

Abstract
An aldo-keto reductase AKR5C3 from Gluconobacter oxydans (designated as Gox0644) is a useful enzyme with various substrates, including aldehydes, diacetyl, keto esters, and α-ketocarbonyl compounds. The crystal structures of AKR5C3 in apoform in complex with NADPH and the D53A mutant (AKR5C3-D53A) in complex with NADPH are presented herein. Structure comparison and site-directed mutagenesis combined with biochemical kinetics analysis reveal that the conserved Asp53 in the AKR5C3 catalytic tetrad has a crucial role in securing active pocket conformation. The gain-of-function Asp53 to Ala mutation triggers conformational changes on the Trp30 and Trp191 side chains, improving NADPH affinity to AKR5C3, which helps increase catalytic efficiency. The highly conserved Trp30 and Trp191 residues interact with the nicotinamide moiety of NADPH and help form the NADPH-binding pocket. The AKR5C3-W30A and AKR5C3-W191Y mutants show decreased activities, confirming that both residues facilitate catalysis. Residue Trp191 is in the loop structure, and the AKR5C3-W191Y mutant does not react with benzaldehyde, which might also determine substrate recognition. Arg192, which is involved in the substrate binding, is another important residue. The introduction of R192G increases substrate-binding affinity by improving hydrophobicity in the substrate-binding pocket. These results not only supplement the AKRs superfamily with crystal structures but also provide useful information for understanding the catalytic properties of AKR5C3 and guiding further engineering of this enzyme.
Keywords: aldo-keto reductase, AKR5C3, crystal structure, site-directed mutagenesis
Introduction
Aldo-keto reductases (AKRs) are a large group of NAD(P)H-dependent oxidoreductases that catalyze the reversible reduction of carbonyl compounds (e.g., aliphatic and aromatic aldehydes/ketones, monosaccharides, steroids, prostaglandins, polycyclic aromatic carbons, and polyketides) found in various species including plants, animals, and prokaryotes.1–4 AKRs usually share less than 30% amino acid sequence identities, but they possess the canonical (α/β)8 TIM-barrel structure and have a common reaction mechanism based on the highly conserved catalytic tetrad of Tyr, Lys, His, and Asp, also known as the DYKH tetrad. The His in the DYKH tetrad reportedly aids in catalytic substrate molecule arrangement and protonation.5 A “push–pull” mechanism has been proposed to highlight Tyr to be bifunctional between His and Lys.6 The assembled Asp-Lys-Tyr protonation system has a salt bridge between Asp and Lys and a hydrogen bond between Lys and Tyr to maintain the cage.7 The function of Asp in the catalytic process is putatively indirect, although the D43N variant of human aldose reductase increases Km-NADPH fivefold and increases Kd-NADPH 5.7-fold.8 AKR generally hosts its active site on the carboxyl-terminal face of the central β-barrel, where several highly variable loops affect substrate specificity.4 Many AKRs have been characterized, and their structure-function relationships have been revealed, enabling enzyme engineering in view of their vital roles in physiological metabolism and their versatile potential in pharmaceutical and organic compound synthesis.9–17
The AKR Gox0644 from Gluconobacter oxydans (G. oxydans), designated as “AKR5C3” according to the AKR nomenclature system (http://www.med.upenn.edu/akr), possesses broad substrate specificities with important biological functions and great biotechnological interests, for example, reduction of aliphatic and aromatic aldehydes involved in aldehyde metabolism and detoxification in G. oxydans, regio- and stereoselective reduction of α-ketoaldehydes, α-diketones, and α-keto esters to the corresponding α-hydroxyaldehyde and α-hydroxyl esters, which are key intermediates in the production of numerous pharmaceuticals and natural products.18,19 AKR5C3 reportedly reduces diacetyl to highly optically pure (3S)-acetoin, which is an important platform chemical.20
To understand the three-dimensional (3D) enzyme structural information and its role in determining specificity and catalysis of this enzyme, we solved the structures of AKR5C3 in apoform and binary complex with NADPH. A distinct NADPH-binding conformation of mutant AKR5C3-D53A/NADPH was also found through crystal structure analysis. Tryptophan residue (Trp191) and arginine residue (Arg192) are the critical determinants for substrate binding to the enzyme. The structural AKR5C3-R192G model and the substrate docking show stronger substrate-binding pocket hydrophobicity.
Results and Discussion
Overall structure of AKR5C3
AKR5C3 showed 51% identity and 72% similarity to a biochemical characterized as 2,5-diketone-d-gluconate reductase from Escherichia coli (E. coli)(DkgA), which could catalyze the reduction of 2,5-diketo-d-gluconate to 2-keto-l-gulonate. Phylogenetic analysis using ClustalW indicated that AKR5C3 diverged from an ancient common ancestor with 2,5-diketo-d-gluconic acid reductases from E. coli.19 The AKR5C3 apoform structure was determined through molecular replacement using E. coli DkgA (AKR5C2)21 as the search model (PDBID: 1MZR) and was refined using space group P212121 to 2.3 Å resolution (Fig. 1). Two molecules were present per asymmetric unit, and Matthews coefficient was ∼2.1 with approximately 45% solvent content. AKR5C3 formed a dimer in one asymmetric unit, and the model comprised residues 9–279. The AKR5C3/NADPH binary complex structure was initially refined to 2.2 Å. The crystallographic statistics are summarized in Table 1.
Figure 1.
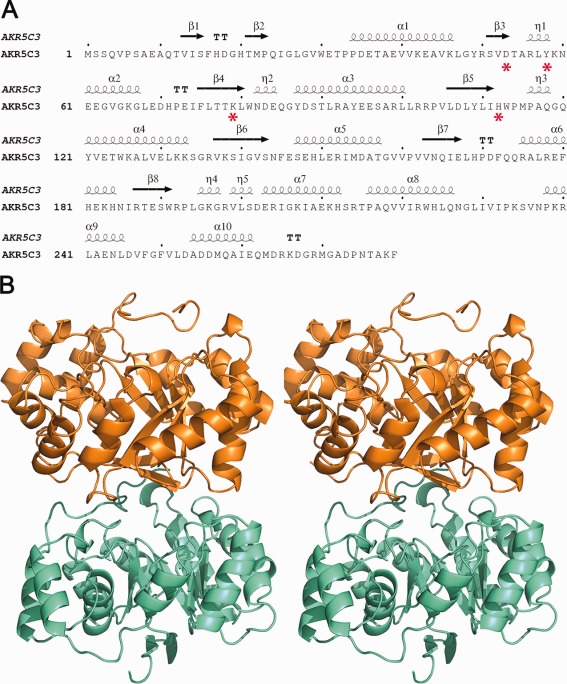
Sequence analysis, secondary structure elements, and overall structure of AKR5C3. (A) Sequence and secondary structure of AKR5C3. The secondary structure diagram for AKR5C3 is shown at the top of the sequence. The α-helices and β-strands are indicated. Conserved catalytic tetrad DYKH are highlighted with red asterisks. (B) Stereo-view cartoon representation of AKR5C3 apoform showing the typical (β/α)8 fold. Two molecules of a dimer are colored in orange and cyan, respectively. An interactive view is available in the electronic version of the article.
