

Abstract
As our understanding of the molecular structures of amyloid fibrils has matured over the past 15 years, it has become clear that, while amyloid fibrils do have well-defined molecular structures, their molecular structures are not uniquely determined by the amino acid sequences of their constituent peptides and proteins. Self-propagating molecular-level polymorphism is a common phenomenon. This article reviews current information about amyloid fibril structures, variations in molecular structures that underlie amyloid polymorphism, and physical considerations that explain the development and persistence of amyloid polymorphism. Much of this information has been obtained through solid state nuclear magnetic resonance measurements. The biological significance of amyloid polymorphism is also discussed briefly. Although this article focuses primarily on studies of fibrils formed by amyloid-β peptides, the same principles apply to many amyloid-forming peptides and proteins.
Keywords: amyloid structure, fibril structure, solid state NMR, Alzheimer’s disease, prion
Introduction
Amyloid fibrils are filamentous self-assembled aggregates of peptides and proteins that contain cross-β structures. A cross-β structure1 is a ribbon-like β-sheet, extending over the length of the fibril, in which β-strands run approximately perpendicular to the fibril growth direction and are linked by inter-strand backbone hydrogen bonds that run approximately parallel to the growth direction. Typically, amyloid fibrils contain two or more cross-β layers, stacked on one another with sidechain-sidechain interactions (see Fig. 1), often in an interdigitating manner.2,3 Evidence for cross-β structures in amyloid fibrils came originally from X-ray fiber diffraction4 and is supported by electron diffraction,5 electron microscopy,6 and solid-state nuclear magnetic resonance7 (SSNMR).
Figure 1.
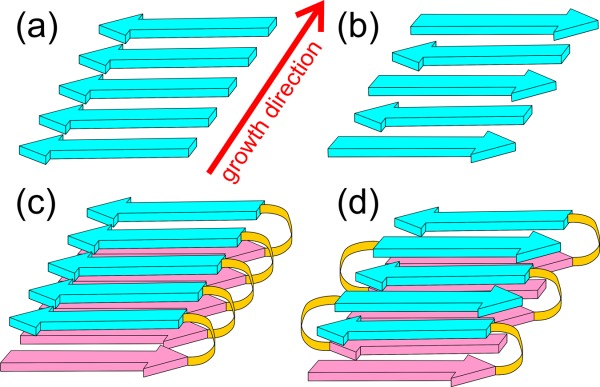
Cartoon representations of cross-β structural motifs. (a,b) Parallel and antiparallel cross-β motifs, as would occur in amyloid fibrils formed by short peptides with a single β-strand segment. The fibrils might contain multiple copies of the same motif, stacked against one another in a direction perpendicular to the growth direction and to the β-strand direction. (c,d) Double-layered parallel and antiparallel cross-β motifs, as would occur in amyloid fibrils formed by longer peptides with two β-strand segments. Again, the fibrils might contain multiple copies of the same motif.
Amyloid fibril formation is of fundamental interest from the standpoint of biophysical chemistry. As emphasized in work by the Cambridge group,8–10 the propensity or ability to form amyloid fibrils is a common, nearly generic, property of polypeptide chains that had not been appreciated until the end of the 20th century. Amyloid fibrils now appear to be a “default state” for polypeptides (unless the chains are too highly charged, contain too many prolines or glycines, or have other features that are not fully understood). That is, amyloid fibrils are a structural state that polypeptides adopt (at sufficiently high concentrations) if they do not have "something better to do," such as fold into the globular structure of an enzyme, participate in a biologically functional supramolecular complex, and so forth. Several examples of biologically functional amyloid structures have also been demonstrated or suggested.11–20
Amyloid formation is also of great current interest in medical research, as amyloid fibrils form in the affected tissue of numerous amyloid diseases,21 including type 2 diabetes and neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and transmissible spongiform encephalopathies (TSEs). It is generally believed that amyloid fibrils play important roles in these diseases, either directly through their inherent cytotoxicity or indirectly through the cytotoxicity of self-assembled structural states that precede fibril formation as kinetic or thermodynamic intermediates. In AD, amyloid fibrils are the targets of imaging agents that hold promise for disease diagnosis and monitoring of disease progression in living patients.22–25
Much of the work on amyloid structure in my own laboratory has focused on the amyloid-β (Aβ) peptides that are associated with AD.26–43 We have also studied fibrils formed by yeast prion proteins,44–49 fibrils formed by the mammalian prion protein PrP associated with TSEs,50 and fibrils formed by the amylin peptide associated with type 2 diabetes.51 Although the discussion below concentrates on Aβ fibrils, many of the same concepts apply to amyloid fibrils in general, as shown by our own work and that of others.11,52–80
One of the intriguing aspects of amyloid formation is that amyloid fibrils are polymorphic at the molecular structural level. Molecular structures of fibrils formed by a given polypeptide can vary, depending on the precise details of growth conditions. Often, multiple distinct structures coexist under a single set of conditions. When we began our studies of Aβ fibrils in 1998, it was not clear that fibrils with distinct morphologies9,81–83 (i.e., appearances) in transmission electron microscope (TEM) images (see Fig. 2) contained distinct molecular structures. The possibility existed that different fibril morphologies might simply be different ways of bundling the same underlying fibril structure, which could be called the "protofilament," with itself.82 However, various experimental results, described below, eventually led us to conclude that (in most cases) molecular structures within amyloid fibrils are not determined uniquely by amino acid sequences. Fibrils with distinct appearances in TEM images contain distinct molecular structures, even at the protofilament level.
Figure 2.

TEM images of negatively-stained Aβ40 fibrils, grown in vitro from synthetic Aβ40 peptides under conditions that produce clear polymorphism. Arrows indicate "striated ribbon" fibrils (red), "slowly twisting" fibrils (blue), and "rapidly twisting" fibrils (yellow).
The following sections describe the experimental evidence for molecular-level structural polymorphism in Aβ fibrils, the specific molecular structural features that distinguish one polymorph from another, our current understanding of the physical basis for the development and persistence of molecular-level structural polymorphism, and current evidence that fibril polymorphism may have consequences in AD and other amyloid diseases.
Evidence for Self-Propagating Molecular Polymorphism in Amyloid Fibrils
Experimental evidence for variable molecular structures in amyloid fibrils formed by the same polypeptide comes from a variety of measurements. First, TEM images, such as those in Figure 2 show a variety of morphologies. It is significant that the morphology of a single fibril does not change along the length of the fibril. Thus, morphological features such as fibril width and twist period (indicated by apparent width modulation along the growth direction in negatively stained TEM images) are self-propagating. Moreover, when a morphologically homogeneous batch of "parent" fibrils is used as the source of "seeds" for the growth of subsequent batches of "daughter" fibrils, the morphology passes from parent to daughter and from daughter to subsequent generations.32 Here, the word "seeds" means short fibril fragments, typically with lengths of order 20–50 nm and typically prepared by sonication of long fibrils. Daughter fibrils are grown by adding monomeric (or at least solubilized) polypeptide molecules to a dilute solution of seeds, typically with a 10- to 50-fold excess of monomeric molecules to molecules in the seeds. Seeds then grow into long fibrils by addition of monomers to their ends.
