

Abstract
Designed armadillo repeat proteins (dArmRP) are promising modular proteins for the engineering of binding molecules that recognize extended polypeptide chains. We determined the structure of a dArmRP containing five internal repeats and 3rd generation capping repeats in three different states by X-ray crystallography: without N-terminal His6-tag and in the presence of calcium (YM5A/Ca2+), without N-terminal His6-tag and in the absence of calcium (YM5A), and with N-terminal His6-tag and in the presence of calcium (His-YM5A/Ca2+). All structures show different quaternary structures and superhelical parameters. His-YM5A/Ca2+ forms a crystallographic dimer, which is bridged by the His6-tag, YM5A/Ca2+ forms a domain-swapped tetramer, and only in the absence of calcium and the His6-tag, YM5A forms a monomer. The changes of superhelical parameters are a consequence of calcium binding, because calcium ions interact with negatively charged residues, which can also participate in the modulation of helix dipole moments between adjacent repeats. These observations are important for further optimizations of dArmRPs and provide a general illustration of how construct design and crystallization conditions can influence the exact structure of the investigated protein.
Keywords: protein structure, armadillo repeat, domain swapping, protein engineering, superhelix
Introduction
The specific recognition of macromolecules is essential for many applications in biochemical research, medical diagnostics and disease treatment. Traditionally, researchers utilized antibodies that were generated by immunization of animals to fulfill these tasks. However, for many applications a renewable resource is desirable, where binders are clearly defined by their protein sequence. With the recent progress of combinatorial biochemistry methods, alternative scaffolds have been successfully explored.1 Among those, scaffolds with modular architectures, such as ankyrin-, HEAT-, and tetratricopeptide repeat proteins are becoming increasingly popular, because the modular topology facilitates a simple adaptation to the size of the targeted protein.2
A common prerequisite for most established techniques to generate target-specific binding modules is the availability of sufficient amounts of stable target protein. Unfortunately, this prerequisite is often the bottle-neck for the production of novel binding modules, because recombinant protein expression can be an elaborate and time-consuming process. To overcome this limitation, we explored the armadillo repeat scaffold to establish a modular system that allows the rational design of peptide recognition modules (reviewed in Ref.3). The armadillo repeat scaffold was selected, because natural armadillo repeat proteins bind their targets in extended anti-parallel conformations with very regular binding topologies. The main chain atoms of the peptide are recognized by a belt of conserved asparagine residues, whereas the peptide side chains are bound in specific pockets between adjacent repeats.
Initially, designed Armadillo Repeat Proteins (dArmRP) were constructed using a consensus design approach based on importin-α and β-catenin sequences in combination with force field-based optimizations of the hydrophobic core.4 The thermodynamic stabilities of these dArmRPs were further optimized using structure-based computational techniques.5,6 dArmRPs possess the overall composition YzMnAz, where Y, M, and A denote the type of the N-terminal, internal-, and C-terminal repeats, respectively [Fig. 1(a)]. The generation number (z) and the number of internal repeats (n) are indicated as subscripts in roman and arabic numbers, respectively. First generation dArmRPs of the YIMnAI series were well-expressed, stable and monomeric proteins (M-repeats are derivatives of the consensus C-type repeats).4,5 However, none of them yielded crystals that diffracted to high resolution. Molecular dynamics calculations suggested modifications in the capping repeats resulting in the 2nd generation series of caps. For YIIM4AII this redesign increased the melting temperature by 9.5°C, compared to its 1st generation ancestor.5
Figure 1.
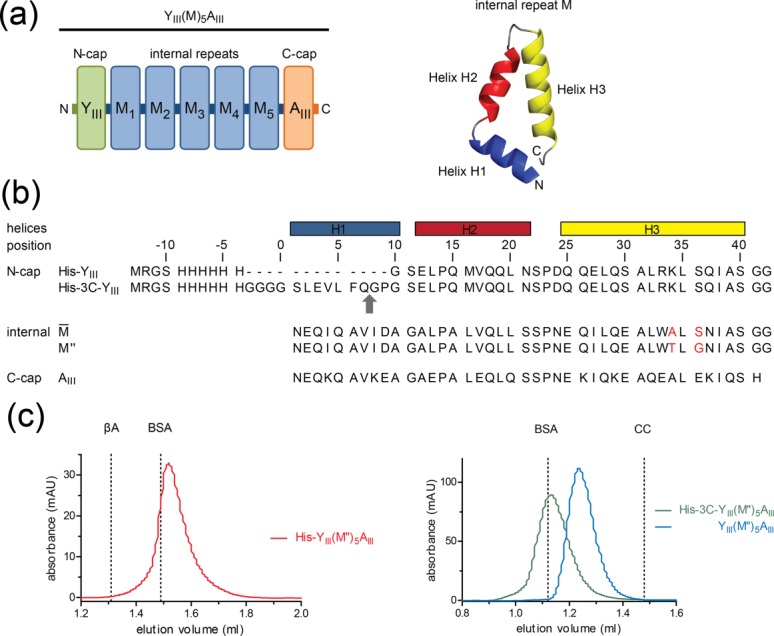
(a) Schematic view of YIIIM5AIII (left) with an N-terminal capping repeat (YIII), five identical internal repeats (M) and a C-cap (AIII). Each internal repeat is composed of three α-helices, designated H1, H2, and H3 (right), except YIII, which is lacking H1. (b) Sequence alignment of N-cap with and without 3C protease cleavage site (the scissile bond is indicated by a grey arrow),  -type internal repeat as described in Refs.5 and6, M″-type internal repeat, and C-cap. The M″-type internal repeat described here differs from the -type repeat by two mutations that are highlighted in red. (c) Gel-filtration chromatograms of His-YIIIM″5AIII (left side) and YIIIM″5AIII before and after cleavage of the His6-tag (right side). Chromatograms are separated because different SEC columns were used (Superdex 200 PC 3.2/30 for the analysis of His-YIIIM″5AIII and Superdex 75 PC 3.2/30 for the analysis of His-3C-YIIIM″5AIII). The protein concentrations are ∼10 μM. β-Amylase (βA, 200 kDa), bovine serum albumin (BSA, 66 kDa), and cytochrome c (CC, 12.4 kDa) were used as molecular weight standards. Under these conditions, the apparent molecular weights for His-YIIIM″5AIII, un-cleaved and cleaved His-3C-YIIIM″5AIII are 54.2 kDa (theoretical Mw 31.0 kDa), 63.0 kDa (theoretical Mw 32.2 kDa), and 39.6 kDa (theoretical Mw 29.9 kDa) and the ratios between apparent and theoretical molecular weights are 1.8, 2.0, and 1.3, respectively. Ratios of ∼1.3 and 2.0 describe the monomeric and dimeric forms of dArmRPs. Notably, all proteins eluted as a monomer on the larger Superdex 200 10/30 column (data not shown).
