

Abstract
For almost half a century, the structure of the full-length Bacillus thuringiensis (Bt) insecticidal protein Cry1Ac has eluded researchers, since Bt-derived crystals were first characterized in 1965. Having finally solved this structure we report intriguing details of the lattice-based interactions between the toxic core of the protein and the protoxin domains. The structure provides concrete evidence for the function of the protoxin as an enhancer of native crystal packing and stability.
Keywords: insecticidal toxin, Bacillus thuringiensis, Cry1A, structure of full length toxin, insecticidal toxin mode of action, natural crystal packing
Introduction
Bacillus thuringiensis (Bt) is a ubiquitous Gram-positive soil bacterium similar to Bacillus cereus or Bacillus subtilis. Bacillus species sporulate to survive harsh conditions; the hardy endospores survive for decades or even centuries.1 The distinguishing feature of Bt is that during sporulation it produces large amounts of insecticidal proteins (parasporins, or Cry proteins) that crystallize within the mother cell, next to the spore. There are thousands of individual toxin proteins discovered to-date, of which the majority belong to the “three-domain” toxin family: insecticidal Bt proteins that contain three structural domains within the toxic core. This is the most populous clan with thousands of known members, of which more than 50% are produced by their parent Bt strains as ∼1200-residue protoxins comprised a proteolytically labile C-terminal segment (sometimes referred to as the protoxin domain) and an N-terminal ∼600-residue segment that encodes the three-domain toxic core.1–3 The role of the protoxin is presently unclear, especially since it is dispensable for toxicity. Cry protein crystals persist together with Bt spores until a suitable insect ingests them. In the gut of the insect (e.g. a Lepidopteran larva) the crystals dissolve and protoxins become activated by proteolysis. Activated toxic core proteins bind cognate receptors on the gut brush border membrane, undergo oligomerization and ultimately form transmembrane pores that cause cell death by osmotic shock.4,5 Toxin-triggered gut injury allows Bt endospores to germinate in the welcoming environment of the insect hemolymph, causing massive infection and the eventual death of the host. Bt continue to thrive in the carcass until its nutrients are depleted, upon which time the pathogen undergoes sporulation and the cycle can repeat.6,7
Various Bt strains have evolved exquisitely specific and highly potent insecticidal proteins active on numerous commercial crop pests. The use of Bt crystal/spore mixtures as effective biopesticides began almost a century ago and more recently transgenic plants bearing Bt Cry protein genes have revolutionized agriculture by reducing the need for hazardous and expensive chemical insecticides.7–9 Changing agricultural practices and the emergence of resistant pest insects define the need for the discovery and design of new and improved Cry proteins—which in turn drives the need for structural and mechanistic studies.10 Despite decades of research, resulting in numerous structures of Cry toxic cores, no structures of full-length Cry protoxins are reported to-date. We have therefore undertaken to solve the structure of a full-length Cry protoxin in order to shed some light on the possible functional relevance of the protoxin domain. We chose Cry1Ac as the target for our investigation because it is one of the first Bt toxins to be discovered, and as such is also the most characterized Cry protein with respect to its biological function and mechanism of action. It is also the first commercialized transgenic insecticide with excellent activity against specific Lepidopteran pests.8
Materials and Methods
It was necessary to mutate the last 14 (out of 16 total) cysteine residues present in Cry1Ac-FL in order to minimize the pernicious aggregation of the solubilized protein. Construction of the mutant gene began with a clone of the full-length Cry1Ac, from which we amplified several PCR fragments with the mutated residues positioned at the ends of PCR fragments and encoded in the primers. We also synthesized a short piece of DNA bearing several internal mutations via primer-overlap extension technique. These fragments were assembled using PCR-based cloning (overlap extension) with the C-terminal His-tag. Modified full-length sequence (Cry1Ac-Δ14C) was cloned into a Bt shuttle vector under the control of sporulation-dependent promoter, and introduced into a plasmid-less Bt strain EG10650 by electroporation. We also constructed a completely cysteine-free version of Cry1Ac (Cry1Ac-Δ16C) by an additional round of PCR mutagenesis starting with Cry1Ac-Δ14C. Finally, a clone of protoxin-only (residues 607-end) portion of Cry1Ac-Δ14C was obtained in the same general manner: a PCR fragment was amplified from the full length gene using primers encoding a His-tag at the C-terminus of the protein; the fragment was cloned into pET28a vector by ligation and the resulting construct was introduced into chemically competent BL21(DE3) Escherichia coli.
