho-ROCK and PKC-CPI-17. Translocation of Rho A from the cytosol to the cell membrane leads to spatial activation of ROCK, which phosphorylates MYPT1. PKC activation following contractile agonist-binding to Gq-coupled receptors results in the phosphorylation of CPI-17 at Thr38, which can phosphorylate MYPT1 potently. Many PKC isoforms including PKCα, PKCδ and PKCε are involved in the phosphorylation of CPI-17, but PKCδ is known to be threefold more specific for CPI-17 than PKCα or ROCK suggesting that PKCδ has an important role in the phosphorylation of CPI-17 (Eto et al., 2001). In addition, the PKC-CPI-17 pathway is considered to be dominant in phenylephrine-induced tonic tension in blood vessels (Kitazawa and Kitazawa, 2012).
PKCδ is a representative member of novel PKC isoforms (PKCδ, ε, ϕ, η). It is ubiquitously expressed and plays a critical role in cytoskeleton maturation, cellular growth, differentiation and apoptosis (Wu-Zhang et al., 2012). A recent study demonstrated that PKCδ is important in angiotensin II-mediated extracellular matrix collagen synthesis and cardiac fibrosis (Chintalgattu and Katwa, 2009), suggesting that it may also be widely involved in other cardiovascular diseases. In contrast to classic PKCs (PKCα, βI, βII, γ), PKCδ is calcium-independent, but is activated by DAG from receptor-mediated hydrolysis of membrane inositol phospholipids (Steinberg, 2004). Direct activation of PKCδ by stimulation of the insulin receptor in skeletal muscle was also observed (Braiman et al., 2001), suggesting that PKCδ is regulated by a distinct mode of action.
In contrast to the remarkable advance in the drug development of ROCK inhibitors, few PKCδ inhibitors are currently under active development. This may be due to a lack of information on the pharmacophore or selective inhibitors for PKCδ, which might be rectified by insights gained from the studies on natural products. Rottlerin, which is a natural compound isolated from Mallotus philippinensis, was originally identified as a specific inhibitor of PKCδ (Gschwendt et al., 1994), but recent reports showed that rottlerin was not effective at inhibiting PKCδ activity (Soltoff, 2007). Instead, it was suggested that rottlerin may have off-target effects and just elicit cellular changes mimicking those caused by the direct inhibition of PKCδ. Emodin inhibited CPI-17 and MYPT1 phosphorylation induced by PDBu as well as that by phenylephrine without affecting PKCα or RhoA activities (data not shown). Considering that PDBu binds to the phorbol ester-binding domain of PKC, causing direct activation of PKC catalytic domain, emodin appears to directly inhibit the catalytic activity of PKCδ, rather than modulating the receptor-binding domain or Gq-mediated DAG production. However, further studies employing purified PKC isoforms are needed to elucidate the selectivity and mode of inhibition of emodin against PKCδ.
Our data show for the first time, that emodin may be a selective and potent inhibitor of PKCδ without affecting PKCα activation. Emodin is widely available in many herbal plants, such as Polygonum multiflorum, Rheum Senna obtusifolia, Fallopia japonica and Rheum palmatum. Various pharmacological effects of emodin have been reported, including anti-diabetic (Feng et al., 2010), anti-tumour (Muto et al., 2007), neuroprotective (Kuo et al., 2009) and anti-inflammatory effects (Meng et al., 2010). These therapeutic activities have been suggested to result from inhibition of 11β-hydroxysteroid dehydrogenase type 1, JAK2 or NF-κB activation. In the present study, we demonstrated that emodin can inhibit PKCδ activity, providing a new mode of action for its therapeutic effects.
Conclusion
In conclusion, our study demonstrated that emodin suppresses tonic tension by suppressing the activity of PKCδ and CPI-17-mediated MLC-phosphatase inhibition (see Figure 6). We believe that this new mode of action for the suppression of tonic tension and structural insights into PKCδ inhibition revealed by emodin may be instrumental for the development of new modulators of tonic tension and for the treatment of hypertensive diseases.
Figure 6.
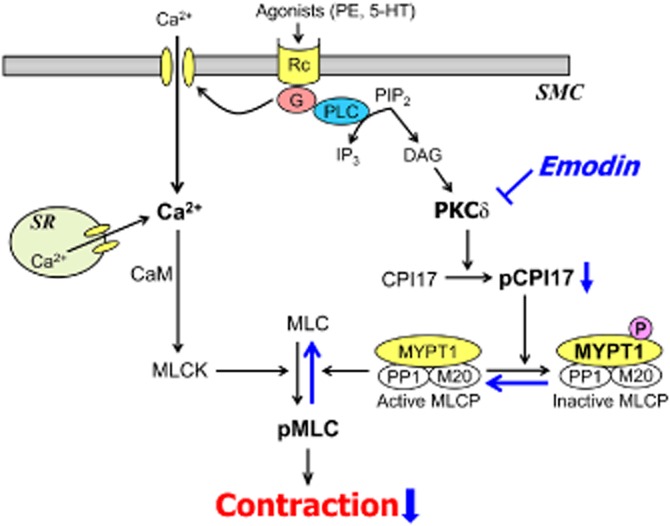
Suggested mechanism for the inhibitory effect of emodin on agonist-induced vasoconstriction.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF), grant funded by the Korean government (MSIP) (No. 2007-0056817).