-type internal repeat as described in Refs.5 and6, M″-type internal repeat, and C-cap. The M″-type internal repeat described here differs from the -type repeat by two mutations that are highlighted in red. (c) Gel-filtration chromatograms of His-YIIIM″5AIII (left side) and YIIIM″5AIII before and after cleavage of the His6-tag (right side). Chromatograms are separated because different SEC columns were used (Superdex 200 PC 3.2/30 for the analysis of His-YIIIM″5AIII and Superdex 75 PC 3.2/30 for the analysis of His-3C-YIIIM″5AIII). The protein concentrations are ∼10 μM. β-Amylase (βA, 200 kDa), bovine serum albumin (BSA, 66 kDa), and cytochrome c (CC, 12.4 kDa) were used as molecular weight standards. Under these conditions, the apparent molecular weights for His-YIIIM″5AIII, un-cleaved and cleaved His-3C-YIIIM″5AIII are 54.2 kDa (theoretical Mw 31.0 kDa), 63.0 kDa (theoretical Mw 32.2 kDa), and 39.6 kDa (theoretical Mw 29.9 kDa) and the ratios between apparent and theoretical molecular weights are 1.8, 2.0, and 1.3, respectively. Ratios of ∼1.3 and 2.0 describe the monomeric and dimeric forms of dArmRPs. Notably, all proteins eluted as a monomer on the larger Superdex 200 10/30 column (data not shown).
Although proteins from the YIIMnAII series were predominantly monomeric in solution, the crystal structures of YIIM3AII and YIIM4AII revealed domain-swapped N-caps.6 To overcome the domain swapping of the N-caps and to improve the thermodynamic stability of the C-caps, 9 and 6 mutations were inserted in the N- and C-caps, respectively. The so obtained 3rd generation N-cap (YIII-type) increased the melting temperature by 4.5°C, over the 2nd generation, but the modifications of the C-cap (AIII-type) decreased it by 5.5°C. The crystal structures of YIIIM3AII and YIIIM3AIII revealed monomeric molecules, suggesting that the redesign obviously reduced the propensity of the N-cap for domain swapping, as desired.6
Domain swapping is defined as the exchange of one or more secondary structural elements among adjacent protein chains (referred to as “domains”), with the consequence that initially monomeric proteins become tightly entangled oligomers.7 An extreme example for domain swapping was observed for RNase A, a naturally monomeric protein that forms four different domain-swapped oligomers (reviewed in Ref.8). Domain swapping has been associated with various processes ranging from the evolution of novel domain folds to pathological disorders caused by the formation of amyloid-fibrils.9–11
Despite its importance for the understanding of amyloid diseases, the molecular events that lead to the transformation from monomeric proteins to domain-swapped oligomers are unclear. In principle, domain-swapped oligomers should not exist, because oligomer formation is coupled to a loss of entropy while most of the enthalpic contributions are maintained, albeit in an intermolecular arrangement. On the other hand, extreme conditions (pH, high protein concentration, denaturants, and temperature) foster domain swapping, because they liberate the swapped secondary structural elements from the protein core by partial protein unfolding. In fact, it was shown that the energy barrier between monomers and domain-swapped dimers is similar to the energy barrier between the folded and unfolded states.12,13 Domain swapping is often observed in combination with particular molecular features, such as high thermal mobility, strained loops, cis/trans isomerization of proline residues or binding of metal ions.8
Domain swapping can also be an issue for protein design. Mutagenesis studies on p13suc1 revealed that the conformation of the hinge region, which connects the swapped domains, is crucial for the equilibrium between swapped and non-swapped states.14 Typically, an increase of the strain in the loop of the monomeric state by loop shortening15,16 or the restriction of its main-chain torsion angle space (e.g., mutation Glu91 to Pro in p13suc1) shifts the equilibrium towards the oligomeric state.14 Domain-swapped dArmRPs were observed previously for 2nd generation N-caps. In the domain swapped YIIM3AII structure a continuous helix was formed by helix 3 of the N-cap and helix 1 of the first internal repeat. The equilibrium was shifted towards the monomer by mutating Asp41 to Gly, which decreases the helix propensity by increasing the torsion angle space of the hinge region between N-cap and internal repeat, combined with eight additional mutations in the interface between adjacent repeats.6 Here we show that dArmRPs with 3rd generation caps can also adopt different states—including a domain-swapped state—depending on the presence or absence of the His6-tag and the exact crystallization conditions. These results show that solvent conditions and the exact construct design can have large effects on the overall structure of the investigated molecules.
Results
Design, expression, and characterization of dArmRPs
The main goal of this study was to investigate the superhelical parameters of internal repeats of dArmRPs. Therefore, we used the 3rd generation capping repeats together with five internal repeats. The internal repeat used in this study, termed M″, differed from the previously established internal repeat (termed  ) at two sites [Fig. 1(b)].5,6 Serine at position 36 was replaced by glycine because it was expected that the serine side chain might disturb the orientation of the Asn37 side chain (superscripted numbers refer to the positions in the repeat). Alanine at position 34 was mutated to threonine to enforce a stronger twist of the superhelix, which might cause improved peptide binding affinities. In contrast to previous studies we used longer dArmRP with more internal repeats for several reasons: more internal repeats allow a better determination of the superhelical parameters because more repeat pairs can be compared. Furthermore, the stacking of internal repeats is not disturbed by long-range effects from the capping repeats. On the other hand, the yield of purified dArmRP decreases with an increasing number of internal repeats. A dArmRP with five internal repeats offers a good compromise.
) at two sites [Fig. 1(b)].5,6 Serine at position 36 was replaced by glycine because it was expected that the serine side chain might disturb the orientation of the Asn37 side chain (superscripted numbers refer to the positions in the repeat). Alanine at position 34 was mutated to threonine to enforce a stronger twist of the superhelix, which might cause improved peptide binding affinities. In contrast to previous studies we used longer dArmRP with more internal repeats for several reasons: more internal repeats allow a better determination of the superhelical parameters because more repeat pairs can be compared. Furthermore, the stacking of internal repeats is not disturbed by long-range effects from the capping repeats. On the other hand, the yield of purified dArmRP decreases with an increasing number of internal repeats. A dArmRP with five internal repeats offers a good compromise.
The dArmRPs used in this study have the general composition YIIIM″5AIII [Fig. 1(a)]. To improve readability by avoiding unnecessary details this dArmRP will be called YM5A unless a more precise nomenclature is necessary. In this context, His-YM5A refers to YIIIM″5AIII with an N-terminal His6-tag and YM5A to the same molecule without it. The suffix “/Ca2+” refers to the crystallization in the presence of Ca2+ ions. After expression and purification by immobilized metal ion affinity chromatography (IMAC), a yield of 80 mg of pure protein was obtained from one liter bacterial culture for all constructs. Cleavable purification tags were removed before proteins were further purified by size-exclusion chromatography (SEC). In SEC the retention volumes of dArmRPs differed depending on protein concentration and the presence or absence of the His6-tag.