Table 1.
Data Collection and Refinement Statistics
| AKR5C3 apoform | AKR5C3 /NADPH binary complex | AKR5C3-D53A/NADPH | |
|---|---|---|---|
| PDBID | 3WBX | 3WBW | 3WBY |
| Space group | P212121 | P212121 | P212121 |
| Cell dimensions | |||
| a (Å) | 55.879 | 55.989 | 55.638 |
| b (Å) | 75.780 | 75.778 | 76.587 |
| c (Å) | 125.585 | 125.604 | 125.624 |
| Protein molecules/ASU | 2 | 2 | 2 |
| Wavelength (Å) | 1.54178 | 0.97916 | 1.079 |
| Resolution (Å) a | 2.3 (2.38–2.3) | 1.85 (1.92–-1.85) | 3.2 (3.26–-3.2) |
| Rsym (%) a | 11.9 (67.3) | 5.2 (38.4) | 16.6 (63.9) |
| I/σ(I) | 23.1 (3.4) | 31.2 (4.3) | 13.5 (3.2) |
| Completeness (%)a | 99.4 (98.9) | 99.6 (94.6) | 100 (100) |
| Redundancya | 13.8 (13.1) | 7.2 (7.3) | 7.0 (7.0) |
| Resolution (Å) | 2.3 | 1.85 | 3.2 |
| No. reflections | 20,012 | 44,086 | 8821 |
| Rwork (Rfree) (%) | 22.4 (30.0) | 21.6 (25.7) | 22.1 (29.1) |
| No. atoms | |||
| Protein | 4366 | 4364 | 4358 |
| NADPH | –- | 96 | 96 |
| Sulfate | 20 | 10 | – |
| Water | 214 | 304 | – |
| B-factors (Å2) | |||
| Protein | 24.339 | 28.954 | 68.323 |
| NADPH | –- | 38.544 | 81.793 |
| Sulfate | 42.672 | 42.107 | – |
| Water | 22.795 | 31.265 | – |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.011 | 0.017 | 0.012 |
| Bond angles (°) | 1.285 | 1.589 | 1.641 |
| % favored (allowed) in Ramachandran plot | 89.8 (10.2) | 92.8 (7.2) | 86.2 (13.6) |
Values for the highest-resolution shell are in parentheses.
The overall AKR5C3 structure resembles canonical AKR structures with a typical TIM-barrel,22 with eight β-strands (β1–β8) in the middle surrounded by eight α-helices (α1–α6 and α8–α9) and interrupted by two extra α-helices (α7 and α10) [Fig. 1(A)]. Three well-ordered short loops linking α2–β4, β5–α4, and β8–α7 are in both structures. These loops are located above the bound NADPH molecule and form an open pocket for substrate binding. The length and the sequence of these loops possibly play important roles in substrate selection.4
NADPH binding-induced structural alteration
The NADPH molecule is located in a deep cavity within the (β/α)8 barrel in the binary structure. The NADPH molecule with anti-conformation is deep inside a channel connected to the central active site cavity in the binary complex structure. The nicotinamide and ribose moieties are stabilized by potential hydrogen bonds and π-stacking interactions from the highly conserved residues Asp53, Tyr58, Lys79, His112, Trp30, Trp113, and Trp19 [Fig. 2(A)]. The well-conserved DYKH tetrad catalytic residues are above the nicotinamide moiety of NADPH, with the side chains pointing toward the nicotinamide moiety. The phosphate group and the NADPH adenine moieties are surrounded by Arg192, Lys234, Asn237, and Arg240. A direct hydrogen bond is formed between the Lys234 side chain and the phosphate group, which proves that NADPH, not NADH, is a natural AKR5C3 cofactor. The Lys234 side chain also fits the crescent groove of the extended NADPH molecule, whereas the Arg240 side chain partially stacks over the adenine moiety of the NADPH molecule to secure tight binding of AKR5C3 and NADPH. Hydrogen bonds were also observed between Asn237 side chain and NADPH phosphate group.
Figure 2.
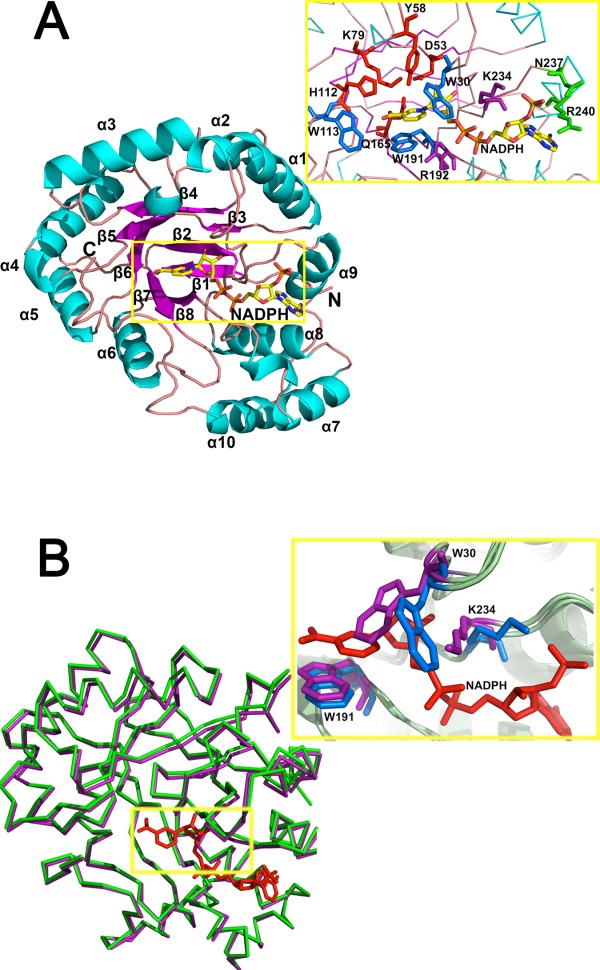
Residues involved in NADPH-binding. (A) Structural details of the (β/α)8 of AKR5C3/NADPH and key residues involved in NADPH-binding pocket. Catalytic tetrad of D53, Y58, K79, and H112 are colored in red, NADPH nicotinamide moiety recognition residues (W30, W113, and W191) are colored in blue, potential NADPH-binding residues (R192 and K234) are colored in magenta, and putative adenine moiety recognition residues (N237 and R240) are colored in green. (B) Superposition of the apoform AKR5C3 and AKR5C3/NADPH binary complex. The ribbon representation of apoform is in green, the binary complex form is in magenta, and NADPH is in red. The detailed NADPH binding-induced conformational alterations are observed in residues W30 and K234. Residues with conformational change are in marine (AKR5C3/NADPH binary complex) and purple (AKR5C3 apoform). An interactive view is available in the electronic version of the article.