In addition to morphological differences, fibrils formed by a given polypeptide can differ in their mass-per-length (MPL) values, which can be measured by various types of dark-field electron microscopy of unstained samples.28,81,84,85 Polymorphs of 40-residue Aβ (Aβ40) fibrils studied in my laboratory exhibit MPL values of roughly 18 kDa/nm and 27 kDa/nm.32,84 Aβ40 fibrils with the same MPL values were also identified in earlier work by Goldsbury et al.81,85 These values have structural significance: given the 4.3 kDa molecular weight of Aβ40 and the 0.47–0.48 nm interstrand spacing along the fibril growth direction in a cross-β structure, a single cross-β structural unit would have MPL ≈9 kDa/nm. The observed MPL values for Aβ40 fibrils therefore indicate molecular structures comprised of either two or three cross-β subunits. MPL values propagate from parent to daughter fibrils in seeded growth experiments.
15N- and 13C-NMR chemical shifts in SSNMR spectra of different Aβ40 fibril polymorphs also differ significantly,32,34,37,38,42 reflecting differences in backbone conformations, sidechain conformations, and supramolecular structure. Morphologically heterogeneous samples, prepared from peptides that contain 15N,13C-labeled residues, exhibit multiple sets of chemical shifts for the labeled residues, as revealed by two-dimensional (2D) SSNMR spectra.40 Only when morphologically homogeneous samples are prepared do the 2D spectra show a single set of sharp crosspeak signals that reflect a single set of chemical shifts (see Fig. 3). Chemical shifts of morphologically homogeneous fibrils are also passed from parents to daughters in seeded growth experiments.
Figure 3.
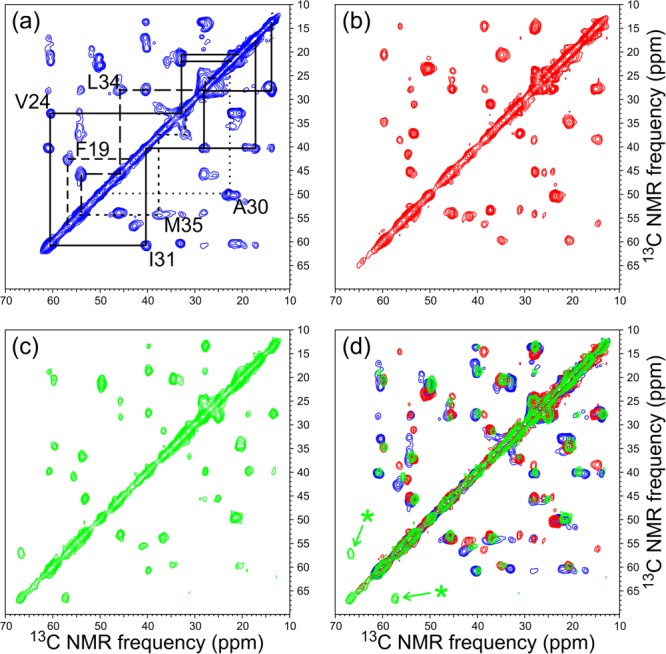
Aliphatic regions of 2D 13C-13C SSNMR spectra of Aβ40 fibrils, prepared with uniform 15N and 13C labeling of F19, V24, G25, A30, I31, L34, and M35. (a) Spectrum of fibrils grown in vitro under conditions that produce fibrils with a predominant "striated ribbon" morphology. Chemical shift assignment paths, connecting the crosspeak signals of each labeled residue (except G25), are shown in solid, dashed, and dotted lines. (b) Spectrum of fibrils grown from seeds extracted from brain tissue of AD Patient 1. (c) Spectrum of fibrils grown from seeds extracted from brain tissue of AD Patient 2. (d) Superposition of 2D spectra from Panels a to c, revealing 13C NMR chemical shift differences that reflect differences in molecular structure. Asterisks indicate signals from S26, which was isotopically labeled in fibrils derived from Patient 2 but not in the other two fibril samples.
Indeed, the observation of relatively sharp crosspeaks in multidimensional SSNMR spectra of amyloid fibrils constitutes the most definitive experimental evidence that individual amyloid fibrils contain well-defined molecular structures, rather than containing disordered arrangements of polypeptide chains into cross-β motifs. The observation of single sets of sharp SSNMR signals from Aβ40 fibril samples with MPL values of 18 kDa/nm and 27 kDa/nm indicates that these structures have approximate two-fold and three-fold rotational symmetry about the fibril growth axis, respectively.34,37,42
Additional evidence for molecular-level polymorphism in Aβ40 fibrils is provided by measurements of thermodynamic and kinetic properties of distinct polymorphs, which reveal significant differences in quasi-equilibrium solubilities43,86 and in rates of fibril extension and shrinkage.43 Mechanical properties of different polymorphs, such as their susceptibility to fragmentation when subjected to shear forces,43 can also vary. Different polymorphs can also exhibit significant differences in dye binding or fluorescence,40,87,88 differences in hydrogen-deuterium exchange rates,86 and differences in X-ray fiber diffraction patterns.89
Underlying Molecular Structural Variations
In what ways to do molecular structures of distinct amyloid fibril polymorphs differ from one another? The most detailed answers come from our studies of Aβ40 fibrils, in which we have developed full molecular structural models from large sets of SSNMR data, supplemented by information from electron microscopy.35,37,41,42 Structural variations similar to those identified in Aβ40 fibrils are likely to occur in other amyloid fibrils, especially in fibrils formed by peptides of similar molecular weight such as amylin.51 Experimental data on structural variations in fibrils formed by α-synuclein,58,61,71,77,79,90 tau,60,91 Sup35NM,48,92–94 and PrP95,96 are also available, although full experimentally-based structural models have not yet been reported in these cases.
Figure 4 shows representations of structural models for three Aβ40 fibril polymorphs, as well as a model for a D23N-Aβ40 "protofibril" (see below). Models in Figure 4(a,b) correspond to polymorphs that were prepared in vitro, as described by Petkova et al.32,35 and by Paravastu et al.37 In both models, residues 1–8 are considered to be structurally disordered and are not shown. Residues 10–22 and 30–40 form β-strands that are separated by a bend or loop in residues 23–29. The two β-strand segments participate in separate in-register parallel β-sheets. Thus, in both models, each Aβ40 molecule adopts a U-shaped, strand-bend-strand conformation, and participates in a double-layered, parallel cross-β structural subunit. The double-layered cross-β subunit is apparently stabilized by hydrophobic interactions among sidechains of the two β-strands (e.g., F19-L34 contacts).
Figure 4.