At semi-preparative scale SEC using a Superdex 200 10/30 column, all proteins were monomeric and no sign of oligomerization was detectable (data not shown). At higher concentration on an analytical SEC system (i.e., 10 μM) His-YM5A eluted as a dimer (MWobs/MWcal = 1.8 − 2.0), whereas YM5A was still monomeric (MWobs/MWcal = 1.3) [Fig. 1(c)]. Elution volumes around MWobs/MWcal = 1.3 have been reported previously for monomeric dArmRPs and were interpreted as an increase of the hydrodynamic radius due to the elongated shape to the molecules.4,6 Higher oligomers than dimers have never been observed under the tested conditions. The accelerated elution of His-YM5A (but not YM5A) at high protein concentration suggests that the His6-tag causes the formation of a weak dimer, that is, with a high dissociation constant.
Structure of YM5A
After purification and elimination of the N-terminal His6-tag, YM5A was crystallized at pH 7.5 using sodium citrate as the precipitating agent with one molecule per asymmetric unit. The structure was refined at 2.1-Å resolution. Data and refinement statistics are summarized in Table 1. The structure comprises a single domain with overall dimensions of 70 × 30 × 30 Å. All residues including Gly8 (non-superscripted numbers refer to the positions in the sequence of the complete protein), which corresponds to the N-terminus that was generated by 3C protease cleavage, are visible in the electron density map (residue numbering according to Ref.6). The polypeptide chain folds into the N-cap (helices Y2 and Y3), five internal armadillo repeats (consisting of helices H1, H2, and H3 each), and the C-cap (helices A1, A2, and A3) [Fig. 2(a)].
Table 1.
Data and Refinement Statistics
| Molecule | YIIIM″5AIII | ||
|---|---|---|---|
| Structure | No His6-tag, no calcium | With His6-tag with calcium | No His6-tag, with calcium |
| Synonym | YM5A | His-YM5A/Ca2+ | YM5A/Ca2+ |
| Data statistics | |||
| Crystallization condition | 1.4 M sodium citrate; 0.1 M HEPES; pH 7.5 | 18% PEG 8000; 0.2 M calcium acetate; 0.1 M sodium cacodylate; pH 6.5 | 25 % PEG 2000-MME; 0.2 M calcium acetate; 0.1 M Tris pH 8.5 |
| Space group | P21212 | I41 | P212121 |
| Number of molecules/AU | 1 | 2 | 4 |
| Unit cell parameters, (Å,°) | 45.53, 105.04, 55.57, 90°, 90°, 90° | 116.05, 116.05, 86.93, 90°, 90°, 90° | 63.87, 95.86, 177.07, 90°, 90°, 90° |
| Resolutiona (Å) | 2.1 (2.2–2.1) | 2.1 (2.2–2.1) | 2.35 (2.40–2.35) |
| Rmerge (%) | 6.2 (62.2) | 11.8 (66.1) | 8.0 (55.1) |
| No. of observations | 102,543 (13209) | 212,948 (30917) | 242,625 (12876) |
| No. of unique reflections | 16,190 (2079) | 33,688 (4908) | 45,974 (2756) |
| I/σ(I) | 18.2 (3.1) | 8.0 (2.6) | 16.0 (3.0) |
| Completeness (%) | 99.7 (99.9) | 99.9 (100.0) | 99.5 (98.9) |
| Refinement statistics | |||
| Resolution range (Å) | 49.12–2.10 | 44.56–2.10 | 46.27–2.35 |
| Rcryst | 19.97 | 17.22 | 18.32 |
| Rfree | 26.26 | 24.77 | 26.69 |
| B-factors | |||
| Wilson B (Å2) | 37.0 | 38.0 | 36.7 |
| Mean B value (Å2) | 34.0 | 52.1 | 39.7 |
| RMSD from ideal values | |||
| Bond lengths (Å) | 0.017 | 0.008 | 0.013 |
| Bond angles (°) | 1.96 | 1.00 | 1.70 |
| Total number of atoms | |||
| Protein | 2086 | 4297 | 8301 |
| Water | 62 | 134 | 210 |
| Calcium | 0 | 8 | 10 |
| Ligands | 0 | 0 | 8 |
Values for highest resolution shells are given in parenthesis.
Figure 2.

(a) Ribbon diagram of YM5A. N-cap, internal repeats, and C-cap are shown in green, cyan and orange, respectively. The Trp33 and Asn37 belts are indicated as side chains. (b) Sketch showing the superhelical parameters given in Table 2. Grey cylinders represent single internal repeats in the absence of Ca2+ ions. Calcium binding causes tilting of the internal repeat (two repeats shown as blue cylinder) and consequently variation of the superhelical parameters (blue arrows). (c) The cavity in the hydrophobic core between repeats M4 and M5 is indicated by grey spheres and the side chains of Pro14 and Leu35* by salmon sticks.
YM5A is bent in such a way that the seven armadillo repeats fold into a right-handed superhelix (Table 2). The superhelix that is generated by the internal repeats has a radius of 14.9 ± 0.7 Å, a height of 86 ± 7 Å per turn and each turn consist of 12.4 ± 0.3 repeats, based on the averaged parameters calculated between pairs of subsequent internal repeats [Fig. 2(b)]. The concave surface of this superhelix is formed by five H3 helices from the internal repeats and helices Y3 and A3 from the caps. Many naturally occurring armadillo repeat proteins, such as importin-α, show very similar superhelical topologies. In most cases, the target peptide binds to the concave side of the superhelix. In YM5A this putative peptide binding site is formed by a belt of Trp33 and Asn37 residues [Fig. 2(a)]. The corresponding tryptophan and asparagine residues directly interact with the cognate peptide in importin-α.17
Table 2.