Expression of the FL Cry1Ac-Δ14C/ Cry1Ac-Δ16C was accomplished by inoculating 1L of C2 sporulation medium11 (5 mg/L chloramphenicol) with a small overnight culture and allowing the large culture to sporulate over the course of 4 days. A crystal-spore mixture was obtained by centrifugation of the fully sporulated and lysed culture. At this stage we discontinued working with Cry1Ac-Δ16C because its crystals dissolved at the end of sporulation. Mostly pure crystal preparation of Cry1Ac-Δ14C was obtained by a discontinuous sucrose gradient centrifugation (5mL crystal spore suspension in water over 15 mL of 65% sucrose on top of 15 mL 80% sucrose in water and centrifuged at 65,000g for 4 hours at 25°C). This step afforded material with negligible protease activity. Since most of the cysteine residues in the protein were mutated away, it was not necessary to use any reducing agents for extraction (normally Cry1Ac and many other 3-domain Cry proteins require reducing agents for solubilization12). Consequently, the issue of cross-linking upon storage did not arise. By trial and error we have found that the crystal size of Cry1Ac-Δ14C was considerably influenced by purification technique—therefore we explored a number of extraction and purification options including extraction in carbonate, borate, and other basic buffers. All purification procedures were carried out at 4°C. The best crystals were obtained from protein that was extracted and purified using ethanolamine, as follows: purified Bt-derived crystals were extracted with 150 mM ethanolamine in water (final pH ∼10.5). Extract was clarified by centrifugation, filtered through a 0.45 micron filter, and applied to an FPLC column packed with Resource-Q15 (GE) resin. The column was washed with 20 mM ethanolamine in water and developed with a gradient of 0–1000 mM NaCl in the same buffer. Full length Cry1Ac-Δ14C eluted around 250 mM NaCl—relevant fractions were pooled and concentrated to 5–8 mg/mL in the same buffer. We have attempted to improve protein quality by size exclusion chromatography (and in the process have discovered that pure full-length protein runs as a dimer), however, the extra time taken by this additional step was detrimental to the final quality and size of the crystals. It was paramount to accomplish extraction and purification within 6–8 hours because protein quality (as perceived from the perspective of forming single crystals of meaningful size) deteriorated rapidly upon storage. We found that the protein tolerated a single round of freezing and thawing (in small aliquots submerged into liquid nitrogen), however, storage of frozen protein at −80°C for over 1 week was detrimental to crystallization. Therefore Cry1Ac-Δ14C was always prepared fresh for individual crystallization campaigns; concentrated using diafiltration and used immediately, or frozen in liquid nitrogen and used within the next 5–7 days.
Expression of the Cry1Ac-Δ14C protoxin was accomplished by growing 1–2L of transformed BL21(DE3) cells in autoinduction medium13 at 25°C, overnight. Frozen cell pellet (25 grams) was lysed at 4°C in 90ml of 3:1 B-PER and Y-PER (Pierce) mixture, supplemented with DNAse (0.1 mg/mL) and Lysozyme (1.5 mg/mL). Protein was purified from clarified lysate first by immobilized affinity chromatography on His-SELECT (Sigma) resin (25 mM TRIS pH 8.0, 10 mM imidazole, 250 mM NaCl wash buffer, same buffer for elution except supplemented with 200 mM imidazole), and then further purified using Resource-Q15 (GE) resin chromatography (25 mM TRIS pH 8.0 base buffer, elution with 0–1000 mM NaCl gradient) and finally polished on size exclusion column (S-200 resin, GE) in 25 mM TRIS, 250 mM NaCl buffer. Protein was concentrated to 6–10 mg/mL and stored at −80°C in small aliquots. Expression and purification of Selenium-labeled protein was accomplished in the same way except methionine-auxotrophic strain of E. coli was used and the autoinduction medium was modified to afford efficient selenomethionine labeling.