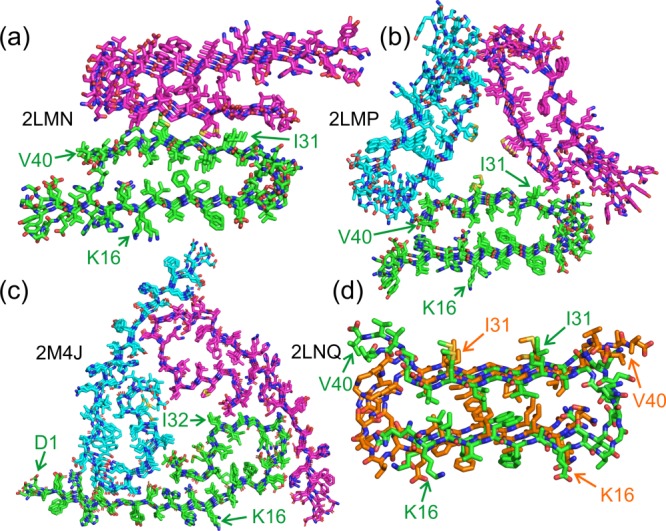
Structural models for Aβ40 fibrils, based on data from SSNMR and electron microscopy, with corresponding Protein Data Bank (PDB) codes. Models are shown in cross-section, with the fibril growth direction perpendicular to the page. In each case, four repeats within the cross-β motifs are shown. Sidechains of K16, I31, or I32, and D1 or V40 are indicated to facilitate interpretation of the structures and identification of N- and C-termini. (a) Model with approximate two-fold symmetry, consisting of two cross-β subunits as in Figure 1(c). Carbon atoms in the two cross-β subunits are colored either green or magenta. Residues 9–40 are shown. This model represents one protofilament within a "striated ribbon" fibril,32,35 as in Figure 2. (b) Model with approximate three-fold symmetry, consisting of three cross-β subunits as in Figure 1(c), colored green, magenta, or cyan. Residues 9–40 are shown. This model represents a "slowly twisting" fibril,37 as in Figure 2. (c) Model with approximate three-fold symmetry, representing fibrils grown from seeds extracted from brain tissue of AD Patient 1,42 as in Figure 3(b). In this case, SSNMR spectra indicate that the entire Aβ40 sequence is structurally ordered, and residues 1–40 are shown. The structural model was subjected to a 100 ps molecular dynamics simulation in water at 37°C to relax the perfect symmetry of the PDB coordinates. (d) Model for protofibrils formed by D23N-Aβ40,41 comprised of one cross-β unit as in Figure 1(d). Carbon atoms of successive peptide chains are colored green or orange to clarify the antiparallel nature of β-sheets within the cross-β unit. Residues 15–40 are shown.
While the models in Figure 4(a,b) share similar cross-β subunit structures, the contacts among subunits are obviously different, producing structures with either approximate two-fold or approximate three-fold rotational symmetry about the fibril growth axis. In addition, the conformations of the bend segments differ in the two models. Specifically, in Figure 4(a), sidechains of D23 and K28 form salt bridges that span the bend segment. These salt bridges, whose presence in the two-fold symmetric fibrils is indicated by quantitative measurements of 15N-13C dipole-dipole couplings between K28 and D23 sidechains,35 are not present in the three-fold symmetric fibrils represented by Figure 4(b).37
The model in Figure 4(c) corresponds to Aβ40 fibrils that were grown by seeding with an extract from brain tissue of a particular AD patients (see below), as described by Lu et al.42 This model resembles the model for three-fold symmetric fibrils grown in vitro (i.e., without brain extract) in Figure 4(b), except that residues 1–8 are not disordered, residues 30–40 do not form a single β-strand, and D23-K28 salt bridges are present.
The model in Figure 4(d) corresponds to protofibrils formed by the disease-associated Asp23-Asn mutant of Aβ40 (D23N-Aβ40). Here, the term "protofibril" refers to relatively short and curvy (i.e., worm-like) fibrils that are thermodynamically metastable with respect to conversion to longer, straighter, "mature" fibrils. Like mature fibrils, protofibrils contain cross-β structures. Under experimental conditions described by Qiang et al.,40,41 protofibrils represent a major portion of the self-assembled material that develops in de novo preparations of D23N-Aβ40 fibrils. The protofibrils appear to nucleate efficiently, but extend more slowly than mature fibrils. By taking advantage of the differences in extension rates, Qiang et al. were able to prepare homogeneous D23N-Aβ40 protofibrils, allowing detailed structural studies by SSNMR and electron microscopy.41
The D23N-Aβ40 protofibril model in Figure 4(d) differs significantly from the Aβ40 fibril models in Figure 4(a–c). MPL measurements indicate that D23N-Aβ40 fibrils contain a single cross-β unit. Solid state NMR spectra suggest that only residues 15–36 are structurally ordered. Although these residues adopt a strand-bend-strand conformation, the two β-strands participate in two separate antiparallel β-sheet layers, with well-defined registries of interstrand hydrogen bonds within the two antiparallel β-sheets that are dictated unambiguously by the SSNMR data.41 Interestingly, the two β-sheets of an antiparallel cross-β unit within D23N-Aβ40 fibrils interact through contacts among the same sets of hydrophobic sidechains (e.g., F19 and L34) as the two β-sheets of parallel cross-β units within Aβ40 fibrils. The similarity of the hydrophobic interactions within the cross-β units may account for the comparable stabilities of protofibrils and fibrils.
D23N-Aβ40 also forms mature fibrils, which contain in-register parallel β-sheets39,41 and presumably have molecular structures similar to those in Figure 4(a–c). Thus, one can think of the antiparallel cross-β structure in Figure 4(d) as being another amyloid fibril polymorph, rather than being a completely separate type of self-assembled entity. It is not yet known whether wild-type Aβ40 protofibrils (or protofibrils formed by other disease-associated mutants of Aβ40) contain antiparallel structures similar to the model in Figure 4(d). Bertini et al. have reported an additional SSNMR-based structural model for a two-fold symmetric Aβ40 fibril polymorph,70 differing from the model in Figure 4(a) primarily in the precise packing of sidechains in the interfaces among parallel β-sheet layers.
To summarize, structural studies of Aβ40 fibrils with distinct appearances in TEM images indicate that amyloid polymorphism can involve differences in the extent of structurally ordered segments, the conformations of non-β-strand segments, the contacts and interactions between β-sheets, the number of cross-β subunits, the overall symmetry, and (if protofibrils are considered to be fibril polymorphs with lower thermodynamic stability) β-sheet organization. Additional structural variations may exist in polymorphs of fibrils formed by other polypeptides.
Physical Principles Behind the Development and Persistence of Amyloid Fibril Polymorphism
De novo amyloid fibril growth by peptides such as Aβ is generally considered to be a nucleated polymerization process.97–102 Many aspects of this process remain uncertain, but a possible scenario is shown in Figure 5. Loosely speaking, starting from a solution of peptide monomers, oligomers of various sizes and with various internal structures form and dissociate in a random, dynamic manner. When oligomers of a critical size appear, they can adopt a structure that is capable of growing into a fibril by subsequent binding of additional monomers. The self-assembly of peptide molecules into such a "critical nucleus" structure can be called a nucleation event. The molecular structures of fibrils are presumably determined by the molecular structures of the critical nuclei. Molecular-level polymorphism of amyloid fibrils then implies that multiple distinct nucleation events and critical nuclei are possible for polypeptides with a given amino acid sequence. Under specific growth conditions (e.g., pH, ionic strength, temperature, polypeptide concentration), certain nucleation events may occur with greater frequency than others, leading to a dependence of the predominant fibril structure on growth conditions.
Figure 5.
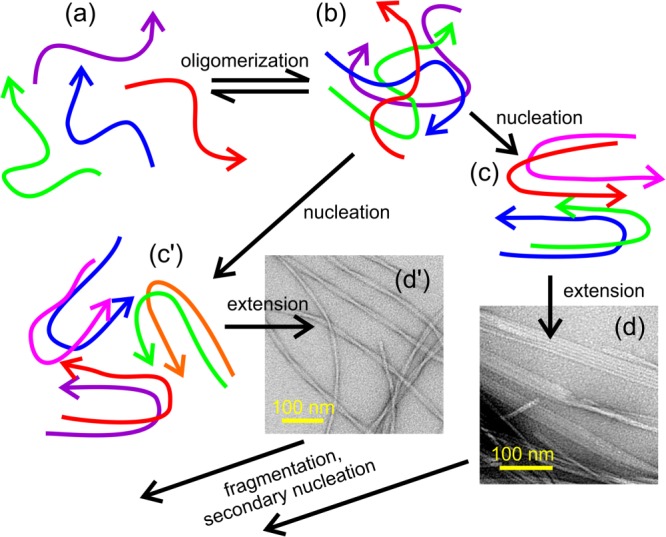
Schematic representation of a possible pathway for amyloid fibril formation by Aβ40 and similar peptides. Starting with conformationally disordered monomers in solution (a), oligomers of various sizes form transiently (b). Occasionally, oligomers adopt "critical nucleus" structures (c,c′) that can grow into fibrils (d,d′) with morphologies and molecular structures that are determined by the nucleation event. Once a small number of fibrils form, subsequent fibril growth can be dominated by fragmentation or secondary nucleation. Amyloid polymorphism can arise when multiple distinct nucleation events coexist. The identities of the predominant polymorphs can vary with growth conditions, due to polymorph-specific dependences of the rates of nucleation, extension, fragmentation, and secondary nucleation on growth conditions.