The AKR5C3 crystal structures in apoform and in complex with NADPH are superimposed, assuming that the mechanistic clues for the cofactor dependence lie in the conformational plasticity of AKR5C3 upon NADPH binding [Fig. 2(B)]. The two structures trace almost identical chain arrangements, but significant conformational changes are at the NADPH-binding pocket and the putative substrate-binding cleft. The NADPH binding rotates the Trp30 side chain ∼90° away from Trp191 and opens the cofactor binding pocket to be accessible to NADPH. By contrast, the Lys234 side chain rotates by ∼60° to tightly fit into the crescent groove of the extended NADPH molecule. Trp30 plasticity suggests a function in NADPH binding, thereby affecting AKR5C3 catalytic activity. Thus, Trp30 was mutated to Ala to avoid any side chain tension. The AKR5C3-W30A variant showed decreased activity toward the tested substrate (Supporting Information Fig. S2). This result is consistent with AKR11B4−Trp23Ala activity. Residue Trp23 in AKR11B4 was proven to be a key residue for NADP+/NADPH and substrate turnover.23
The conserved catalytic tetrad DYKH
Similar to other AKR members, DYKH tetrad joins synchrony to catalyze the reduction of carbonyl substrates. Tyr58 is flanked in the AKR5C3 active sites, and is hydrogen-bonded to Asp53 and Lys79, whereas His112 is next to Lys79 and is closely located to the bound NADPH. The phenolic hydroxyl group of Tyr58 provides general acid catalytic assistance to carbonyl group reduction [Fig. 3(A)]. The four residues were separately mutated to Ala to decrease steric hindrance and partially disrupt the hydrogen-bond network to confirm whether the conserved catalytic tetrad residues are involved in substrate catalysis. The second group of mutants was designed such that the size of side chains would minimally deviate, namely D53N, Y58F, K79R, and H112F. The site-directed mutagenesis of these residues confirms their role in catalysis because neither mutant exhibited decreased or detectable benzaldehyde reduction activity, except for the D53A mutant [Fig. 3(B)]. The D53A mutant exhibited slightly increased benzaldehyde catalysis (27% increase) [Fig. 3(B)]. The other five substrates (glyceraldehyde, glutaraldehyde, octaldehyde, o-chlorobenzaldehyde, and butanedione) were catalyzed by the variant AKR5C3-D53A with higher activities (by 0.5∼2-fold improvement) than the wild-type enzyme [Fig. 4(C)].
Figure 3.
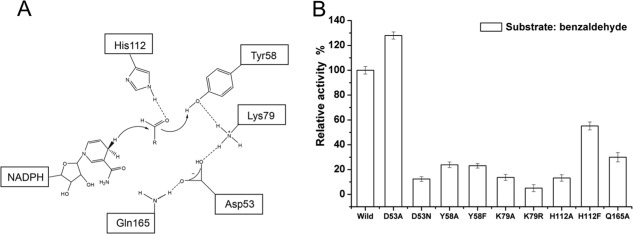
The proposed catalytic mechanisms and reductive activity determined for AKR5C3 mutants. (A) Schematic representation of NADPH-dependent reduction of the carbonyl group. Hydride transfer occurs from the pro-R-hydrogen of NADPH to the carbonyl group of substrate, whereas C carbonyl is polarized by both Tyr58 and His112. Dashed lines show hydrogen bond network. (B) The relative activity of AKR5C3 and its mutants using benzaldehyde as substrate. The enzyme activity was measured as described in “Materials and methods”. The reaction mixture contains 0.05M potassium phosphate (pH 6.5), 1 mM NADPH and 2.5 mM substrate, initiated at 30°C by the addition of NADPH. One unit enzyme activity was defined as the amount of enzyme activity catalyzing the conversion of 1.0 µmol pyridine nucleotide per minute at optimum temperature. The 100% activity of wild AKR5C3 with benzaldehyde was 2.91 U mg−1.
Figure 4.
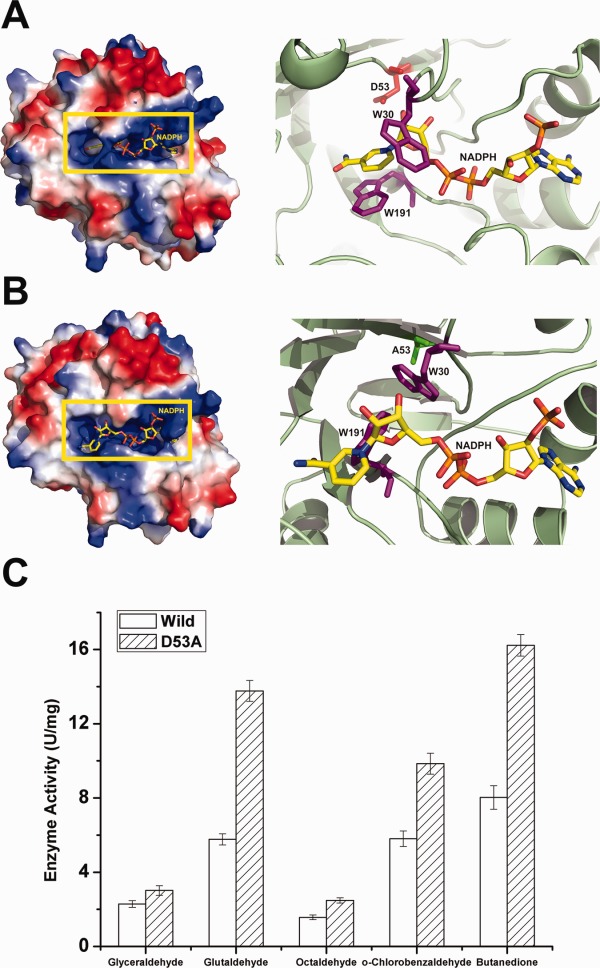
Structural observations of AKR5C3-D53A/NADPH and reduction activity assays. (A) Electrostatic potential surface view of AKR5C3. D53 is shown in red and side chains of residues W30 and W191 are shown in magenta. (B) Electrostatic potential surface view of AKR5C3-D53A. A53 is shown in green and side chains of residues W30 and W191 are shown in magenta. Compared with the corresponding residues in wild AKR5C3 binary complex, the side chains of W30 and W191 in AKR5C3-D53A/NADPH showed a rotation and the bound NADPH molecule rearranged the nicotimide moiety. (C) Enzyme activity of AKR5C3 and mutant AKR5C3-D53A. An interactive view is available in the electronic version of the article.