Superhelical Parameters as Defined in Figure 2b
| YM5A | His-YM5A/Ca2+ | YM5A/Ca2+ | |
|---|---|---|---|
| Repeats per turn | 12.4 ± 0.3 | 14.0 ± 0.5 | 13.5 ± 0.6 |
| Height of turn (Å) | 86.4 ± 9.0 | 97.2 ± 5.5 | 97.4 ± 11.5 |
| Radius (Å) | 14.9 ± 0.7 | 17.1 ± 1.7 | 16.3 ± 0.6 |
| 2 Omega (°) | 29.0 ± 0.7 | 25.8 ± 0.9 | 26.7 ± 1.1 |
The hydrophobic core is well packed by interdigitating side chains of aliphatic residues and can be subdivided into two clusters (Supporting Information Fig. 1). The first cluster involves mainly residues from helices H1 and H3. Here Ile4, Val7, Ile8, and Leu13 interact with Ile38 and Ala39*. The second cluster involves residues from helices H2 and H3, namely residues Leu20, Leu28, Ala31, Leu32, and Leu35, which interact with Ala12#, Leu16#, Leu19#, and Ile27# (* and # indicate the positions in the previous and following repeats, respectively). All repeats contain two proline residues. Pro14 and Pro23 are located at the N-terminus of helix H2 and in the H2/H3 loop, respectively. Particularly Pro14 interrupts the regular H-bond pattern of the α-helix and introduces a short loop between helices H1 and H2. Because of subtle side-chain rearrangements the kink between H1 and H2 creates cavities in the hydrophobic core of some repeats. These cavities are located in direct proximity to the side chain of Leu35* [Fig. 2(c)].
The analysis of YM5A revealed elevated B-factors for the capping repeats compared to the five internal repeats (Table 3). The same trend was previously observed for its YIIIM3AIII homolog.6 However, for YM5A the average B-factor of the C-cap is significantly higher than the average B-factor of the N-cap, whereas for YIIIM3AIII the opposite distribution was observed. To investigate whether the elevated B-factor of the C-cap is a consequence of crystal packing, we analyzed the crystal contacts. Crystals are tightly packed (VM = 2.1 Å3 Da−1) and every molecule forms six crystal contacts. The largest crystal contact buries a surface area of 1591 Å2. This crystal contact is generated by a twofold rotation axis, which places the N-cap of YM5A on the concave surface of the symmetry-related molecule. The crystal contact involving the N-cap contains several hydrogen bonds between symmetry-related molecules, whereas the C-cap is only weakly fixed in the crystal lattice. Therefore, the different average B-factors of the caps are most likely a consequence of their involvement in crystal contacts (Table 3).
Table 3.
Average B-factors of Armadillo Repeats
| Chain | N-cap | N-cap (swapped)a | Internal | C-cap | C-cap (swapped)a | |
|---|---|---|---|---|---|---|
| YM5A | A | 35.91 | 30.21 | 50.41 | ||
| His-YM5A/Ca2+ | A | 59.12 | 47.72 | 68.67 | ||
| B | 58.29 | 47.49 | 67.96 | |||
| YM5A/Ca2+ | A | 69.65 | 33.85 | 52.49 | ||
| B | 50.91 | 28.44 | 64.19 | |||
| C | 40.50 | 35.91 | 81.88 | |||
| D | 72.87 | 32.53 | 41.41 |
All values given in Å2.
The following domain-swapped caps form the sequestered cap domain: N-cap, chain B with C-cap, chain A and N-cap, chain C with C-cap, chain D.
Structure of His-YM5A/Ca2+
In this construct, YM5A was expressed with an N-terminal His6-tag and no 3C protease cleavage side. It was crystallized in the presence of 0.2 M calcium acetate (Table 1). The structure was refined at 2.1-Å resolution and forms a dimer in the asymmetric unit. The structure of the armadillo domain is very similar to the domain in the absence of the His6-tag. Just as there, the N-cap binds to the concave surface of the neighboring molecule. However, in the presence of the His6-tag the armadillo domain is tilted and the extended N-terminal His6-tag occupies the peptide binding site on the symmetry-related molecule.
In the His-YM5A/Ca2+/His6-tag complex the main chain of the armadillo domain and the cognate His6-tag run in the same direction [Fig. 3(a)]. The binding of the His6-tag is dominated by π−π stacking interactions between the imidazole and indole rings of His4/Trp117† and His6/Trp159† (where † indicates the position in the symmetry-related molecule). Furthermore, a salt bridge is formed between the side chains of His6 and Glu156† [Fig. 3(b)]. In contrast to many naturally occurring armadillo/peptide complexes, the belt of asparagine residues does not participate in binding the His6-tag, because the tag polypeptide backbone crosses the asparagine belt at an angle of ∼30°. In natural armadillo/peptide complexes the peptide runs almost antiparallel to the asparagine belt. A similar interaction between the His6-tag and side chains from the peptide binding site was observed previously in the His-YIIM4AII crystal structure.6
Figure 3.
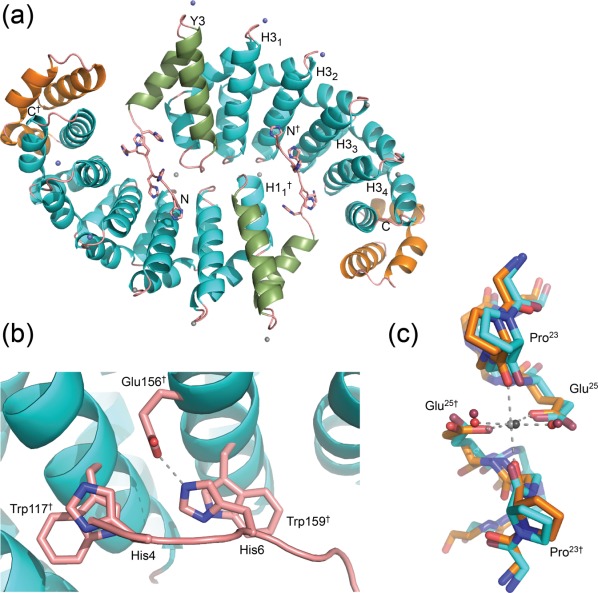
(a) Ribbon diagram of His-YM5A/Ca2+ with bound calcium. The Ca2+ ions are shown as spheres (spheres are shown with reduced radii). Blue spheres indicate Ca2+ ions that belong to symmetry related molecules and magenta spheres indicate Ca2+ ions sitting on the twofold axis (see text). All other Ca2+ ions are grey. (b) Close-up view of the interaction between the His6-tag and side chains from the peptide binding site of the second subunit. The salt bridge is shown as a dashed line. (c) Superposition of calcium binding sites in His-YM5A/Ca2+ (light blue carbon atoms) and YM5A/Ca2+ (orange carbon atoms) with bound Ca2+ ions (grey) and water molecules (red). Bright colored atoms belong to YM5A/Ca2+ and darker atoms to His-YM5A/Ca2+.