Large, visually perfect crystals of the Cry1Ac-Δ14C protoxin-only form [Fig. 1(A)] were readily obtained from a number of conditions in the standard screens; and without exception all of the crystals diffracted at best to 4–6 Å resolution even at the insertion device synchrotron beamlines. After several rounds of optimization and crystal improvement we were able to collect several 3.2–3.5 Å datasets from crystals grown in 2.2-µL hanging drops at 8 mg/mL protein mixed in 1.2:1 ratio with the reservoir solution containing 230 mM LiCl, 17% PEG3500, 100 mM HEPES pH 7.6, and 55 mM NDSB-201. We were able to collect similar quality data from selenium-labeled protein crystals as well as a few crystals derivatized with heavy atom compounds. A crude structure was solved using selenium SAD data combined with mercury, lanthanum, and platinum derivatives-with over 80% solvent there was considerable disorder that spanned entire domains and as the result of this it was impossible to reliably assign most of the side chains.
Figure 1.
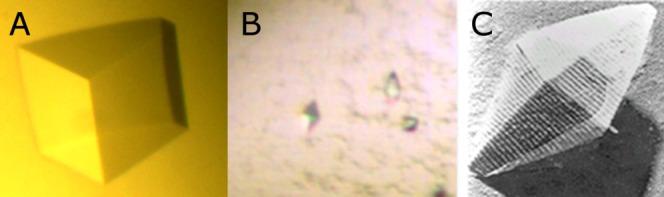
(A) A crystal of cysteine-free Cry1Ac protoxin fragment (approximately 0.2 mm in length), (B) crystals of Cry1Ac-Δ14C (approximately 0.03 mm in length), (C) electron microphotograph of a Bt-derived Cry1Ac crystal (approximately 0.5 microns in length)—previously unpublished image courtesy of Dr. Thomas Malvar (Monsanto) reproduced with permission.
Crystals of full-length Cry1Ac-Δ14C appeared in 1–2 days post-setup in numerous conditions from commercial and in-house screens, set up as 0.2 µL sitting drops. Unlike the protoxin-only construct the FL protein tends to produce showers of tiny bipyramidal crystals (5–8 microns in size) which are visually quite similar to the microscopic crystals observed in sporulated Bt preparations [Fig. 1(B,C)]. After considerable struggle and multiple adjustments to the purification process we were able to gradually increase the size of the crystals into the useful range with the rare few reaching approximately 25 × 40 × 60 µm size. We observed that crystallization occurred within the first 24–48 hours post-setup and in no cases any new crystals were found after 48 hours, indicating that protein stability was the main limiting factor (the same observation was made with respect to purification improvement process). Final crystallization setup was as follows: 600 nL of 13 mg/mL protein in 20 mM ethanolamine buffer with 0.35M NaCl was mixed with 600 nL of 700 mM sodium-potassium tartrate, 100 mM BIS-TRIS propane pH 7.84 and 120 nL of 9 mM n-decyl thiomaltoside, and the resulting 1.2 µL sitting drop was allowed to equilibrate against 80 µL of the tartrate/BIS-TRIS propane reservoir solution at 19°C. Crystals were cryoprotected using glycerol mixed with the reservoir solution (25% glycerol), snap-frozen in liquid nitrogen and shipped to the SER-CAT beamline of the Advanced Photon Source at Argonne National Laboratory for data collection (0.25° oscillation, 8 seconds exposure at 45% beam transmittance with 10 µm capillary collimator). Data were integrated using HKL2000 software,14 and the structure was solved using molecular replacement (Phaser15) using two independent search fragments: the Cry1A N-terminal domain structure solved in-house previously and the abovementioned partial crude structure of the protoxin fragment. Refinement and rebuilding using Refmac16 and Coot17 resulted in a good quality final model of the full-length protein. Salient data collection, refinement, and model quality parameters are summarized in Supporting Information Table 1. The structure of the full-length Cry1Ac-Δ14C was deposited with the PDB/RCSB under accession code 4W8J.