The Gln165 residue located in β7 near the active sites interacts with Asp53 via hydrogen bond. It was noted that Gln165 is conserved in the vast majority of bacterial AKRs. The decreased activity toward benzaldehyde of the variant AKR5C3-Q165A indicates the effect of the position of the Gln165 side chain. The Gln165 hydrogen bonded with Asp53 helps stabilize the catalytic tetrad. Therefore, a hydrogen bond network of Gln165-Asp53-Lys79-Tyr58 was proposed to facilitate hydrogen donation to the carbonyl oxygen of the bound substrate from Tyr58.
Catalytic residue Asp53 and the Crystal structure of AKR5C3-D53A/NADPH
The crystal structure of AKR5C3-D53A in complex with NADPH was determined to investigate how Ala mutation on the catalytic Asp residue affects catalysis. The overall AKR5C3-D53A conformation is almost identical to the wild type, but significant conformational changes exist on the Trp30 and Trp191 side chains. The Trp30 side chain adopts a rotamer from that of AKR5C3/NADPH or that of AKR5C3 at free form [Fig. 4(A–B)]. The Trp30 side chain in the AKR5C3-D53A/NADPH mutant structure rotates ∼90° away from the original position in the AKR5C3/NADPH binary complex structure. The Trp191 side chain remarkably rotates ∼90° toward the Trp30 side chain. Thus, the pocket used for the binding of nicotinamide and NADPH ribose moieties is occupied by the Trp30 and Trp191 side chains. The nicotinamide and ribose moieties of NADPH subsequently rotate 60° away from the original binding pocket and partially π-stacks over the Trp191 and Arg192 side chains in the mutant AKR5C3-D53A structure. The significant conformational changes on Trp30 and Trp191 side chains and the nicotinamide and ribose moieties of NADPH significantly changed the shape, size, and location of the substrate-binding pocket. The structure shows that the affinity between NADPH and enzyme should be largely affected, thus facilitating substrate catalysis.
To elucidate the reason for the increases in activity from a biochemical perspective, the apparent kinetic parameters of the Km-NADPH and Km-Butanedione pairs were measured according to Lineweaver–Burk plots (Supporting Information Fig. S3) to determine why activity increases from a biochemical perspective. The Km value of NADPH and the Km of the substrates are largely affected (Table 2). The Km-value of AKR5C3-D53A for NADPH is significantly reduced from 0.36 mM (wild type) to 0.04 mM, indicating about ninefold stronger binding affinity. Although kcat decreased to 104.22 s−1, kcat/Km is 2.68-fold compared to that of the wild AKR5C3, showing that the catalytic efficiency of cofactor NADPH improved. The Km-butanedione of AKR5C3-D53A slightly increased by 1.38-fold compared to that of AKR5C3. The turnover number (kcat) of AKR5C3-D53A increased from 42.80 s−1 to 77.82 s−1, and thus, catalytic efficiency (kcat/Km) increased by 1.31-fold. AKRs follow an ordered bi–bi reaction kinetic mechanism within which the NAD(P)(H) cofactor binds ahead of the substrate and leaves last.19 The major rate-limiting step of the catalysis is often cofactor binding and release rate instead of substrate recognition.24,25 These observations confirm that cofactor turnover is the major rate determinant in AKR5C3 activity. The AKR5C3-D53A/NADPH crystal structure shows that the contribution of Alanine in Asp53 changes the conformation of the Trp30 and Trp191 side chains, thus reorienting the nicotinamide and ribose moieties of NADPH. Asp53 does not specifically go in contact with Trp30 or Trp191, but ionic Asp53 is predicted to tightly hold the oxidized cofactor through Trp30 and Trp191 in the binding pocket, which could be the driving force of the catalysis.
Table 2.