Glossary
- DAB
3,3'-diaminobenzidine
- ET-1
endothelin-1
- ICS
intracellular calcium store
- KR solution
Krebs-Ringer solution
- MLC20
myosin light chain
- MYPT1
myosin-targeting subunit
- NCEs
new chemical entities
- PDBu
phorbol 12,13-dibutyrate
- pMLC
phospho-MLC
- ROCK
RhoA–Rho-associated PK
- SOCC
store-operated calcium channel
- TCA
trichloroacetic acid
- VOCC
voltage-operated calcium channel
- VSMC
vascular smooth muscle cell
Author contributions
K.M.L., J.H.K. and K.K. designed the experiments, analysed the data and wrote the paper; J.Y.N. and S.K. performed additional experiments for the revision; J.M.P and M.Y.L. performed calcium experiments in live cells; O.N.B. analysed the data and edited the manuscript. J.H.C. supervised the study.
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12804
Figuer 1 Effects of emodin on phenylephrine-induced activation of PKCα and PKCδ. Phosphorylation of PKCα was determined after aortic rings were treated with or without emodin and phenylephrine. To confirm the specificity of antibodies against PKCα and PKCδ, the whole blot was obtained with molecular weight markers.
References
- Aksoy MO, Mras S, Kamm KE, Murphy RA. Ca2+, cAMP, and changes in myosin phosphorylation during contraction of smooth muscle. Am J Physiol. 1983;245:C255–C270. doi: 10.1152/ajpcell.1983.245.3.C255. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol. 2013a;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1862. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braiman L, Alt A, Kuroki T, Ohba M, Bak A, Tennenbaum T, et al. Insulin induces specific interaction between insulin receptor and protein kinase C delta in primary cultured skeletal muscle. Mol Endocrinol. 2001;15:565–574. doi: 10.1210/mend.15.4.0612. [DOI] [PubMed] [Google Scholar]
- Brozovich FV. Myosin light chain phosphatase: it gets around. Circ Res. 2002;90:500–502. doi: 10.1161/01.res.0000014224.43774.03. [DOI] [PubMed] [Google Scholar]
- CDC. Vital signs: prevalence, treatment, and control of hypertension – United States, 1999–2002 and 2005–2008. MMWR Morb Mortal Wkly Rep. 2011;60:103–108. [PubMed] [Google Scholar]
- Chiba Y, Ueno A, Shinozaki K, Takeyama H, Nakazawa S, Sakai H, et al. Involvement of RhoA-mediated Ca2+ sensitization in antigen-induced bronchial smooth muscle hyperresponsiveness in mice. Respir Res. 2005;6:4. doi: 10.1186/1465-9921-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintalgattu V, Katwa LC. Role of protein kinase C-delta in angiotensin II induced cardiac fibrosis. Biochem Biophys Res Commun. 2009;386:612–616. doi: 10.1016/j.bbrc.2009.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ G, Wingard C. Calcium sensitization as a pharmacological target in vascular smooth-muscle regulation. Curr Opin Investig Drugs. 2005;6:920–933. [PubMed] [Google Scholar]
- DeFeo TT, Morgan KG. Calcium-force relationships as detected with aequorin in two different vascular smooth muscles of the ferret. J Physiol. 1985;369:269–282. doi: 10.1113/jphysiol.1985.sp015900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, Kitazawa T, Yazawa M, Mukai H, Ono Y, Brautigan DL. Histamine-induced vasoconstriction involves phosphorylation of a specific inhibitor protein for myosin phosphatase by protein kinase C alpha and delta isoforms. J Biol Chem. 2001;276:29072–29078. doi: 10.1074/jbc.M103206200. [DOI] [PubMed] [Google Scholar]
- Feng Y, Huang SL, Dou W, Zhang S, Chen JH, Shen Y, et al. Emodin, a natural product, selectively inhibits 11beta-hydroxysteroid dehydrogenase type 1 and ameliorates metabolic disorder in diet-induced obese mice. Br J Pharmacol. 2010;161:113–126. doi: 10.1111/j.1476-5381.2010.00826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, et al. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart. 2005;91:391–392. doi: 10.1136/hrt.2003.029470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Godoy MA, Rattan S. Role of rho kinase in the functional and dysfunctional tonic smooth muscles. Trends Pharmacol Sci. 2011;32:384–393. doi: 10.1016/j.tips.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, et al. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- Hajjar I, Kotchen JM, Kotchen TA. Hypertension: trends in prevalence, incidence, and control. Annu Rev Public Health. 2006;27:465–490. doi: 10.1146/annurev.publhealth.27.021405.102132. [DOI] [PubMed] [Google Scholar]