The B-factors of His-YM5A/Ca2+ are similarly distributed as in the structure without His6-tag. The internal repeats possess lower average B-factors than the caps and the average B-factor of the C-cap is higher than for the N-cap (Table 3). The elevated flexibility of the N-cap partially explains the conformational differences between both structures. The superposition of YM5A with and without His6-tag reveals different conformations of the N-cap, which are propagated towards the internal repeats (Supporting Information Fig. 2). The superposition suggests that the binding of the N-terminal His6-tag causes a counter-clockwise rotation of the N-terminal repeats compared to the structure in the absence of the His6-tag. This rotation could have implications for the superhelical parameters. With a radius of 17.1 ± 1.7 Å and 14.0 ± 0.5 Å repeats per turn, the superhelix of His-YM5A/Ca2+ is wider than in the absence of the His6-tag and requires additional 1.5 repeats to complete a full superhelical turn.
During the refinement we observed strong positive difference electron density at the N-terminal end of helix H3. Because His-YM5A was crystallized in the presence of 0.2 M calcium acetate we interpreted this difference electron density as a Ca2+ ion. This assumption was corroborated by the octahedral coordination of the metal ion and by the fact that the B-factors of the ion and Glu25 were similar. Besides the interactions of the His6-tag the binding of Ca2+ ions seems to play an equally important role in stabilizing the crystal lattice. The His-YM5A/Ca2+ dimer binds eight Ca2+ ions [Fig. 3(a)]. Six Ca2+ ions bind into well-defined binding sites at the N-terminal ends of H3 helices and connect molecules that are related by crystallographic symmetry. These ions are distributed in a peculiar stoichiometry. Each His-YM5A/Ca2+ chain is connected to its symmetry mate by three Ca2+ ions, where two ions occupy binding sites between helices Y3/H34† and H31/H33† (the subscript refers to the internal repeat number and the † to the symmetry mate) and one ion is located exactly on a two-fold axis between helices H32/H32†. Each Ca2+ ion is octahedrally coordinated by Pro23-O and Glu25-OE1 (Gln25-OE1 in Y3) from both subunits and two water ions [Fig. 3(c)]. Therefore, the N-cap and the internal repeats M1 to M4 participate in calcium binding, whereas the binding sites in M5 and the C-cap remain unoccupied. Two further Ca2+ ions connect the molecules within the asymmetric unit. Here, each Ca2+ ion is bound between the C-terminus of H32 and the N-terminus of H11†.
Structure of YM5A/Ca2+
To investigate whether the altered superhelical parameters are caused by the presence of the His6-tag or the binding of Ca2+ ions we analyzed the structure of YM5A without His6-tag but in the presence of calcium (Table 1). This structure was refined at 2.35-Å resolution and revealed four polypeptide chains in the asymmetric unit. The crystallographic tetramer shows a basket-shaped arrangement of domain-swapped subunits [Fig. 4(a)]. Two YM5A/Ca2+ molecules are connected by a head-to-tail stacking of internal repeats, forming an extended armadillo domain dimer with 10 internal repeats. The C-cap of the N-terminal subunit and the N-cap of the C-terminal subunit are extruded from the hydrophobic core. Instead of serving as capping repeats they interact and form a handle-like extension at the center of the armadillo domain. This domain is designated the sequestered cap domain. Two domain-swapped dimers are connected in an anti-parallel orientation by 10 Ca2+ ions that bind to the N-termini of the H3 helices. The calcium binding modes seen here are identical to the binding modes seen in the His-YM5A/Ca2+ structure [Fig. 3(c)]. The superhelical parameters of the domain-swapped YM5A/Ca2+ structure are also very similar to the His-YM5A/Ca2+ structure (Table 2).
Figure 4.

(a) Structure of the YM5A/Ca2+ tetramer. The domain-swapped dimer of chain A and B is shown in surface representation (N-caps, internal repeats and C-caps are colored in green, blue and orange), whereas chain C and D are shown as ribbons (colored in light blue). Internal repeats of domain swapped dimers are specified by light and dark colors according to their chains. Chain names are indicated as subscripts at the N- and C-terminal labels. Ca2+ ions are shown as grey spheres. (b) Sketch showing the stoichiometry of the domain-swapping reaction as observed in the crystal. (c) Superposition of one YM5A/Ca2+ chain (chain B, dark blue carbons and chain D, light blue carbons) on YM5A (salmon carbons). Hydrogen bonds in YM5A/Ca2+ and YM5A are shown as grey and yellow dotted lines, respectively. The directions of helix dipole moments are indicated.
Again, internal repeats showed lower average B-factors than the capping repeats. The average B-factors of capping repeats differ significantly among each other, whereas the average B-factors of internal repeats are very similar for all four chains (Table 3). The average B-factors of the interacting caps in the sequestered cap domains are almost identical (Y(chain B) and A(chain A) ≅ 41 Å2; Y(chain C) and A(chain D) ≅ 52 Å2) and significantly lower than the values for the repeats that act as true caps for the armadillo domains. This observation is surprising, because the extruded caps form a separate structural domain, which seems to be only loosely attached to the central armadillo domain.
A superposition of His-YM5A/Ca2+ on the sequestered cap domain reveals that the packing interactions between Y and A† are almost identical to the packing interactions between Y and M1 (or M5 and A) in the non-swapped armadillo domain [Supporting Information Fig. 3(a)]. The only exception here concerns the residue at position 34. In the sequestered cap domain Leu35 from the N-cap is contacted by Ala34 from the C-cap, whereas in the intact armadillo domain Leu35 interacts with Thr34. Because the side chain of threonine is bulkier than alanine, the side chain of Leu35 adopts different rotamer conformations in the intact armadillo and in the sequestered cap domains.
The inter-chain interface between M5 and M1† is also very similar to the intra-chain M:M interfaces, which is shown by the low RMSD of 1.06 Å for all atoms (0.5 Å for Cα atoms) for the superposition of residues 127–210 (repeats M3 and M4) on 211–251 (M5) and 42†–84† (M1†). In both interfaces most side chain conformations of hydrophobic core residues and some water molecules are conserved. Major differences are only seen for Gly41 and Gly42 from the loops [Supporting Information Fig. 3(b)]. In the non-swapped armadillo structures Gly41 and Gly42 adopt main-chain conformations that fall into the β-sheet region of the Ramachandran plot, whereas in the domain-swapped structure Gly41 and Gly42 from M5 and Gly42 from the N-cap adopt α-helical main chain conformations. Gly41 from the N-cap adopts a main chain conformation that is only allowed for glycine residues (ϕ = 76°, Ψ = −173°). In the domain-swapped structure the linker between the M5-repeat from the armadillo domain and the sequestered C-cap domain is formed by a continuous α-helix. Gly41 and Gly42 participate in the H-bond network of this helix. However, this helix is kinked, because Ile248-O and Gly252-N interact via a water-mediated H-bond [Supporting Information Fig. 3(b)]. This water molecule occupies the position of Gly41 in the non-swapped armadillo structures. The B-factors suggest that domain swapping has no effect on the rigidity of Gly41 and Gly42.