Results and Discussion
Full-length Cry1Ac can be readily extracted from Bt spore-crystal mixtures under alkaline conditions in the presence of reducing agents,12 however, the resulting soluble protein properties leave much to be desired from the crystallographer’s perspective: soluble Cry1Ac-FL rapidly and irreversibly aggregates upon storage; it undergoes intermolecular disulfide cross-linking; and it is gradually digested by contaminating proteases. Some of these issues can be alleviated by judicious manipulation of extraction and purification conditions; however, it was necessary to engineer a multiple-cysteine mutant of the toxin in order to alleviate aggregation and cross-linking of the protein. There are a total of sixteen cysteine residues in Cry1Ac-FL, in two clusters-two residues at the very N-terminus of the protein and 14 more within the protoxin portion—in contrast, the toxic core of the protein is cysteine-free. We chose to replace the 14 cysteines within the protoxin region first, because the N-terminal two cysteines are within the 20-some residue section of the protoxin that is often proteolyzed during purification and so we hoped that the N-terminal cysteines are less likely to cause an issue in solution. We also produced the entirely cysteine-free construct Cry1Ac-Δ16C which, like Cry1Ac-Δ14C and the native protein, expressed copious amounts of full-length material, however at the end of sporulation Bt-formed crystals of Cry1Ac-Δ16C dissolved, whereas the Cry1Ac-Δ14C crystals remained intact—therefore further work with the cysteine-free Cry1Ac-Δ16C was discontinued.
In general, Cry1Ac-Δ14C mutant was indistinguishable from the original in terms of expression, native crystallinity, or toxicity (data not shown). We were able to produce multiple batches of this protein that were stable and homogenous enough for crystallization. We also expressed, purified, and crystallized the cysteine-free protoxin section of the protein, then solved its structure via isomorphous replacement with heavy atoms which allowed us to use molecular replacement to solve the full-length structure.
The first 31 residues of Cry1Ac-FL appear to be disordered (or perhaps proteolyzed off during purification or crystallization); interpretable electron density is observed beginning with Gly-32. The C-terminus of the protein is ordered up to the very last Glu-1178, however, there are several surface-exposed regions of the protein that are disordered [Supporting Information Fig. 1]. Overall the structure contains seven distinct domains (DI–DVII): the three canonical toxin core domains (D-I through D-III) and four protoxin domains (D-IV through D-VII). Cry1Ac-FL is sickle-shaped [Fig. 2] with the toxic core as handle and the protoxin domains as the blade. Toxic core structure (DI-II-III) is not significantly different from the published Cry1Aa toxic core18 (PDB ID 1CIY, 75% sequence identity, 0.9Å Cα rmsd over 551 aligned residues). Local differences are observed in the D-II loops 371–378, 438–447, and D-III loops 503–512, 558–564, 580–586, 591–598—this is in agreement with the high level of local sequence diversity in these loops. Domains IV and VI are alpha-bundles that resemble structural/interaction domains such as spectrin19 [PDB ID: 1CUN] or bacterial fibrinogen-binding complement inhibitor20 [PDB ID: 2GOM] [Fig. 3(A)]. Domains V and VII are beta-rolls (similar to D-II or D-III) that closely resemble carbohydrate-binding modules (CBM) found in sugar hydrolases21,22 (2ZEW, 2XON), however, it is difficult to guess which particular carbohydrates (if any) may serve as their ligands because residues on the putative sugar-binding interfaces are conserved neither in sequence nor in local structure [Fig. 3(B)].
Figure 2.