Once nucleation events have occurred, subsequent fibril growth can be affected by other processes that are also sensitive to growth conditions. One of these is fragmentation, induced by shear forces (vortexing, stirring, pipetting, etc.), by deliberate sonication, or perhaps by biological processes in human tissue. Assuming that fibrils grow at their ends, the rate of increase of fibril mass by extension of existing fibrils is proportional to the number of fibril ends, which doubles in each fragmentation event. When two or more polymorphs coexist in the early stages of fibril growth, due to coexistence of two or more types of nucleation events, the polymorph that fragments at the greatest rate will tend to predominate at later stages.
In fact, differences in susceptibility to fragmentation by shear forces account, at least in part, for our experimental observation that Aβ40 fibrils with two-fold symmetry [Fig. 4(a)] predominate when fibrils are grown in vitro with agitation of the Aβ40 solution, while fibrils with three-fold symmetry [Fig. 4(b)] predominate when fibrils are grown without agitation, all other conditions being equal.32 Atomic force microscope (AFM) images show that long Aβ40 fibrils with two-fold symmetry fragment more rapidly than long Aβ40 fibrils with three-fold symmetry when the fibril-containing solutions are deliberately stirred.43
A recent study of Aβ42 fibril growth kinetics by Cohen et al. indicates that, in the absence of shear forces, these kinetics can be influenced strongly by a secondary nucleation process in which new fibrils nucleate on the sides of existing fibrils, leading to a rate of increase of fibril mass that is proportional to the existing mass, rather than being proportional to the number of fibril ends.103 Such a secondary nucleation process may be especially important when shear forces are too low to cause fragmentation.
The considerations outlined above explain how polymorphism arises. But why does polymorphism persist? Fibril samples with distinct MPL values, distinct SSNMR spectra, and distinct appearances in TEM images can be prepared and studied for many months or even years. Why do the fibrils in these samples not gradually convert to a single structure that is most thermodynamically stable?
Our recent study of polymorph-specific Aβ40 fibril growth kinetics and thermodynamics helps explain the persistence of polymorphism.43 In this study, AFM images were used to quantify the time-dependences of fibril length distributions in the presence or absence of excess soluble Aβ40, that is, in conditions under which short fibril fragments extend or long fibrils shrink [see Fig. 6(a,b)]. Fibrils were grown either from two-fold symmetric or three-fold symmetric seeds, allowing the properties of the two polymorphs to be measured separately. Fibril extension rates were found to be proportional to soluble peptide concentration [S], that is, equal to ke[S] with ke being the extension rate constant. Values of ke were 8.7 ± 0.1 × 10−3 nm/µM-s and 6.1 ± 0.2 × 10−3 nm/µM-s for two-fold and three-fold fibrils, respectively. The ratio of shrinkage rate ks (in units of nm/s) to ke defines a quasi-equilibrium solubility [S]qe = ks/ke for each polymorph. For both Aβ40 fibril polymorphs, values of [S]qe were found to be about 0.5 µM at 24°C in 10 mM phosphate buffer at pH 7.4, but the best-fit value of [S]qe for two-fold symmetric fibrils was smaller by ∼0.05 µM. Values of [S]qe derived from AFM measurements were in good agreement with direct measurements of soluble peptide concentrations by ultraviolet absorption. The small difference in quasi-equilibrium solubilities implies a chemical potential difference for Aβ40 in the two polymorphs of 0.8 kcal/mol or less.
Figure 6.
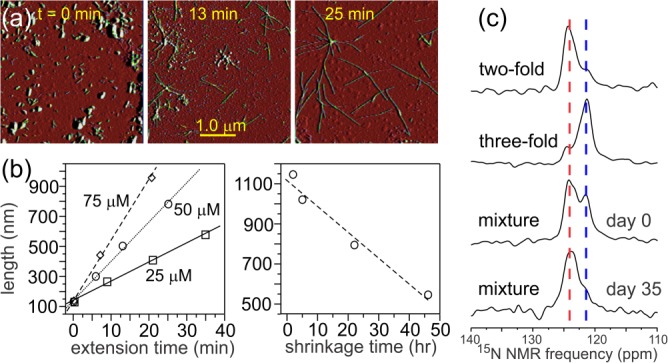
Methods for investigating polymorph-specific kinetics and thermodynamics of amyloid fibril formation. (a) Visualization of fibril extension by AFM. Starting with short Aβ40 fibril fragments (i.e., seeds) in a solution of Aβ40, aliquots were taken for AFM imaging at the indicated times. Here, the seeds had the two-fold symmetric structure in Figure 4(a), soluble Aβ40 concentration was 50 µM, and the solution temperature was 24°C. (b) Time-dependence of average fibril length, determined from AFM images, in extension experiments at the indicated concentrations of soluble Aβ40 (left) and in shrinkage experiments, in which long fibrils were placed in a solution devoid of soluble Aβ40 (right). (c) Direct demonstration by 15N SSNMR that Aβ40 fibrils with the two-fold symmetric structure in Figure 4(a) are thermodynamically more stable than Aβ40 fibrils with the three-fold symmetric structure in Figure 4(b). An approximate 1:1 mixture of the two structures was prepared on Day 0, with 15N labels at I32. By Day 35, most of the 15N SSNMR signal from three-fold symmetric fibrils had disappeared.
The low quasi-equilibrium solubilities of the two polymorphs, the small difference in solubilities, and the small shrinkage rates (ks values of 3.4 ± 0.4 × 10−3 nm/s and 2.7 ± 0.2 × 10−3 nm/s for two-fold and three-fold symmetric fibrils, respectively43) explain the persistence of polymorphism. Once fibrils have formed, even if they are not the most stable polymorphs, the remaining concentration of soluble peptide is too low for spontaneous nucleation of new fibrils to occur at an appreciable rate. If the fibrils are structurally heterogeneous, comprised of a mixture of more stable and less stable polymorphs, the mass of the less stable fibrils will decrease with time, while the mass of the more stable fibrils will increase. However, less stable fibrils do not convert to more stable fibrils by internal structural rearrangements, because such rearrangements would require multiple conformational changes within a densely packed fibril core, breakage of multiple hydrogen bonds, and so forth. Interconversion of polymorphs can only occur by shrinkage (i.e., dissolution) of less stable polymorphs and extension of more stable polymorphs. The rate of interconversion is then limited by the number of fibril ends (where shrinkage and extension occur) and by the differences in kinetic parameters. Specifically, when fibril structures A and B are present in a 1:1 ratio, the steady-state rate of transfer of monomers from less stable fibrils (A) to more stable fibrils (B) can be expressed as 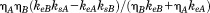 , where ηA and ηB are the numbers of monomers per nm in structures A and B. This expression, together with the experimentally determined kinetic parameters, implies a time scale on the order of 10 days for the less stable fibrils to disappear if their lengths are initially 100 nm. Longer fibrils or smaller differences in [S]qe lead to longer time scales.