- Ichikawa K, Ito M, Hartshorne DJ. Phosphorylation of the large subunit of myosin phosphatase and inhibition of phosphatase activity. J Biol Chem. 1996;271:4733–4740. doi: 10.1074/jbc.271.9.4733. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Kitazawa K. Size-dependent heterogeneity of contractile Ca2+ sensitization in rat arterial smooth muscle. J Physiol. 2012;590:5401–5423. doi: 10.1113/jphysiol.2012.241315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Masuo M, Somlyo AP. G protein-mediated inhibition of myosin light-chain phosphatase in vascular smooth muscle. Proc Natl Acad Sci U S A. 1991;88:9307–9310. doi: 10.1073/pnas.88.20.9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Eto M, Woodsome TP, Brautigan DL. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J Biol Chem. 2000;275:9897–9900. doi: 10.1074/jbc.275.14.9897. [DOI] [PubMed] [Google Scholar]
- Kuo TC, Yang JS, Lin MW, Hsu SC, Lin JJ, Lin HJ, et al. Emodin has cytotoxic and protective effects in rat C6 glioma cells: roles of Mdr1a and nuclear factor kappaB in cell survival. J Pharmacol Exp Ther. 2009;330:736–744. doi: 10.1124/jpet.109.153007. [DOI] [PubMed] [Google Scholar]
- Lee MY, Song H, Nakai J, Ohkura M, Kotlikoff MI, Kinsey SP, et al. Local subplasma membrane Ca2+ signals detected by a tethered Ca2+ sensor. Proc Natl Acad Sci U S A. 2006;103:13232–13237. doi: 10.1073/pnas.0605757103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. Executive summary: heart disease and stroke statistics – 2010 update: a report from the American Heart Association. Circulation. 2010;121:948–954. doi: 10.1161/CIRCULATIONAHA.109.192666. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng G, Liu Y, Lou C, Yang H. Emodin suppresses lipopolysaccharide-induced pro-inflammatory responses and NF-kappaB activation by disrupting lipid rafts in CD14-negative endothelial cells. Br J Pharmacol. 2010;161:1628–1644. doi: 10.1111/j.1476-5381.2010.00993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto A, Hori M, Sasaki Y, Saitoh A, Yasuda I, Maekawa T, et al. Emodin has a cytotoxic activity against human multiple myeloma as a Janus-activated kinase 2 inhibitor. Mol Cancer Ther. 2007;6:987–994. doi: 10.1158/1535-7163.MCT-06-0605. [DOI] [PubMed] [Google Scholar]
- Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981–2002. J Nat Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics – 2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamanca DA, Khalil RA. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem Pharmacol. 2005;70:1537–1547. doi: 10.1016/j.bcp.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh S, Kreutz R, Wilm C, Ganten D, Pfitzer G. Augmented agonist-induced Ca(2+)-sensitization of coronary artery contraction in genetically hypertensive rats. Evidence for altered signal transduction in the coronary smooth muscle cells. J Clin Invest. 1994;94:1397–1403. doi: 10.1172/JCI117475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, et al. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res. 2003;92:411–418. doi: 10.1161/01.RES.0000059987.90200.44. [DOI] [PubMed] [Google Scholar]
- Seok YM, Baek I, Kim YH, Jeong YS, Lee IJ, Shin DH, et al. Isoflavone attenuates vascular contraction through inhibition of the RhoA/Rho-kinase signaling pathway. J Pharmacol Exp Ther. 2008;326:991–998. doi: 10.1124/jpet.108.138529. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Seto M, Katsumata N, Amano M, Kozai T, Yamawaki T, et al. Rho-kinase-mediated pathway induces enhanced myosin light chain phosphorylations in a swine model of coronary artery spasm. Cardiovasc Res. 1999;43:1029–1039. doi: 10.1016/s0008-6363(99)00144-3. [DOI] [PubMed] [Google Scholar]
- Soltoff SP. Rottlerin: an inappropriate and ineffective inhibitor of PKCdelta. Trends Pharmacol Sci. 2007;28:453–458. doi: 10.1016/j.tips.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Srinivas G, Babykutty S, Sathiadevan PP, Srinivas P. Molecular mechanism of emodin action: transition from laxative ingredient to an antitumor agent. Med Res Rev. 2007;27:591–608. doi: 10.1002/med.20095. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem J. 2004;384:449–459. doi: 10.1042/BJ20040704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Wu-Zhang AX, Murphy AN, Bachman M, Newton AC. Isozyme-specific interaction of protein kinase Cdelta with mitochondria dissected using live cell fluorescence imaging. J Biol Chem. 2012;287:37891–37906. doi: 10.1074/jbc.M112.412635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhou D, Guo J, Ren Z, Zhou L, Wang S, et al. Effect of fasudil hydrochloride, a protein kinase inhibitor, on cerebral vasospasm and delayed cerebral ischemic symptoms after aneurysmal subarachnoid hemorrhage. Neurol Med Chir (Tokyo) 2006;46:421–428. doi: 10.2176/nmc.46.421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figuer 1 Effects of emodin on phenylephrine-induced activation of PKCα and PKCδ. Phosphorylation of PKCα was determined after aortic rings were treated with or without emodin and phenylephrine. To confirm the specificity of antibodies against PKCα and PKCδ, the whole blot was obtained with molecular weight markers.