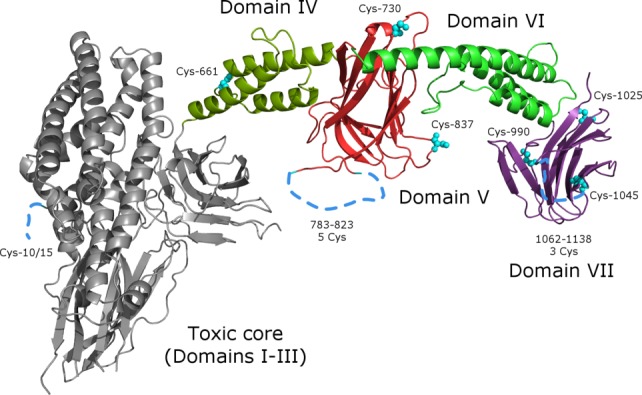
Overall structure of Cry1Ac monomer, colored by domain: domains I through III (the toxic core) are light grey domain IV is dark green, domain V—red, domain VI—green, domain VII—violet. This and other figures were made using PyMol.28 Cysteine residues are highlighted and numbered; disordered regions are shown as dotted lines. Use this link to access the interactive version of this figure
Figure 3.
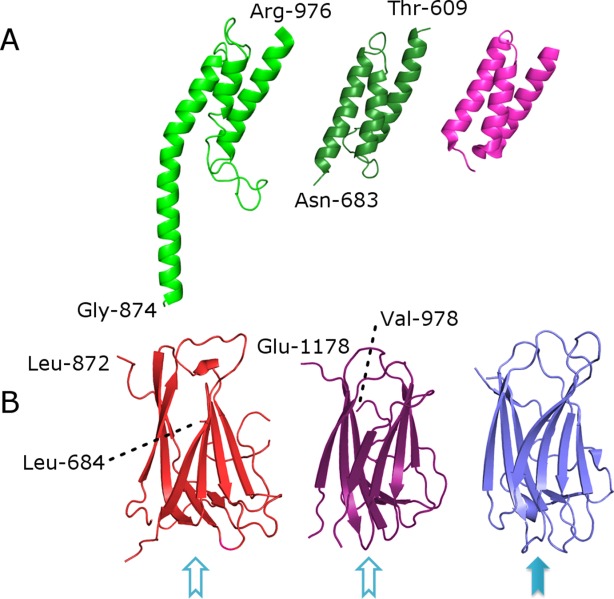
Detailed view of individual domains and their comparison with known structures. (A) domain VI (green), domain IV (dark green), and complement inhibitor PDB ID 2GOM (pink), (B) domain V (red), domain VII (violet) and carbohydrate-binding domain (PDB ID 2ZEW, blue). Solid blue arrow and empty arrows indicate known and putative sugar-binding sites, respectively.
The asymmetric unit of the crystal contains a monomer of Cry1Ac-FL which forms a dimer with a symmetry mate [Fig. 4(A)]. This dimer is likely the same one observed in solution via light scattering and gel filtration (data not shown). The dimer interface buries 2300 Å2 of protein surface per monomer—only 5% of the 45,400 Å2 total—this is not surprising since the liberated toxic core is a soluble monomeric protein in its own right. Dimer interface is predominantly hydrophilic in nature; the protoxin domains of one monomer neatly cup the toxic core of the other one. Other than the dimer interface there are eight more crystal contact regions totalling 4300 Å2. Overall 6600 Å2 (14%) of monomer solvent-accessible surface is buried by crystal packing interactions.
Figure 4.
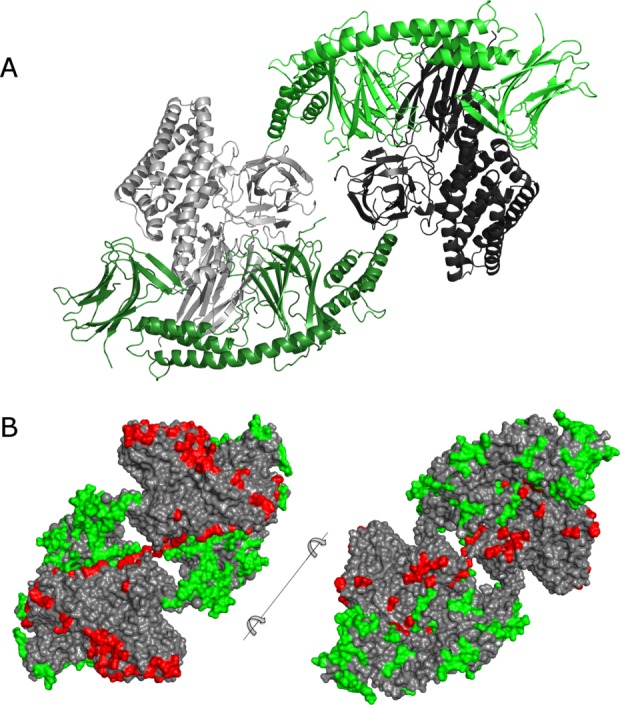
(A) The structure of Cry1Ac dimer. Protoxin domains are green, toxic cores are grey. Use this link to access the interactive version of this figure. (B) Lattice packing of Cry1Ac—interfaces that involve protoxin domains and those involving toxic cores are highlighted in green and red, respectively.