, where ηA and ηB are the numbers of monomers per nm in structures A and B. This expression, together with the experimentally determined kinetic parameters, implies a time scale on the order of 10 days for the less stable fibrils to disappear if their lengths are initially 100 nm. Longer fibrils or smaller differences in [S]qe lead to longer time scales.
As shown by SSNMR spectra in Figure 6(c), evolution from a mixture of two-fold symmetric and three-fold symmetric Aβ40 fibrils toward a pure sample of the more stable two-fold symmetric fibrils can be observed experimentally over a 35-day period, provided that the mixture has been sonicated in order to make all fibrils less than 100 nm in length initially.43 When the fibrils have lengths greater than 1 µm and when the two polymorphs are not initially present in comparable numbers, structural interconversion is not observed.
Biological Implications of Amyloid Polymorphism
Although not the main focus of this article, it should be mentioned that amyloid polymorphism can have biological implications, some of which are well established, while others are current subjects of research.
In TSEs, self-propagating variations in disease characteristics such as incubation period and patterns of aggregated PrP deposition are called "strains" or "variants". Measurements of proteolysis patterns and resistance to denaturation indicate that PrP aggregates (at least some of which are amyloid fibrils) in brain tissue infected with different TSE prion strains have somewhat different molecular structures.104,105 Structural variations within distinct TSE prion strains can also account for the strain-dependence of barriers to prion transmission between animal species.106
Studies of yeast prion proteins show that yeast prions also have variants.107 In the well-studied case of [PSI+] prions, which are amyloid fibrils formed by Sup35p or its N-terminal portion, distinct variants produce "strong" and "weak" phenotypes attributable to variations in the stability of the fibrils, arising from molecular structural variations.92,108
Several lines of evidence suggest that phenomena analogous to prion strains may exist in diseases such as AD and PD, i.e., that molecular structural variations within Aβ fibrils, tau fibrils, or α-synuclein fibrils may be correlated with (perhaps cause) variations in the severity, clinical symptoms, progression rate, neuropathology, or other characteristics of AD or PD. One can imagine that such effects might arise from variations in the inherent cytotoxicity of different fibril polymorphs (perhaps due to variations in the identities and conformations of surface-exposed amino acid sidechains), variations in their ability to bind metal ions that play roles in oxidative damage74 or to induce inflammation,109 variations in their self-propagation efficiency within tissue (perhaps due to variations in susceptibility to fragmentation by biological processes), or variations in other chemical, mechanical, or biological properties.
In the case of Aβ, Petkova et al. have shown that different Aβ40 fibril polymorphs can have somewhat different levels of toxicity in neuronal cell cultures.32 Experiments on the induction of Aβ plaque formation in transgenic mice by injection of exogenous Aβ aggregates have shown that different sources of Aβ aggregates produce different distributions and quantities of plaques.110–112 Our recent study of Aβ40 fibrils produced by seeded growth from amyloid extracted from human brain tissue showed that two different AD patients (Patients 1 and 2 in Fig. 3) can develop different predominant fibril polymorphs in their brains.42 Interestingly, the two patients in this study had significantly different clinical histories. This study also showed that the fibril structures in a given patient can be surprisingly homogeneous throughout the cerebral cortex, despite the inherently polymorphic nature of Aβ40 aggregation. Further experiments are certainly required before correlations between molecular structural variations in Aβ fibrils and variations in characteristics of AD can be considered statistically significant. If such correlations prove to be significant, then it may become important to develop diagnostic imaging agents that interact preferentially with specific fibril structures, as well as drugs that inhibit formation of specific fibril structures.
Acknowledgments
Work described in this article was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, a component of the National Institutes of Health.
References
- Astbury WT, Beighton E, Parker KD. The cross-b configuration in supercontracted proteins. Biochim Biophys Acta. 1959;35:17–25. doi: 10.1016/0006-3002(59)90330-0. [DOI] [PubMed] [Google Scholar]
- Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-b spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, Madsen AO, Riekel C, Eisenberg D. Atomic structures of amyloid cross-b spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- Eanes ED, Glenner GG. X-ray diffraction studies on amyloid filaments. J Histochem Cytochem. 1968;16:673–677. doi: 10.1177/16.11.673. [DOI] [PubMed] [Google Scholar]
- Baxa U, Cheng NQ, Winkler DC, Chiu TK, Davies DR, Sharma D, Inouye H, Kirschner DA, Wickner RB, Steven AC. Filaments of the Ure2p prion protein have a cross-b core structure. J Struct Biol. 2005;150:170–179. doi: 10.1016/j.jsb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Serpell LC, Smith JM. Direct visualisation of the b-sheet structure of synthetic Alzheimer’s amyloid. J Mol Biol. 2000;299:225–231. doi: 10.1006/jmbi.2000.3650. [DOI] [PubMed] [Google Scholar]
- Oyler NA, Tycko R. Absolute structural constraints on amyloid fibrils from solid state NMR spectroscopy of partially oriented samples. J Am Chem Soc. 2004;126:4478–4479. doi: 10.1021/ja031719k. [DOI] [PubMed] [Google Scholar]
- Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson CM. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc Natl Acad Sci USA. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez JL, Guijarro JL, Orlova E, Zurdo J, Dobson CM, Sunde M, Saibil HR. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. Embo J. 1999;18:815–821. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPhee CE, Dobson CM. Formation of mixed fibrils demonstrates the generic nature and potential utility of amyloid nanostructures. J Am Chem Soc. 2000;122:12707–12713. [Google Scholar]