Kinetic Constants of AKR5C3, AKR5C3-D53A, and AKR5C3-R192G
| Apparent kinetic parameters | Km-NADPH (mM) | kcat (s−1) | kcat/Km (s−1 mM−1) | Km-Butanedione (mM) | kcat (s−1) | kcat/Km (s−1 mM−1) |
|---|---|---|---|---|---|---|
| AKR5C3 | 0.36 | 349.54 | 970.94 | 4.62 | 42.80 | 9.26 |
| AKR5C3-D53A | 0.04 | 104.22 | 2605.50 | 6.39 | 77.82 | 12.18 |
| AKR5C3-R192G | 0.67 | 18.67 | 27.87 | 1.08 | 75.49 | 69.90 |
Trp191 and Arg192 are key residues in substrate-binding pocket
The substrate-binding pocket in the AKR5C3 structure is formed by the tetrad catalytic residues at the top; conserved Trp30, Trp113, and C-terminal tail (Phe279) bracketed on both sides; and conserved Trp191 and variable Arg192 at the bottom [Fig. 5(B), right panel]. The modeled butanedione molecule is partially sandwiched between the π-electron atmosphere formed by the nicotinamide moiety of NADPH, Trp191, and Arg192. Residues Trp191 and Arg192 are in the loop structure, which presumably contributes to substrate recognition. We searched all the sequences of bacterial AKRs in database (http://www.med.upenn.edu/akr/tree.shtml) whose structure are reported and found Trp191 to be relatively conserved residue, while Arg192 a variable residue [Fig. 5(A)]. Tyr is another conserved residue in the Trp191 relative position, whereas Gly and Ser are two common residues in the Arg192 relative position. The catalytic characterization shows that AKR5C3 shares a relatively similar substrate spectrum (carbonyl compound reduction) with the AKR11B4 from G. oxydans.18,19,26 AKR5C3 displayed higher activities with 2,3-diones, α-keto esters, and 2,5-diketo-gluconate than aldehydes, whereas AKR11B4 was less active with these keto substrates, but displayed preference for aliphatic, branched, and aromatic aldehydes. Neither Trp191 nor Arg192 in AKR5C3 are conserved in AKR11B4. The Trp191 of AKR5C3 is replaced by Tyr208 in the AKR11B4 structure, whereas Arg192 is replaced by Gly209. AKR5C3 structure was superimposed on AKR11B4 structure (3N2T, Z score 27.3, r.m.s.d. 2.3 Å, 253 Cα atoms) to investigate the difference of substrate binding pocket23 using the DaliLite server. AKR5C3 and AKR11B4 superimposed well at the (β/α)8 barrel portion, but AKR5C3 has shorter flexible loops surrounding the putative substrate-binding pocket than the AKR11B4 structure [Fig. 5(B), left panel]. The location of the C-terminal tail in the AKR5C3 structure is partially occupied by two flexible loops in AKR11B4. The AKR5C3 C-terminus participates in substrate binding pocket formation, whereas the AKR11B4 C-terminus is swung away from the pocket. These differences in C-terminal tail orientations noticeably affect the sizes of the substrate-binding pockets of AKR5C3 and AKR11B4. AKR11B4 has a slightly bigger substrate-binding pocket than that of AKR5C3.
Figure 5.
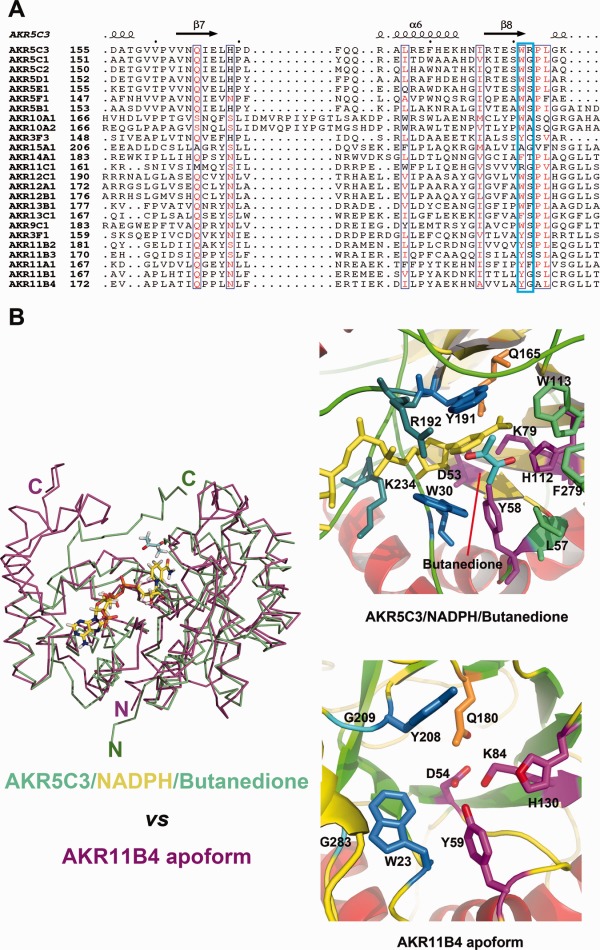
Key residues W191 and R192 involved in substrate catalysis. (A) Sequences (involved in substrate binding or recognition) alignment between AKR5C3 and other bacterial AKRs. The listed bacterial AKRs are from http://www.med.upenn.edu/akr/tree.shtml. W191 is relatively conserved while R192 is much variable. AKR11B4 and AKR5C3 shared a similar substrate spectrum but neither W191 nor R192 is conserved. (B) Structural superimposition of AKR5C3/NADPH/Butanedione (colored in green) and AKR11B4 in apoform (colored in magenta). The bound NADPH is highlighted in stick and colored in yellow. The right panel is cartoon presentation of the detailed substrate-binding pocket. The catalytic tetrad residues are colored in magenta, NADPH recognition residues are colored in blue, conserved Gln residue is colored in orange, whereas the putative aromatic cage residues for substrate binding are colored in marine. Substrate butanedione is colored in cyan.
Based on the structural differences, we made single mutants to replace Trp191 and Arg192 with their corresponding residue at AKR11B4, respectively. The π-electron atmosphere disruption at the substrate-binding pocket of AKR5C3 was expected to contribute to substrate preference. AKR5C3-W191Y single mutant exhibited decreased activity with glutaldehyde, octaldehyde, or benzaldehyde (Supporting Information Fig. S2). Almost no activity was detected toward benzaldehyde when Trp191 was mutated to Tyr, suggesting that Trp191 is a critical and strictly conservative residue for benzaldehyde reduction. The structure observation and the previous description suggest that Trp191 supports NADPH binding, thus affecting catalytic activity. The AKR5C3-W191Y mutant also displayed decreased activity with glutaraldehyde or octaldehyde (Supporting Information Fig. S2). This result, the structure observation, and the previous description suggest that Trp191 plays a key roles in catalytic activity, which supports NADPH binding and π-π packing formation against the aromatic ring of substrates (e.g., benzaldehyde). The removal of the bulky Arg192 side chain by mutation was also expected to create a large cavity suitable for substrates with long aliphatic chains. The AKR5C3-R192G mutant expectedly increased reductive activity to the tested substrates, and the activity increase rate toward long-chain aliphatic aldehydes (glutaldehyde and octaldehyde) was higher than that of the other aldehydes or ketone [Fig. 6(C)]. The structural insights into AKR5C3-R192G were based on a 3D model generated through homology modeling using Discovery Studio based on the existing AKR5C3/NADPH crystal structure [Fig. 6(A,B)]. The bound butanedione substrate fits into a pre-formed hydrophobic aromatic cage formed by several aromatic rings and hydrophobic residues, including Trp81, Trp113, Phe279, and Leu57. The stronger hydrophobicity of the substrate-binding pocket in AKR5C3-R192G benefits catalysis. The kinetic data of AKR5C3-R192G and its comparison with the wild enzyme are consistent with the model structure-based predictions. The Km constants of AKR5C3-R192G for butanedione decreased significantly when compared to that of AKR5C3, thus indicating an over fourfold stronger binding affinity. Although the Km-NADPH increased, the kcat value of butanedione exhibited a 1.76-fold to that of AKR5C3 and therefore the catalytic efficiency (kcat/Km) increased 7.55-fold. A small-scale reduction of 2,3-butanedione was performed through simultaneous regeneration of NADPH using glucose and GDH. The concentration of acetoin increased rapidly during the first 15 min in AKR5C3-R192G mediated catalysis. The catalysis course of AKR5C3, AKR5C3-D53A, and AKR5C3-R192G were presented in Supporting Information Figure S4.