Ordinarily, protein crystal packing has little biological meaning since most proteins are not crystalline in their native state. The situation is different for Cry1Ac-FL since it is produced by Bt as geometrically perfect bipyramidal crystals.1,3,6 Macroscopic crystals produced in vitro are visually identical to microscopic ones made by Bt—could it be that they are in fact the same? In 1965 Holmes and Monro23 conducted powder X-ray diffraction and electron microscopy studies of protein crystals produced by sporulating Bt. They stated that the crystals belong to a tetragonal space group P41212 with unit axis dimensions of a = b = 90 Å, c = 269 Å—the same as the crystals discussed in the present article (P41212, a = b = 87.3 Å, c = 266.4 Å). At the time the nomenclature of Bt Cry proteins was not established and sequences of Bt Cry proteins were not available—therefore we are not able to ascertain that the authors indeed studied the exact same protein used in this paper. Nevertheless since Cry1Ac and its close relatives Cry1Aa and Cry1Ab are very common proteins found in many Bt strains, and since it is extremely unlikely that the same space group and unit cell parameters are obtained randomly, we conclude that the protein crystals studied by Holmes and Monro in 1965 are either identical, or very close to the ones described in this paper—meaning that we were very fortunate to obtain the very same crystal form of the protein by re-crystallization as is naturally produced by sporulating Bt.
Early inquiries into the role of the protoxin domain24,25 and the weight of accumulated evidence to-date suggest that the protoxin domain is dispensable for insecticidal activity—its function likely has a lot to do with crystal formation, stability, and selective solubilization in the insect gut. Cysteine-rich protoxin sequences stabilize mature Bt crystals via intermolecular disulphide cross-linking—in the alkaline, reducing environment of the insect gut the cross-links dissociate, releasing protoxin dimers that undergo proteolysis into mature toxin cores.26,27 Notably, the protoxin region and the cysteine residues at the N-terminus are nearly perfectly conserved across a very large clade of Cry1-like proteins [Supporting Information Fig. 2]. Many of the cysteine residues in the protoxin domain are located on flexible loops that are positioned within mutual proximity in the crystal. Given that many of the cysteine residues are positioned within large flexible loops there can be more than one version of disulfide network within the crystal; a plausible list of bridging interactions can be formulated as follows: the N-terminal cysteines (Cys-10 & Cys-15) can bridge with Cys-1045, Cys-661, Cys-990, or Cys-1025, the latter two also can bridge with some of the five cysteines in the 783–823 disordered region, which is very likely cross-linking to a symmetry mate of itself [Supporting Information Fig. 3(A)] further stabilizing the crystallographic dimer that we believe to be the oligomeric form of the full-length protein in solution. A large disordered region 1062–1138 contains three cysteine residues; their cross-linking partners are presently unclear. Proximity of symmetry-related cysteine residues in the crystal, in particular those of the 783–823 region and the N-terminus supports the theory that the protoxin domain stabilizes mature Bt crystals via disulfide cross-linking; it is likely that the N-terminal Cys-10/15, the cysteines in the 783–823 region together with Cys-1025, Cys-990, and Cys-1045 form the three-dimensional network of cross-links across the crystal [Supporting Information Fig. 3(B)].