- Ritter C, Maddelein ML, Siemer AB, Luhrs T, Ernst M, Meier BH, Saupe SJ, Riek R. Correlation of structural elements and infectivity of the HET-s prion. Nature. 2005;435:844–848. doi: 10.1038/nature03793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006;4:100–107. doi: 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer ND, Schmidt JC, Chapman MR. The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc Natl Acad Sci USA. 2007;104:12494–12499. doi: 10.1073/pnas.0703310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewmaker F, McGlinchey RP, Thurber KR, McPhie P, Dyda F, Tycko R, Wickner RB. The functional curli amyloid is not based on in-register parallel b-sheet structure. J Biol Chem. 2009;284:25065–25076. doi: 10.1074/jbc.M109.007054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu KN, McGlinchey RP, Wickner RB, Tycko R. Segmental polymorphism in a functional amyloid. Biophys J. 2011;101:2242–2250. doi: 10.1016/j.bpj.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewmaker F, McGlinchey RP, Wickner RB. Structural insights into functional and pathological amyloid. J Biol Chem. 2011;286:16533–16540. doi: 10.1074/jbc.R111.227108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JX, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, Chan FKM, Wu H. The Rip1/Rip3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macindoe I, Kwan AH, Ren Q, Morris VK, Yang WR, Mackay JP, Sunde M. Self-assembly of functional, amphipathic amyloid monolayers by the fungal hydrophobin EAS. Proc Natl Acad Sci USA. 2012;109:E804–E811. doi: 10.1073/pnas.1114052109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Han TNW, Xie SH, Shi K, Du XL, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei JM, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ader C, Frey S, Maas W, Schmidt HB, Gorlich D, Baldus M. Amyloid-like interactions within nucleoporin FG hydrogels. Proc Natl Acad Sci USA. 2010;107:6281–6285. doi: 10.1073/pnas.0910163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchettini JC, Kelly JW. Therapeutic strategies for human amyloid diseases. Nat Rev Drug Discov. 2002;1:267–275. doi: 10.1038/nrd769. [DOI] [PubMed] [Google Scholar]
- Kung HF, Choi SR, Qu WC, Zhang W, Skovronsky D. 18F stilbenes and styrylpyridines for PET imaging of Ab plaques in Alzheimer’s disease: A miniperspective. J Med Chem. 2010;53:933–941. doi: 10.1021/jm901039z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher AS, Chen KW, Liu XF, Roontiva A, Thiyyagura P, Ayutyanont N, Joshi AD, Clark CM, Mintun MA, Pontecorvo MJ, Doraiswamy PM, Johnson KA, Skovronsky DM, Reiman EM. Using positron emission tomography and florbetapir 18F to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol. 2011;68:1404–1411. doi: 10.1001/archneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- Mathis CA, Wang YM, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang YM, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Antzutkin ON, Balbach JJ, Leapman RD, Rizzo NW, Reed J, Tycko R. Multiple quantum solid state NMR indicates a parallel, not antiparallel, organization of b-sheets in Alzheimer’s b-amyloid fibrils. Proc Natl Acad Sci USA. 2000;97:13045–13050. doi: 10.1073/pnas.230315097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, Dyda F, Reed J, Tycko R. Amyloid fibril formation by a b(16–22), a seven-residue fragment of the Alzheimer’s b-amyloid peptide, and structural characterization by solid state NMR. Biochemistry. 2000;39:13748–13759. doi: 10.1021/bi0011330. [DOI] [PubMed] [Google Scholar]
- Antzutkin ON, Leapman RD, Balbach JJ, Tycko R. Supramolecular structural constraints on Alzheimer’s b-amyloid fibrils from electron microscopy and solid state nuclear magnetic resonance. Biochemistry. 2002;41:15436–15450. doi: 10.1021/bi0204185. [DOI] [PubMed] [Google Scholar]
- Balbach JJ, Petkova AT, Oyler NA, Antzutkin ON, Gordon DJ, Meredith SC, Tycko R. Supramolecular structure in full-length Alzheimer’s b-amyloid fibrils: evidence for a parallel b-sheet organization from solid state nuclear magnetic resonance. Biophys J. 2002;83:1205–1216. doi: 10.1016/S0006-3495(02)75244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzutkin ON, Balbach JJ, Tycko R. Site-specific identification of non-b-strand conformations in Alzheimer’s b-amyloid fibrils by solid state NMR. Biophys J. 2003;84:3326–3335. doi: 10.1016/S0006-3495(03)70057-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R. Solid state NMR reveals a ph-dependent antiparallel b-sheet registry in fibrils formed by a b-amyloid peptide. J Mol Biol. 2004;335:247–260. doi: 10.1016/j.jmb.2003.10.044. [DOI] [PubMed] [Google Scholar]
- Petkova AT, Leapman RD, Guo ZH, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s b-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- Sciarretta KL, Gordon DJ, Petkova AT, Tycko R, Meredith SC. Ab40-lactam(D23/K28) models a conformation highly favorable for nucleation of amyloid. Biochemistry. 2005;44:6003–6014. doi: 10.1021/bi0474867. [DOI] [PubMed] [Google Scholar]
- Paravastu AK, Petkova AT, Tycko R. Polymorphic fibril formation by residues 10–40 of the Alzheimer’s b-amyloid peptide. Biophys J. 2006;90:4618–4629. doi: 10.1529/biophysj.105.076927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Yau WM, Tycko R. Experimental constraints on quaternary structure in Alzheimer’s b-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu ZM, Shi Y, Callaway DJE, Tycko R. Molecular alignment within b-sheets in Ab14–23 fibrils: solid state NMR experiments and theoretical predictions. Biophys J. 2007;92:594–602. doi: 10.1529/biophysj.106.091017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s b-amyloid fibrils. Proc Natl Acad Sci USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. Seeded growth of b-amyloid fibrils from Alzheimer’s brain-derived fibrils produces a distinct fibril structure. Proc Natl Acad Sci USA. 2009;106:7443–7448. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R, Sciarretta KL, Orgel J, Meredith SC. Evidence for novel b-sheet structures in Iowa mutant b-amyloid fibrils. Biochemistry. 2009;48:6072–6084. doi: 10.1021/bi9002666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Yau WM, Tycko R. Structural evolution of Iowa mutant b-amyloid fibrils from polymorphic to homogeneous states under repeated seeded growth. J Am Chem Soc. 2011;133:4018–4029. doi: 10.1021/ja109679q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Yau WM, Luo YQ, Mattson MP, Tycko R. Antiparallel b-sheet architecture in Iowa mutant b-amyloid fibrils. Proc Natl Acad Sci USA. 2012;109:4443–4448. doi: 10.1073/pnas.1111305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Molecular structure of b-amyloid fibrils in Alzheimer’s disease brain tissue. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Kelley K, Tycko R. Polymorph-specific kinetics and thermodynamics of b-amyloid fibril growth. J Am Chem Soc. 2013;135:6860–6871. doi: 10.1021/ja311963f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewmaker F, Wickner RB, Tycko R. Amyloid of the prion domain of Sup35p has an in-register parallel b-sheet structure. Proc Natl Acad Sci USA. 2006;103:19754–19759. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxa U, Wickner RB, Steven AC, Anderson DE, Marekov LN, Yau WM, Tycko R. Characterization of b-sheet structure in Ure2p1–89 yeast prion fibrils by solid state nuclear magnetic resonance. Biochemistry. 2007;46:13149–13162. doi: 10.1021/bi700826b. [DOI] [PubMed] [Google Scholar]
- Shewmaker F, Ross ED, Tycko R, Wickner RB. Amyloids of shuffled prion domains that form prions have a parallel in-register b-sheet structure. Biochemistry. 2008;47:4000–4007. doi: 10.1021/bi7024589. [DOI] [PubMed] [Google Scholar]