Figure 6.
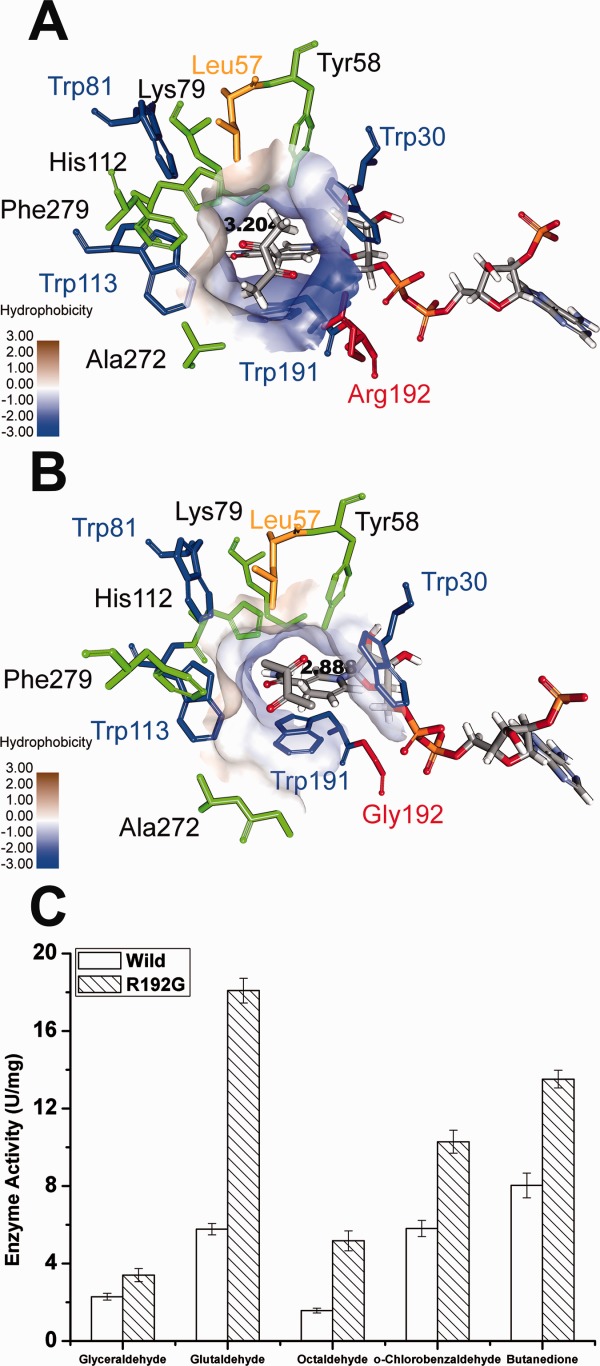
Structural insights into the AKR5C3-R192G/NADPH/ butanedione model and the improved reductive activity of AKR5C3-R192G. (A) The substrate-binding pocket was putatively identified upon butanedione docking in AKR5C3/NADPH modeled structure. (B) The identical view of (A) is shown with AKR5C3-R192G/NADPH/ butanedione model. The residues involved in the substrate-binding pocket were shown and marked. Hydrophobicity surface of substrate-binding pocket was demonstrated respectively in AKR5C3/NADPH crystal structure (A) and AKR5C3-R192G/NADPH (B). (C) Enzyme activity of AKR5C3-R192G.
In summary, we solved the crystal structures of AKR5C3 in apo and cofactor bound forms, as well as the AKR5C3-D53A/NADPH binary complex. Structural comparison between AKR5C3/NADPH and AKR5C3-D53A/NADPH identified a conformational alteration of Trp30 and Trp191. The D53A mutation-induced conformational change included 60° rotation of the nicotinamide and ribose moieties of NADPH away from the original position and partial π-stacking over the Trp191 and Arg192 side chains, which contributed to the improved activity. The highly conserved Trp191 replaced by Tyr resulted in an inactive form with benzaldehyde, which showed that the π-electron atmosphere facilitated AKR5C3 to catalyze aromatic substrate reduction. The variable Arg192 was a key residue involved in the substrate-binding pocket, and the mutant AKR5C3-R192G showed better reaction turnover than the improved hydrophobicity in its substrate-binding pocket. The AKR5C3 crystal structures at different conformations provided mechanistic details of cofactor-dependent AKR activity and new functional sights into Asp53 and Arg192 residues.
Materials and Methods
Site-directed mutagenesis
AKR5C3 (Gox0644) open reading frame (ORF) was amplified from G. oxydans genomic DNA via PCR, cloned into pET28b (Novagen) vector, and expressed into pSE380 vector (Novagen) using an N-terminal 6×His-tag from pET28b. Site-directed mutagenesis studies were performed through inverse PCR (iPCR) using mutation primers according to KOD-Plus-Mutagenesis Kit (TOYOBO, Osaka, Japan).
Protein expression and purification
Recombinant E. coli BL21 (DE3) containing AKR5C3 mutant plasmids were induced by 0.4 mM isopropyl β-d-thiogalactoside for 12 h at 20°C. The bacterial culture was harvested through centrifugation at 10,000g for 10 min at 4°C and resuspended in buffer (20 mM phosphate buffer, pH 7.4). The cells were sonicated for 10 min under 300 W and centrifuged at 10,000g for 20 min to remove debris. Proteins were purified using Ni2+ affinity column followed by HiLoad Superdex S-75 26/60 column.