Furthermore, it appears that the crystal packing of Cry1Ac-FL is naturally optimized against contacts between symmetry-related toxic cores (230 inter-atomic contacts) and in favor of contacts that involve protoxin domain residues (645 contacts involving protoxin domains and toxic cores and 1568 contacts between protoxin domains only) [Fig. 4(B)]. Given that protoxin domains are highly conserved across related Bt toxin groups [Supporting Information Fig. 2], it is possible that the conserved protoxin domains have evolved to package diverse toxin cores such that the multiple related toxins may be packed into a single crystal, thus ensuring synchronous and co-localized delivery of these toxins to the target insect. While it is difficult to prove this theory conclusively—one such proof could consist of isolating a single protein crystal and showing the presence of several different Cry1 family members within—we feel that indirect evidence is in agreement: many naturally occurring strains of Bt carry several well-expressed Cry1-family toxin genes—yet they commonly form single crystal in each sporulating cell.
In conclusion, the structure of the full length Cry1Ac answers a half-century riddle and poses a score of new questions: why do two of the protoxin domains share fold with CBMs? Is the role of helical domains purely structural? Do other protoxin-bearing Cry protein families (e.g. Cry7 or Cry9, etc.) have similar structural arrangements? Why have some Cry protein families evolved away from protoxin domains (Cry2, Cry3, etc.)? We hope that the structure presented in this article will help us, and others, to answer these questions and pave the way to deeper understanding of Bt insect pathogenesis.
Acknowledgments
Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31–109-Eng-38. We would also like to acknowledge the data collection services provided by Richard Walter and Gina Rainieri of Shamrock Structures LLC. We also would like to thank Julia Maret, T.K. Ball, and Byron Olsen for the critical review of the manuscript and Thomas Malvar for his gift of Bt crystal electron micrograph.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- Schnepf E, Crickmore N, Van Rie J, Lereclus D, Baum J, Feitelson J, Zeigler DR, Dean DH. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol Mol Biol Rev. 1998;62:775–806. doi: 10.1128/mmbr.62.3.775-806.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crickmore N, Zeigler DR, Feitelson J, Schnepf E, Van Rie J, Lereclus D, Baum J, Dean DH. Revision of the nomenclature for the Bacillus thuringiensis pesticidal crystal proteins. Microbiol Mol Biol Rev. 1998;62:807–813. doi: 10.1128/mmbr.62.3.807-813.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Maagd RA, Bravo A, Crickmore N. How Bacillus thuringiensis has evolved specific toxins to colonize the insect world. Trends Genet. 2001;17:193–199. doi: 10.1016/s0168-9525(01)02237-5. [DOI] [PubMed] [Google Scholar]
- Gomez I, Pardo-Lopez L, Munoz-Garay C, Fernandez LE, Perez C, Sanchez J, Soberon M, Bravo A. Role of receptor interaction in the mode of action of insecticidal Cry and Cyt toxins produced by Bacillus thuringiensis. Peptides. 2007;28:169–173. doi: 10.1016/j.peptides.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Soberon M, Fernandez LE, Perez C, Gill SS, Bravo A. Mode of action of mosquitocidal Bacillus thuringiensis toxins. Toxicon. 2007;49:597–600. doi: 10.1016/j.toxicon.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Aronson AI, Beckman W, Dunn P. Bacillus thuringiensis and related insect pathogens. Microbiol Rev. 1986;50:1–24. doi: 10.1128/mr.50.1.1-24.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanahuja G, Banakar R, Twyman RM, Capell T, Christou P. Bacillus thuringiensis: a century of research, development and commercial applications. Plant Biotechnol J. 2011;9:283–300. doi: 10.1111/j.1467-7652.2011.00595.x. [DOI] [PubMed] [Google Scholar]