- Wickner RB, Dyda F, Tycko R. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register b-sheet structure. Proc Natl Acad Sci USA. 2008;105:2403–2408. doi: 10.1073/pnas.0712032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewmaker F, Kryndushkin D, Chen B, Tycko R, Wickner RB. Two prion variants of Sup35p have in-register parallel b-sheet structures, independent of hydration. Biochemistry. 2009;48:5074–5082. doi: 10.1021/bi900345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryndushkin DS, Wickner RB, Tycko R. The core of Ure2p prion fibrils is formed by the N-terminal segment in a parallel cross-b structure: Evidence from solid state NMR. J Mol Biol. 2011;409:263–277. doi: 10.1016/j.jmb.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R, Savtchenko R, Ostapchenko VG, Makarava N, Baskakov The α-helical C-terminal domain of full-length recombinant PrP converts to an in-register parallel b-sheet structure in PrP fibrils: Evidence from solid state nuclear magnetic resonance. Biochemistry. 2010;49:9488–9497. doi: 10.1021/bi1013134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca S, Yau WM, Leapman R, Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: Constraints from solid state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torok M, Milton S, Kayed R, Wu P, McIntire T, Glabe CG, Langen R. Structural and dynamic features of Alzheimer’s Ab peptide in amyloid fibrils studied by site-directed spin labeling. J Biol Chem. 2002;277:40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- Der-Sarkissian A, Jao CC, Chen J, Langen R. Structural organization of a-synuclein fibrils studied by site-directed spin labeling. J Biol Chem. 2003;278:37530–37535. doi: 10.1074/jbc.M305266200. [DOI] [PubMed] [Google Scholar]
- Margittai M, Langen R. Template-assisted filament growth by parallel stacking of tau. Proc Natl Acad Sci USA. 2004;101:10278–10283. doi: 10.1073/pnas.0401911101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Margittai M, Chen J, Langen R. Investigation of a-synuclein fibril structure by site-directed spin labeling. J Biol Chem. 2007;282:24970–24979. doi: 10.1074/jbc.M700368200. [DOI] [PubMed] [Google Scholar]
- Benzinger TLS, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, Meredith SC. Propagating structure of Alzheimer’s b-amyloid(10–35) is parallel b-sheet with residues in exact register. Proc Natl Acad Sci USA. 1998;95:13407–13412. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG. High-resolution molecular structure of a peptide in an amyloid fibril determined by magic angle spinning NMR spectroscopy. Proc Natl Acad Sci USA. 2004;101:711–716. doi: 10.1073/pnas.0304849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Molecular-level secondary structure, polymorphism, and dynamics of full-length a-synuclein fibrils studied by solid state NMR. Proc Natl Acad Sci USA. 2005;102:15871–15876. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel PCA, Lewandowski JR, Griffin RG. Solid state NMR study of amyloid nanocrystals and fibrils formed by the peptide GNNQQNY from yeast prion protein Sup35p. J Am Chem Soc. 2007;129:5117–5130. doi: 10.1021/ja068633m. [DOI] [PubMed] [Google Scholar]
- Andronesi OC, von Bergen M, Biernat J, Seidel K, Griesinger C, Mandelkow E, Baldus M. Characterization of Alzheimer’s-like paired helical filaments from the core domain of tau protein using solid state NMR spectroscopy. J Am Chem Soc. 2008;130:5922–5928. doi: 10.1021/ja7100517. [DOI] [PubMed] [Google Scholar]
- Heise H, Celej MS, Becker S, Riede D, Pelah A, Kumar A, Jovin TM, Baldus M. Solid state NMR reveals structural differences between fibrils of wild-type and disease-related A53T mutant a-synuclein. J Mol Biol. 2008;380:444–450. doi: 10.1016/j.jmb.2008.05.026. [DOI] [PubMed] [Google Scholar]
- Helmus JJ, Surewicz K, Nadaud PS, Surewicz WK, Jaroniec CP. Molecular conformation and dynamics of the Y145Stop variant of human prion protein. Proc Natl Acad Sci U S A. 2008;105:6284–6289. doi: 10.1073/pnas.0711716105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verel R, Tomka IT, Bertozzi C, Cadalbert R, Kammerer RA, Steinmetz MO, Meier BH. Polymorphism in an amyloid-like fibril-forming model peptide. Angew Chem-Int Edit. 2008;47:5842–5845. doi: 10.1002/anie.200800021. [DOI] [PubMed] [Google Scholar]
- Walsh P, Simonetti K, Sharpe S. Core structure of amyloid fibrils formed by residues 106–126 of the human prion protein. Structure. 2009;17:417–426. doi: 10.1016/j.str.2008.12.018. [DOI] [PubMed] [Google Scholar]
- Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO. Structural conversion of neurotoxic amyloid-b(1–42) oligomers to fibrils. Nat Struct Mol Biol. 2010;17:561–U556. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayro MJ, Maly T, Birkett NR, MacPhee CE, Dobson CM, Griffin RG. High-resolution MAS NMR analysis of PI3-SH3 amyloid fibrils: Backbone conformation and implications for protofilament assembly and structure. Biochemistry. 2010;49:7474–7484. doi: 10.1021/bi100864t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelouchina GT, Platt GW, Bayro MJ, Radford SE, Griffin RG. Magic angle spinning NMR analysis of b2-microglobulin amyloid fibrils in two distinct morphologies. J Am Chem Soc. 2010;132:10414–10423. doi: 10.1021/ja102775u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GT Debelouchina, GW Platt, MJ Bayro, SE Radford, RG Griffin. Intermolecular alignment in b2-microglobulin amyloid fibrils. J Am Chem Soc. 2010;132:17077–17079. doi: 10.1021/ja107987f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmus JJ, Surewicz K, Surewicz WK, Jaroniec CP. Conformational flexibility of Y145Stop human prion protein amyloid fibrils probed by solid state nuclear magnetic resonance spectroscopy. J Am Chem Soc. 2010;132:2393–2403. doi: 10.1021/ja909827v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I, Gonnelli L, Luchinat C, Mao JF, Nesi A. A new structural model of Ab(40) fibrils. J Am Chem Soc. 2011;133:16013–16022. doi: 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]
- Comellas G, Lemkau LR, Nieuwkoop AJ, Kloepper KD, Ladror DT, Ebisu R, Woods WS, Lipton AS, George JM, Rienstra CM. Structured regions of a-synuclein fibrils include the early-onset Parkinson’s disease mutation sites. J Mol Biol. 2011;411:881–895. doi: 10.1016/j.jmb.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmus JJ, Surewicz K, Apostol MI, Surewicz WK, Jaroniec CP. Intermolecular alignment in Y145Stop human prion protein amyloid fibrils probed by solid state NMR spectroscopy. J Am Chem Soc. 2011;133:13934–13937. doi: 10.1021/ja206469q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski JR, van der Wel PCA, Rigney M, Grigorieff N, Griffin RG. Structural complexity of a composite amyloid fibril. J Am Chem Soc. 2011;133:14686–14698. doi: 10.1021/ja203736z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathy S, Long F, Miller Y, Xiao YL, McElheny D, Thurber K, Ma BY, Nussinov R, Ishii Y. Molecular-level examination of Cu2+ binding structure for amyloid fibrils of 40-residue Alzheimer’s Ab by solid state NMR spectroscopy. J Am Chem Soc. 2011;133:3390–3400. doi: 10.1021/ja1072178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Schumacher MC, Mueller H, Nand D, Klaukien V, Heise H, Riedel D, Wolf G, Behrmann E, Raunser S, Seidel R, Engelhard M, Baldus M. Structural characterization of polyglutamine fibrils by solid state NMR spectroscopy. J Mol Biol. 2011;412:121–136. doi: 10.1016/j.jmb.2011.06.045. [DOI] [PubMed] [Google Scholar]
- Bedrood S, Li YY, Isas JM, Hegde BG, Baxa U, Haworth IS, Langen R. Fibril structure of human islet amyloid polypeptide. J Biol Chem. 2012;287:5235–5241. doi: 10.1074/jbc.M111.327817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkau LR, Comellas G, Kloepper KD, Woods WS, George JM, Rienstra CM. Mutant protein A30P a-synuclein adopts wild-type fibril structure, despite slower fibrillation kinetics. J Biol Chem. 2012;287:11526–11532. doi: 10.1074/jbc.M111.306902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoop CL, Mishra R, Kar K, Kodali R, Wetzel R, van der Wel PCA. Structural and motional investigations of polyglutamine-containing amyloid fibrils by magic-angle-spinning solid state NMR. Biophys J. 2013;104:181A–181A. [Google Scholar]