Discussion
Precise knowledge of three-dimensional structure is key to understanding the biological function of proteins, for inhibitor design and protein engineering.18 However, biological macromolecules are flexible structural entities and their three-dimensional structures are therefore affected by crystal lattice forces. It can be rather difficult to distinguish the impact of the crystal lattice on the true structure of the protein in solution, but for the development of a peptide-binding module we need to determine the exact superhelical parameters of YM5A in a setting resembling as closely as possible the equilibrium conformation in solution, because the conformation of the extended target peptide and the spatial distribution of binding sites on the dArmRP have to match as precisely as possible.
We identified two parameters, which could have a major impact on those parameters: the presence of dispensable purification tags and the crystallization condition. Therefore, we determined the crystal structures of YM5A in three states: (i) with His6-tag and in the presence of calcium, (ii) without His6-tag also in the presence of calcium, and (iii) without His6-tag and in the absence of calcium. The crystal structures with calcium are peculiar, because in both cases significant structural alterations compared to the calcium-free structure were observed. When the N-terminal His6-tag was not removed prior to crystallization, the tag partially occupied the peptide binding site and thereby crosslinked two adjacent molecules, whereas after the removal of the His6-tag, some of the capping repeats are sequestered from their expected positions and form a separate domain. Despite these significant structural alterations, the superhelical parameters of the calcium-bound structures (with and without His6-tag) are almost identical, suggesting that neither the binding of the His6-tag to the neighboring molecule nor the swapping of caps affect the conformation of the superhelix.
In contrast, the binding of Ca2+ ions to the N-termini of the H3 helices seems to have a major impact on the superhelix, because the parameters of the calcium-free structure are clearly different from those in the presence of calcium (RMSDs for the pairwise superposition of internal repeats of His-YM5A/Ca2+ vs. YM5A/Ca2+, YM5A vs. His-YM5A/Ca2+, and YM5A vs. YM5A/Ca2+ are 0.7 Å, 1.1 Å and 1.3 Å, respectively). The binding of Ca2+ ions entails that approximately one additional armadillo repeat is needed to finish one superhelical turn (Table 2). Therefore, the superhelix is shorter and narrower in the absence of calcium [Fig. 2(b)].
The impact of calcium on the superhelical parameters can be explained by the observation that in the absence of Ca2+ ions the side chain of Glu25 binds the N-terminus of the H3 helix in the following repeat via the side chain of Asn24 [Fig. 4(c)]. Therefore, Glu25 bridges adjacent repeats and serves as an N-terminal helix cap, because the negative charge of Glu25 compensates the positive dipole moment of H3. The positive charge of the Ca2+ ion competes with this inter-repeat interaction. After adding calcium, the inter-repeat interaction is destroyed, because the side chain of Glu25 binds directly to the Ca2+ ion in the crystal contact. The lack of this inter-repeat interaction in the calcium-bound structures could explain the structural alteration of the superhelix.
Unfortunately the crystal lattice forces that act on the protein structure through the binding of Ca2+ ions are difficult to measure, but it can be assumed that those forces are rather strong. Because of the repetitive character of the armadillo domain there are as many putative calcium binding sites as there are repeats. Furthermore, the calcium binding sites are distributed with a crystal-like regularity, causing an additive effect of these lattice forces on the structure of the protein. These additive effects are often seen in full-consensus repeat proteins. For example in the case of the full-consensus designed ankyrin repeat protein NI3C, three bound sulfate ions contributed significantly to the thermodynamic stability of the protein.19 Thus, 10 Ca2+ ions, which connect four YM5A/Ca2+ chains, could exert lattice forces that are sufficiently strong to cause the swapping of caps.
Besides calcium-induced lattice forces, the rigidity of the swapped domains seems to be equally important for the resulting structure. In the domain-swapped YM5A/Ca2+ structure the inter-molecular interactions seen in the M5:M1† and A:Y† interfaces are almost identical to the intra-molecular Y:M1 and M5:A interactions. That the inter-molecular interactions are energetically equal or perhaps even stronger than the intra-molecular interactions is supported by low B-factors, because the caps in the sequestered cap domain possess average B-factors that are even lower than the equivalent B-factors in the armadillo domain (Table 3). This observation could be explained by strained loops between the caps and the armadillo domain. If the β-strand conformation in the loops of the non-swapped caps is energetically less favorable than the α-helix conformation of the loops in the swapped structure, the β→α transition would contribute significantly to the stabilization of the swapped state. Indeed, it was shown previously that the reduction of the α-helix propensity of the loop between the N-cap and the first internal repeat (e.g., replacement of Asp41 by Gly) in combination with eight additional mutations in the interface between the N-cap and the first internal repeat abrogated domain swapping between YIIM3AII molecules with 2nd generation caps.6
When the His6-tag was not removed prior to crystallization, the binding of the His6-tag to the neighboring subunit keeps the N-cap in place, thereby preventing the swapping of domains. The observation that His-YM5A/Ca2+ forms a clear dimer at elevated protein concentrations, whereas YM5A does not, suggests that the interactions seen in the crystal structure are also present in solution. Thus, the His6-tag seems to exert a stabilizing effect on the His-YM5A/Ca2+ structure in the presence of calcium, when calcium otherwise facilitates domain swapping. Because His-YM5A/Ca2+ shows the same superhelical parameters like the domain-swapped YM5A/Ca2+ it can be assumed that domain swapping is not a consequence of the structural rearrangements in the superhelix. Furthermore, the His6-tag binds in direct vicinity to the peptide binding site and might compete with putative target peptides. Liberating the peptide binding site by His6-tag cleavage should thus improve the likelihood to obtain structures of dArmRPs/peptide complexes.
The thermodynamics of domain swapping has been investigated for the Cyanovirin-N protein, where it was shown that the transition energy between domain-swapped species is of the same order of magnitude as unfolding.13 These results are in agreement with previous studies on p13suc1 where complete unfolding was required to obtain the domain swapped dimer.14 Partial unfolding would also be required for the domain swapping of YM5A/Ca2+, but in contrast to many other swapped structures where a single domain is exchanged between subunits, the swapped structure presented above requires the existence of two partially unfolded species. To form the sequestered cap domain and to permit the head-to-tail stacking of internal repeats molecules with unfolded N- and C-caps have to exist simultaneously in solution.