- Perlak FJ, Deaton RW, Armstrong TA, Fuchs RL, Sims SR, Greenplate JT, Fischhoff DA. Insect resistant cotton plants. Biotechnology. 1990;8:939–943. doi: 10.1038/nbt1090-939. [DOI] [PubMed] [Google Scholar]
- Perlak FJ, Oppenhuizen M, Gustafson K, Voth R, Sivasupramaniam S, Heering D, Carey B, Ihrig RA, Roberts JK. Development and commercial use of Bollgard cotton in the USA—early promises versus today’s reality. Plant J. 2001;27:489–501. doi: 10.1046/j.1365-313x.2001.01120.x. [DOI] [PubMed] [Google Scholar]
- Pardo-Lopez L, Soberon M, Bravo A. Bacillus thuringiensis insecticidal three-domain Cry toxins: mode of action, insect resistance and consequences for crop protection. FEMS Microbiol Rev. 2013;37:3–22. doi: 10.1111/j.1574-6976.2012.00341.x. [DOI] [PubMed] [Google Scholar]
- Baum JA, Kakefuda M, Gawron-Burke C. Engineering Bacillus thuringiensis bioinsecticides with an indigenous site-specific recombination system. Appl Environ Microbiol. 1996;62:4367–4373. doi: 10.1128/aem.62.12.4367-4373.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson L, Mazza A, Sangadala S, Adang MJ, Brousseau R. Polydispersity of Bacillus thuringiensis Cry1 toxins in solution and its effect on receptor binding kinetics. Biochim Biophys Acta. 2002;1594:266–275. doi: 10.1016/s0167-4838(01)00312-0. [DOI] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Express Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW, Sweet RM, editors. Methods in enzymology. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Grochulski P, Masson L, Borisova S, Pusztai-Carey M, Schwartz JL, Brousseau R, Cygler M. Bacillus thuringiensis CryIA(a) insecticidal toxin: crystal structure and channel formation. J Mol Biol. 1995;254:447–464. doi: 10.1006/jmbi.1995.0630. [DOI] [PubMed] [Google Scholar]
- Grum VL, Li D, MacDonald RI, Mondragon A. Structures of two repeats of spectrin suggest models of flexibility. Cell. 1999;98:523–535. doi: 10.1016/s0092-8674(00)81980-7. [DOI] [PubMed] [Google Scholar]
- Hammel M, Sfyroera G, Ricklin D, Magotti P, Lambris JD, Geisbrecht BV. A structural basis for complement inhibition by Staphylococcus aureus. Nature Immunol. 2007;8:430–437. doi: 10.1038/ni1450. [DOI] [PubMed] [Google Scholar]
- Bae B, Ohene-Adjei S, Kocherginskaya S, Mackie RI, Spies MA, Cann IK, Nair SK. Molecular basis for the selectivity and specificity of ligand recognition by the family 16 carbohydrate-binding modules from Thermoanaerobacterium polysaccharolyticum ManA. J Biol Chem. 2008;283:12415–12425. doi: 10.1074/jbc.M706513200. [DOI] [PubMed] [Google Scholar]
- Cid M, Pedersen HL, Kaneko S, Coutinho PM, Henrissat B, Willats WG, Boraston AB. Recognition of the helical structure of beta-1,4-galactan by a new family of carbohydrate-binding modules. J Biol Chem. 2010;285:35999–36009. doi: 10.1074/jbc.M110.166330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes KC, Monro RE. Studies on the structure of parasporal inclusions from Bacillus thuringiensis. J Mol Biol. 1965;14:572–581. doi: 10.1016/s0022-2836(65)80205-4. [DOI] [PubMed] [Google Scholar]
- Luthy P, Ebersold HR. The entomocidal toxins of Bacillus thuringiensis. Pharmacol Therap. 1981;13:257–283. doi: 10.1016/0163-7258(81)90003-6. [DOI] [PubMed] [Google Scholar]
- Hofte H, Whiteley HR. Insecticidal crystal proteins of Bacillus thuringiensis. Microbiol Rev. 1989;53:242–255. doi: 10.1128/mr.53.2.242-255.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastidar PG, Nickerson KW. Interchain crosslinks in the entomocidal Bacillus thuringiensis protein crystal. FEBS Lett. 1979;108:411–414. doi: 10.1016/0014-5793(79)80575-x. [DOI] [PubMed] [Google Scholar]
- Couche GA, Pfannenstiel MA, Nickerson KW. Structural disulfide bonds in the Bacillus thuringiensis subsp. israelensis protein crystal. J Bacteriol. 1987;169:3281–3288. doi: 10.1128/jb.169.7.3281-3288.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System. Version. 2014;1:7. Schrödinger, LLC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.