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Bockmann A, Meier BH, Melki R. Structural and functional characterization of two a-synuclein strains. Nat Commun. 2013;4 doi: 10.1038/ncomms3575. [PAGE #S] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Melckebeke H, Wasmer C, Lange A, Ab E, Loquet A, Bockmann A, Meier BH. Atomic-resolution three-dimensional structure of HET-s(218–289) amyloid fibrils by solid state NMR spectroscopy. J Am Chem Soc. 2010;132:13765–13775. doi: 10.1021/ja104213j. [DOI] [PubMed] [Google Scholar]
- Goldsbury CS, Wirtz S, Muller SA, Sunderji S, Wicki P, Aebi U, Frey P. Studies on the in vitro assembly of Ab1–40: implications for the search for Ab fibril formation inhibitors. J Struct Biol. 2000;130:217–231. doi: 10.1006/jsbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- Jimenez JL, Nettleton EJ, Bouchard M, Robinson CV, Dobson CM, Saibil HR. The protofilament structure of insulin amyloid fibrils. Proc Natl Acad Sci USA. 2002;99:9196–9201. doi: 10.1073/pnas.142459399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhardt J, Sachse C, Hortschansky P, Grigorieff N, Fandrich M. Ab(1–40) fibril polymorphism implies diverse interaction patterns in amyloid fibrils. J Mol Biol. 2009;386:869–877. doi: 10.1016/j.jmb.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Thurber KR, Shewmaker F, Wickner RB, Tycko R. Measurement of amyloid fibril mass-per-length by tilted-beam transmission electron microscopy. Proc Natl Acad Sci U S A. 2009;106:14339–14344. doi: 10.1073/pnas.0907821106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsbury C, Frey P, Olivieri V, Aebi U, Muller SA. Multiple assembly pathways underlie amyloid-b fibril polymorphisms. J Mol Biol. 2005;352:282–298. doi: 10.1016/j.jmb.2005.07.029. [DOI] [PubMed] [Google Scholar]
- Kodali R, Williams AD, Chemuru S, Wetzel R. Ab(1–40) forms five distinct amyloid structures whose b-sheet contents and fibril stabilities are correlated. J Mol Biol. 2010;401:503–517. doi: 10.1016/j.jmb.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Lopresti BJ, Debnath ML, Holt DP, Wang YM, Huang GF, Shao L, Lefterov I, Koldamova R, Ikonomovic M, DeKosky ST, Mathis CA. Amyloid deposits in transgenic PS1/APP mice do not bind the amyloid PET tracer, PIB, in the same manner as human brain amyloid. Neurobiol Aging. 2004;25:S232–S233. [Google Scholar]
- Nilsson KPR, Aslund A, Berg I, Nystrom S, Konradsson P, Herland A, Inganas O, Stabo-Eeg F, Lindgren M, Westermark GT, Lannfelt L, Nilssonl LNG, Hammarstrom P. Imaging distinct conformational states of amyloid-b fibrils in Alzheimer’s disease using novel luminescent probes. ACS Chem Biol. 2007;2:553–560. doi: 10.1021/cb700116u. [DOI] [PubMed] [Google Scholar]
- McDonald M, Box H, Bian W, Kendall A, Tycko R, Stubbs G. Fiber diffraction data indicate a hollow core for the Alzheimer’s Ab 3-fold symmetric fibril. J Mol Biol. 2012;423:454–461. doi: 10.1016/j.jmb.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gath J, Bousset L, Habenstein B, Melki R, Bockmann A, Meier BH. Unlike twins: An NMR comparison of two a-synuclein polymorphs featuring different toxicity. PLoS One. 2014;9 doi: 10.1371/journal.pone.0090659. [PAGES] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daebel V, Chinnathambi S, Biernat J, Schwalbe M, Habenstein B, Loquet A, Akoury E, Tepper K, Muller H, Baldus M, Griesinger C, Zweckstetter M, Mandelkow E, Vijayan V, Lange A. β-sheet core of tau paired helical filaments revealed by solid state NMR. J Am Chem Soc. 2012;134:13982–13989. doi: 10.1021/ja305470p. [DOI] [PubMed] [Google Scholar]
- Toyama BH, Kelly MJS, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature. 2007;449:233–U238. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- Luckgei N, Schutz AK, Bousset L, Habenstein B, Sourigues Y, Gardiennet C, Meier BH, Melki R, Bockmann A. The conformation of the prion domain of Sup35p in isolation and in the full-length protein. Angew Chem-Int Edit. 2013;52:12741–12744. doi: 10.1002/anie.201304699. [DOI] [PubMed] [Google Scholar]
- Frederick KK, Debelouchina GT, Kayatekin C, Dorminy T, Jacavone AC, Griffin RG, Lindquist S. Distinct prion strains are defined by amyloid core structure and chaperone binding site dynamics. Chem Biol. 2014;21:295–305. doi: 10.1016/j.chembiol.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EM, Surewicz WK. Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell. 2005;121:63–72. doi: 10.1016/j.cell.2005.01.034. [DOI] [PubMed] [Google Scholar]
- Cobb NJ, Apostol MI, Chen SG, Smirnovas V, Surewicz WK. Conformational stability of mammalian prion protein amyloid fibrils is dictated by a packing polymorphism within the core region. J Biol Chem. 2014;289:2643–2650. doi: 10.1074/jbc.M113.520718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin A, Chung DS, Benedek GB, Kirschner DA, Teplow DB. On the nucleation and growth of amyloid b-protein fibrils: detection of nuclei and quantitation of rate constants. Proc Natl Acad Sci USA. 1996;93:1125–1129. doi: 10.1073/pnas.93.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin A, Teplow DB, Kirschner DA, Benedek GB. Kinetic theory of fibrillogenesis of amyloid b-protein. 1997;94:7942–7947. doi: 10.1073/pnas.94.15.7942. Proc Natl Acad Sci USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SM, Ferrone FA, Wetzel R. Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. Proc Natl Acad Sci USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SIA, Vendruscolo M, Dobson CM, Knowles TPJ. Nucleated polymerization with secondary pathways. II. Determination of self-consistent solutions to growth processes described by non-linear master equations. J Chem Phys. 2011;135:065106. doi: 10.1063/1.3608917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIA Cohen, M Vendruscolo, CM Dobson, TPJ andKnowles. Nucleated polymerization with secondary pathways. III. Equilibrium behavior and oligomer populations. J Chem Phys. 2011;135:065107. doi: 10.1063/1.3608918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SIA, Vendruscolo M, Welland ME, Dobson CM, Terentjev EM, Knowles TPJ. Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J Chem Phys. 2011;135:065105. doi: 10.1063/1.3608916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SIA, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TPJ. Proliferation of amyloid-b42 aggregates occurs through a secondary nucleation mechanism. 2013;110:9758–9763. doi: 10.1073/pnas.1218402110. Proc Natl Acad Sci USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- Safar J, Wille H, Itrri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrPSc molecules with different conformations. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- Wickner RB, Shewmaker F, Edskes H, Kryndushkin D, Nemecek J, McGlinchey R, Bateman D, Winchester CL. Prion amyloid structure explains templating: How proteins can be genes. FEMS Yeast Res. 2010;10:980–991. doi: 10.1111/j.1567-1364.2010.00666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer’s disease. Neurobiol Dis. 2010;37:503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral b-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Ab seeds are potent inducers of cerebral b-amyloid deposition. J Neurosci. 2011;31:14488–14495. doi: 10.1523/JNEUROSCI.3088-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, Prusiner SB, Giles K. Purified and synthetic Alzheimer’s amyloid b (Ab) prions. Proc Natl Acad Sci USA. 2012;109:11025–11030. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]