Unfortunately the folding/unfolding mechanisms of armadillo repeat proteins are unknown, but for the Sel1-like repeat protein HcpB it was shown that the refolding of internal repeats is much faster than the proper formation of the N-cap. Thus, the folding of the capping repeat was the rate-limiting step upon oxidative refolding of HcpB.20 Provided that the folding of dArmRPs follows a similar mechanism like HcpB a sufficient amount of YM5A/Ca2+ molecules with partially unfolded caps might exist simultaneously in solution. This assumption requires that dArmRPs with displaced caps and solvent accessible hydrophobic cores are fairly stable molecules that possess lifetimes that are long enough to allow the dimerization of partially unfolded molecular species. The capping repeats being of lower stability have also been observed for ankyrin repeat proteins.21–24
Indeed it was shown recently that dArmRPs can be assembled from two fragments, where one carries only an N-cap and two internal repeats, the other one an internal repeat and a C-cap.25 NMR studies showed that the association is practically identical as in covalently connected repeats, indicating that the ArmRPs have a very strong ability to stack in a precise geometrical manner. This may explain why both caps can stack with each other, and the “uncapped” internal repeats can stack as if they were covalently connected in YM5A/Ca2+. Furthermore, even the isolated C-terminal dArmRP fragment lacking the N-cap was stable enough to permit structural investigation by NMR.25 Therefore, it can be assumed that for YM5A two open conformations with displaced N- and C-caps are in equilibrium with a closed conformation where the caps are shielding the hydrophobic core. Nonetheless, our studies show that the interaction energy between caps and internal repeats may still need further improvements, which could be achieved by reverting the design from the 3rd generation to the more stable 2nd generation C-cap6 or by releasing the strain in the hinge region by extending the loops that connect the caps with the internal repeats.
Interestingly, neither YM5A dimers nor higher order oligomers have been discovered in SEC after His6-tag cleavage, suggesting that oligomerization by domain swapping is a dynamic process that requires even higher protein concentration than easily achievable in solution. Provided that the lifetime of partially unfolded molecules (i.e., with one of the caps removed from the internal repeats) is relatively short, elevated protein concentrations would enhance the likelihood that two molecules with different open conformations interact to form the domain-swapped molecule that we observe in the crystal structure. Whether the equilibrium between open and closed conformations is a consequence of imperfect molecular design or whether it is an intrinsic and unavoidable property of many solenoid proteins remains to be seen in the future.
Materials and Methods
General molecular biology methods
Unless stated otherwise, experiments were performed as described previously.4 The cloning and expression vector for genes without cleavable His6-tag was pPANK (GenBank accession number AY327140), a pQE30 (Qiagen, Switzerland) derivative lacking the BpiI and BsaI sites. For His6-tag cleavage, the vector p148_3C was used. The vector p148_3C, also a derivative of vector pQE30, has an N-terminal MRGS-His6-tag followed by a GGGGS linker, a 3C-cleavage site and a removable selection marker SacB from the pDNR-DUAL vector system (GenBank: DQ666273.1, Clontech, CA). Cloning and expression was performed in E. coli XL1-blue cells (Stratagene, CA).
Gene assembly and protein expression
The DNA module of the internal M″-repeats was generated by PCR amplification of the -type internal repeat5 by primer pQE30_for and M4_34T_36G_rev (a complete list of all oligonucleotides used is given in Supporting Information Table 1). Oligonucleotides were purchased from Microsynth AG (Balgach, Switzerland). Full-length gene assembly for the protein containing an N-terminal capping repeat (YIII), five internal repeats (M″) and a C-terminal capping repeat (AIII), was performed by a single multi-fragment ligation step before insertion into pPANK or p148_3C. Expression was also done as described previously4, with the following modification: 2YT medium was used and the expression time after induction was increased to 4 h. Proteins were purified by suspending cells in lysis buffer (50 mM Tris, pH 7.6, 500 mM NaCl, 20 mM imidazole) supplemented with 1 mg mL−1 lysozyme and lysed by sonication (Branson Sonifier 250, USA). Lysed material was treated with 10 µg mL−1 of DNase and the insoluble material was separated by centrifugation at 20,000g for 30 min. The supernatant was purified by IMAC on a nickel-loaded NTA resin (Qiagen, Germany), equilibrated with lysis buffer. Columns were washed extensively with the lysis buffer and proteins were eluted with elution buffer (lysis buffer, supplemented with 250 mM imidazole). The elution buffer was exchanged against cleavage buffer (50 mM Tris, pH 7.6, 300 mM NaCl) using a PD-10 column (GE Healthcare Life Sciences, United Kingdom). To eliminate the His6-tag, proteins with cleavable tags were dialyzed for 12 h at 4°C with purified 3C protease at a dArmRP:3C protease ratio of 50:1 against cleavage buffer. Uncut proteins were removed by a second IMAC column. Size exclusion chromatography on a Superdex 200 HiLoad 26/60 column using an ÄKTA explorer system (GE Healthcare Life Sciences, United Kingdom) was used to prepare protein samples for crystallization. The protein was concentrated in crystallization buffer (10 mM Tris, pH 7.6, 100 mM NaCl) to 20 mg mL−1 using Amicon Ultra centrifugation filters (Millipore, USA). Prior to crystallization, samples were filtered through a 0.22 μm Milex® filter (Millipore). Analytical SEC was performed on an ÄKTA micro system with Superdex 75 or Superdex 200 columns (PC 3.2/30, GE Healthcare Life Sciences) in crystallization buffer.
Crystallization and structure determination
Sparse-matrix screens from Hampton Research (California) and Molecular Dimensions (Suffolk, UK) were used to identify initial crystallization conditions. Sitting-drop vapor-diffusion experiments were pipetted into 96-well Corning plates (Corning Incorporated, NY) using a Phoenix crystallization robot (Art Robbins Instruments). Prior to crystallization, YM5A and YM5A/Ca2+ were supplemented with a 1.5-fold molar excess of (Lys-Arg)5 peptide (peptide was dissolved in water and changed the volume of the sample by 1%). Protein solutions were mixed with reservoir solutions at 1:1, 1:2, or 2:1 ratios (200–300 nL final volume) and the mixtures were equilibrated against 50 μL of reservoir solution at 4°C. Reservoir conditions are summarized in Table 1. After washing the crystals in reservoir solutions that were supplemented with glycerol (10% for YM5A and YM5A/Ca2+; 15% for His-YM5A/Ca2+) and 10-fold molar excess of peptide (only for YM5A and YM5A/Ca2+) crystals were flash-cooled in liquid nitrogen. For ArmRPs which were subjected to crystallization in a mixture with the peptide, only the protein alone crystallized from the mixture.
Data were collected at beam lines X06SA and X06DA at the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland) using a Pilatus Detector (Dectris, Baden, Switzerland) and a wavelength of 1.0 Å. Diffraction data from His-YM5A/Ca2+ crystals were processed using programs MOSFLM26 and SCALA27, whereas data from YM5A and YM5A/Ca2+ crystals were processed with XDS.28 Structures were solved by molecular replacement using program PHASER.29 Models for molecular replacement were prepared as follows. For His-YM5A/Ca2+ a homology model was generated based on the structure of YIIIM3AIII (PDB id: 4DB9).6 For YM5A and YM5A/Ca2+ the refined His-YM5A/Ca2+ structure was used as a template.