Catalytic activity assay
The substrate solution (concentration: 100 mM) was supplemented with dimethyl sulfoxide to improve substrate solubility. The final dimethyl sulfoxide concentration [2.5% (v/v)] had no detectable effect on AKR5C3 activity, as shown when the same protocol was tested in glyoxal catalytic assay (data not shown). The reaction mixture contained 50 mM potassium phosphate (pH 6.5), 1 mM NADPH, and 2.5 mM substrate and enzyme. Reactions were initiated by adding cofactor at 30°C. The enzyme activity was determined through spectrophotometric NADPH quantification (A340) within 1 min.27 Because NADPH easily oxidizes, the A340 was calibrated by the negative control which included the substrate and NADPH but without enzyme. One unit of catalytic activity was defined as the amount of enzyme to consume 1 μmol NADPH per minute. The apparent kinetic parameter was determined by constructing a Lineweaver–Burk plot with incremental cofactor concentrations (0.01–2 mM) and glutaraldehyde (2.5 mM) as substrate. All measurements were performed in triplicate based on the corresponding negative control, which ensured enzyme activity data.
A GC system was used to measure substrate (2,3-butanedione) decrease and product (acetoin) formation to determine positive mutant function. The 10 mL reaction mixture contained 20 mM phosphate buffer (pH 7.4), 3 g/L 2,3-butanedione, 6 g/L glucose, crude AKR5C3 and GDH. The mixtures were shaken at 30°C for 30 min. Samples were taken every 15 min and centrifuged at 12,000 rpm. The supernatant was extracted using ethyl acetate (1:1, v:v), and analysis was performed using a gas chromatographer (Agilent Technologies 7890A GC system) fitted with a flame ionization detector. Samples and chemical standards were analyzed on a DB-WAX capillary column (30 m × 0.25 mm × 0.25 μm, Agilent Technologies). The injector temperature was 215°C. The oven temperature was maintained at 50°C for 1.5 min, then rose to 180°C at a rate of 25°C/min, and held for 1 min. The detector temperature was 250°C.
Crystallization and data collection
AKR5C3 crystals were grown by hanging drop vapor diffusion at 20°C. A 2.0 μL hanging drop typically contains 1.0 μL of protein (∼15 mg/mL) mixed with 1.0 μL of reservoir containing 29.5% PEG3350, 190 mM Li2SO4, and 100 mM Bis-Tris (pH 6.3) equilibrated over 1 mL of reservoir solution. The crystals grew to maximum size of 0.2 × 0.1 × 0.1 mm3 over 6 h. Both AKR5C3/NADPH and AKR5C3-D53A/NADPH complex crystals were obtained at the same condition by mixing different ratios of the enzyme and NADPH for 3 days. The crystals were directly flash frozen in liquid nitrogen (100 K) for data collection. A total of 360 frames of 1° oscillation were collected at 0.98 Å wavelength for AKR5C3/NADPH binary complex, which were subsequently processed using HKL2000 (www.hkl-xray.com). The AKR5C3/NADPH crystals belong to the space group P212121, with the following unit cell dimensions: a = 55.989 Å, b = 75.778 Å, c = 125.604 Å, and α = β = γ = 90°, with two molecules per asymmetric unit (Table 1). The crystallographic statistics of AKR5C3, AKR5C3/NADPH, and AKR5C3-D53A/NADPH are shown in Table 1.
Structure determination
The AKR5C3 crystal structure was determined through molecular replacement using the 2,5-diketo-d-gluconic acid reductase (AKR5C2) crystal structure from E. coli as the search model (PDBID: 1MZR). The model was built using O program (http://xray.bmc.uu.se/alwyn), and was refined using REFMAC/CCP4 (www.ccp4.ac.uk). Structure determination and model validation were performed using the MOLREP and PROCHECK software, respectively. The R-free set containing 5% of the reflections was randomly chosen. The model comprised 9–279 residues. Docking was performed using CDDOCKER28 of the Discovery Studio version 3.5 program package (Accelrys, Inc.).
Acknowledgments
The authors are grateful to S. Huang and J. He of Shanghai Synchrotron Radiation Facility (SSRF, U17) and to H. Robinson of X29A NSLS Brookhaven National Laboratory for their assistance in data collection.
Database: Coordinates and the structure factors have been deposited to Protein Data Bank with accession codes 3WBX (AKR5C3 in apoform), 3WBW (AKR5C3/NADPH binary complex) and 3WBY (AKR5C3-D53A/NADPH binary complex).
Glossary
- AKRs
aldo-keto reductases
- NADPH
nicotinamide adenine dinucleotide phosphate.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- Jez JM, Penning TM. The aldo-keto reductase (AKR) superfamily: an update. Chem Biol Interact. 2001;130:499–525. doi: 10.1016/s0009-2797(00)00295-7. [DOI] [PubMed] [Google Scholar]
- Sanli G, Dudley JI, Blaber M. Structural biology of the aldo-keto reductase family of enzymes. Cell Biochem Biophys. 2003;38:79–101. doi: 10.1385/CBB:38:1:79. [DOI] [PubMed] [Google Scholar]
- Hyndman D, Bauman DR, Heredia VV, Penning TM. The aldo-keto reductase superfamily homepage. Chem Biol Interact. 2003;143–144:621–631. doi: 10.1016/s0009-2797(02)00193-x. [DOI] [PubMed] [Google Scholar]
- Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J. 1997;326:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzer R, Wilson DK, Nidetzky B. Catalytic mechanism and substrate selectivity of aldo-keto reductases: insights from structure-function studies of Candida tenuis xylose reductase. IUBMB Life. 2008;58:499–507. doi: 10.1080/15216540600818143. [DOI] [PubMed] [Google Scholar]
- Schlegel BP, Jez JM, Penning TM. Mutagenesis of 3α-hydroxysteroid dehydrogenase reveals a “push-pull” mechanism for proton transfer in aldo-keto reductases. Biochemistry. 1998;37:3538–3548. doi: 10.1021/bi9723055. [DOI] [PubMed] [Google Scholar]
- Bohren KM, Grimshaw CE, Lai CJ, Harrison DH, Ringe D, Petsko GA, Gabbay KH. Tyrosine-48 is the proton donor and histidine-110 directs substrate stereochemical selectivity in the reduction reaction of human aldose reductase: enzyme kinetics and crystal structure of the Y48H mutant enzyme. Biochemistry. 1994;33:2021–2032. doi: 10.1021/bi00174a007. [DOI] [PubMed] [Google Scholar]
- Tarle I, Borhani D, Wilson D, Quiocho F, Petrash J. Probing the active site of human aldose reductase. Site-directed mutagenesis of Asp-43, Tyr-48, Lys-77, and His-110. J Biol Chem. 1993;268:25687–25693. [PubMed] [Google Scholar]
- Banner DW, Bloomer AC, Petsko GA, Phillips DC, Pogson CI, Wilson IA, Corran PH, Furth AJ, Milman JD, Offord RE, Priddle JD, Waley SG. Structure of chicken muscle triose phosphate isomerase determined crystallographically at 2.5 Å resolution: using amino acid sequence data. Nature. 1975;255:609–614. doi: 10.1038/255609a0. [DOI] [PubMed] [Google Scholar]
- Pawlowski JE, Penning TM. Overexpression and mutagenesis of the cDNA for rat liver 3 alpha-hydroxysteroid/dihydrodiol dehydrogenase. Role of cysteines and tyrosines in catalysis. J Biol Chem. 1994;269:13502–13510. [PubMed] [Google Scholar]
- Barski OA, Gabbay KH, Grimshaw CE, Bohren KM. Mechanism of human aldehyde reductase: characterization of the active site pocket. Biochemistry. 1995;34:11264–11275. doi: 10.1021/bi00035a036. [DOI] [PubMed] [Google Scholar]
- Várnai P, Richards WG, Lyne PD. Modelling the catalytic reaction in human aldose reductase. Proteins. 1999;37:218–227. doi: 10.1002/(sici)1097-0134(19991101)37:2<218::aid-prot7>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Habrych M, Rodriguez S, Stewart JD. Purification and Identification of an Escherichiacoli β-Keto Ester Reductase as 2, 5-Diketo-d-gluconate Reductase YqhE. Biotechnol Prog. 2002;18:257–261. doi: 10.1021/bp0101841. [DOI] [PubMed] [Google Scholar]
- Moore JC, Pollard DJ, Kosjek B, Devine PN. Advances in the enzymatic reduction of ketones. Acc Chem Res. 2007;40:1412–1419. doi: 10.1021/ar700167a. [DOI] [PubMed] [Google Scholar]
- Di Costanzo L, Drury JE, Penning TM, Christianson DW. Crystal structure of human liver Δ4-3-ketosteroid 5β-reductase (AKR1D1) and implications for substrate binding and catalysis. J Biol Chem. 2008;283:16830–16839. doi: 10.1074/jbc.M801778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucher FDR, Cantin L, Luu-The V, Labrie F, Breton R. The crystal structure of human Δ4-3-ketosteroid 5β-reductase defines the functional role of the residues of the catalytic tetrad in the steroid double bond reduction mechanism. Biochemistry. 2008;47:8261–8270. doi: 10.1021/bi800572s. [DOI] [PubMed] [Google Scholar]
- Scoble J, McAlister AD, Fulton Z, Troy S, Byres E, Vivian JP, Brammananth R, Wilce MC, Le Nours J, Zaker-Tabrizi L. Crystal structure and comparative functional analyses of a Mycobacterium aldo-keto reductase. J Mol Biol. 2010;398:26–39. doi: 10.1016/j.jmb.2010.02.021. [DOI] [PubMed] [Google Scholar]
- Chen M, Lin J, Ma Y, Wei D. Characterization of a novel NADPH-dependent oxidoreductase from Gluconobacter oxydans. Mol Biotechnol. 2010;46:176–181. doi: 10.1007/s12033-010-9283-4. [DOI] [PubMed] [Google Scholar]
- Schweiger P, Gross H, Deppenmeier U. Characterization of two aldo–keto reductases from Gluconobacter oxydans 621H capable of regio-and stereoselective α-ketocarbonyl reduction. Appl Microbiol Biotechnol. 2010;87:1415–1426. doi: 10.1007/s00253-010-2607-2. [DOI] [PubMed] [Google Scholar]
- Gao C, Zhang L, Xie Y, Hu C, Zhang Y, Li L, Wang Y, Ma C, Xu P. Production of (3S)-acetoin from diacetyl by using stereoselective NADPH-dependent carbonyl reductase and glucose dehydrogenase. Bioresource Technol. 2013;137:111–115. doi: 10.1016/j.biortech.2013.02.115. [DOI] [PubMed] [Google Scholar]
- Jeudy S, Monchois V, Maza C, Claverie JM, Abergel C. Crystal structure of Escherichia coli DkgA, a broad-specificity aldo-keto reductase. Proteins. 2006;62:302–307. doi: 10.1002/prot.20710. [DOI] [PubMed] [Google Scholar]
- Farber GK, Petsko GA. The evolution of α/β barrel enzymes. Trends Biochem Sci. 1990;15:228–234. doi: 10.1016/0968-0004(90)90035-a. [DOI] [PubMed] [Google Scholar]
- Richter N, Breicha K, Hummel W, Niefind K. The three-dimensional structure of AKR11B4, a glycerol dehydrogenase from Gluconobacter oxydans, reveals a tryptophan residue as an accelerator of reaction turnover. J Mol Biol. 2010;404:353–362. doi: 10.1016/j.jmb.2010.09.049. [DOI] [PubMed] [Google Scholar]
- Kubiseski TJ, Hyndman DJ, Morjana NA, Flynn T. Studies on pig muscle aldose reductase. Kinetic mechanism and evidence for a slow conformational change upon coenzyme binding. J Biol Chem. 1992;267:6510–6517. [PubMed] [Google Scholar]
- Grimshaw CE, Shahbaz M, Putney C. Mechanistic basis for nonlinear kinetics of aldehyde reduction catalyzed by aldose reductase. Biochemistry. 1990;29:9947–9955. doi: 10.1021/bi00494a027. [DOI] [PubMed] [Google Scholar]
- Richter N, Neumann M, Liese A, Wohlgemuth R, Eggert T, Hummel W. Characterisation of a recombinant NADP-dependent glycerol dehydrogenase from Gluconobacter oxydans and its application in the production of L-glyceraldehyde. Chembiochem. 2009;10:1888–1896. doi: 10.1002/cbic.200900193. [DOI] [PubMed] [Google Scholar]
- Rella R, Raia Ca, Pensa M, Pisani Fm, Gambacorta A, Rosa M, Rossi M. A novel archaebacterial NAD+ dependent alcohol dehydrogenase. Eur J Biochem. 1987;167:475–479. doi: 10.1111/j.1432-1033.1987.tb13361.x. [DOI] [PubMed] [Google Scholar]
- Wu G, Robertson DH, Brooks CL, Vieth M. Detailed analysis of grid-based molecular docking: a case study of CDOCKER-A CHARMm-based MD docking algorithm. J Comp Chem. 2003;24:1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.