The His-YM5A/Ca2+ structure was refined against measured intensities using the program PHENIX,30 whereas the structures of YM5A and YM5A/Ca2+ were refined against structure factor amplitudes using the program REFMAC5.31 For manual model building we used the program COOT.32 Water molecules were added to well-defined difference electron density peaks at H-bond distance from the protein. No (Lys-Arg)5 peptides were identified in the final electron density maps. Figures were prepared using program PYMOL.33
Analysis of superhelical parameters
Superhelical parameters were determined by analyzing the geometry of internal repeat pairs using the generalized helix description as it has been implemented in the make_symmdef_file.pl script from the Rosetta symmetry framework.34 As input structures we used the Cα-atom coordinates from 41 residues of two consecutive internal repeats (the flexible residues at position 23 were excluded). Curvature parameters as depicted in Table 2 were first generated for each pair of internal repeats (M1:M2, M2:M3, M3:M4, and M4:M5) and for each molecule found within the asymmetric unit and then averaged. The angle 2·Ω (°) describes the angle between the centers of mass of two consecutive internal repeats [Fig. 2(a)].
Acknowledgments
The authors thank Céline Stutz-Ducommun and Beat Blattmann from the high-throughput crystallization center and the staff from beamlines X06SA and X06DA from the Swiss Light Source for skilful technical support. Additional thanks goes to Dr. Johannes Schilling and Simon Hansen for helpful discussion and assistance for crystallization.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- Binz HK, Plückthun A. Engineered proteins as specific binding reagents. Curr Opin Biotechnol. 2005;16:459–469. doi: 10.1016/j.copbio.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Boersma YL, Plückthun A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr Opin Biotechnol. 2011;22:849–857. doi: 10.1016/j.copbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Reichen C, Hansen S, Plückthun A. Modular peptide binding: from a comparison of natural binders to designed armadillo repeat proteins. J Struct Biol. 2014;185:147–162. doi: 10.1016/j.jsb.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Parmeggiani F, Pellarin R, Larsen AP, Varadamsetty G, Stumpp MT, Zerbe O, Caflisch A, Plückthun A. Designed armadillo repeat proteins as general peptide-binding scaffolds: consensus design and computational optimization of the hydrophobic core. J Mol Biol. 2008;376:1282–1304. doi: 10.1016/j.jmb.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Alfarano P, Varadamsetty G, Ewald C, Parmeggiani F, Pellarin R, Zerbe O, Plückthun A, Caflisch A. Optimization of designed armadillo repeat proteins by molecular dynamics simulations and NMR spectroscopy. Protein Sci. 2012;21:1298–1314. doi: 10.1002/pro.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhurantakam C, Varadamsetty G, Grütter MG, Plückthun A, Mittl PR. Structure-based optimization of designed Armadillo-repeat proteins. Protein Sci. 2012;21:1015–1028. doi: 10.1002/pro.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 1995;4:2455–2468. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Eisenberg D. 3D domain swapping: as domains continue to swap. Protein Sci. 2002;11:1285–1299. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Li W, Johnson DJ, Huntington JA. Crystal structure of a stable dimer reveals the molecular basis of serpin polymerization. Nature. 2008;455:1255–1258. doi: 10.1038/nature07394. [DOI] [PubMed] [Google Scholar]
- Zerovnik E, Stoka V, Mirtic A, Guncar G, Grdadolnik J, Staniforth RA, Turk D, Turk V. Mechanisms of amyloid fibril formation—focus on domain-swapping. FEBS J. 2011;278:2263–2282. doi: 10.1111/j.1742-4658.2011.08149.x. [DOI] [PubMed] [Google Scholar]
- Liu C, Zhao M, Jiang L, Cheng PN, Park J, Sawaya MR, Pensalfini A, Gou D, Berk AJ, Glabe CG, Nowick J, Eisenberg D. Out-of-register beta-sheets suggest a pathway to toxic amyloid aggregates. Proc Natl Acad Sci USA. 2012;109:20913–20918. doi: 10.1073/pnas.1218792109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerala R, Zerovnik E. Accessing the global minimum conformation of stefin A dimer by annealing under partially denaturing conditions. J Mol Biol. 1999;291:1079–1089. doi: 10.1006/jmbi.1999.3045. [DOI] [PubMed] [Google Scholar]
- Liu L, Byeon IJ, Bahar I, Gronenborn AM. Domain swapping proceeds via complete unfolding: a 19F- and 1H-NMR study of the Cyanovirin-N protein. J Am Chem Soc. 2012;134:4229–4235. doi: 10.1021/ja210118w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Schymkowitz JW, Wilkinson HR, Itzhaki LS. Three-dimensional domain swapping in p13suc1 occurs in the unfolded state and is controlled by conserved proline residues. Proc Natl Acad Sci USA. 2001;98:5596–5601. doi: 10.1073/pnas.101542098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green SM, Gittis AG, Meeker AK, Lattman EE. One-step evolution of a dimer from a monomeric protein. Nat Struct Biol. 1995;2:746–751. doi: 10.1038/nsb0995-746. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Head JG, Barker JJ, Brady RL. Engineering an intertwined form of CD2 for stability and assembly. Nat Struct Biol. 1998;5:778–782. doi: 10.1038/1816. [DOI] [PubMed] [Google Scholar]
- Conti E, Uy M, Leighton L, Blobel G, Kuriyan J. Crystallographic analysis of the recognition of a nuclear localization signal by the nuclear import factor karyopherin alpha. Cell. 1998;94:193–204. doi: 10.1016/s0092-8674(00)81419-1. [DOI] [PubMed] [Google Scholar]
- Blomberg R, Kries H, Pinkas DM, Mittl PR, Grütter MG, Privett HK, Mayo SL, Hilvert D. Precision is essential for efficient catalysis in an evolved Kemp eliminase. Nature. 2013;503:418–421. doi: 10.1038/nature12623. [DOI] [PubMed] [Google Scholar]
- Merz T, Wetzel SK, Firbank S, Plückthun A, Grütter MG, Mittl PR. Stabilizing ionic interactions in a full-consensus ankyrin repeat protein. J Mol Biol. 2008;376:232–240. doi: 10.1016/j.jmb.2007.11.047. [DOI] [PubMed] [Google Scholar]
- Devi VS, Sprecher CB, Hunziker P, Mittl PR, Bosshard HR, Jelesarov I. Disulfide formation and stability of a cysteine-rich repeat protein from Helicobacter pylori. Biochemistry. 2006;45:1599–1607. doi: 10.1021/bi052352u. [DOI] [PubMed] [Google Scholar]
- Interlandi G, Wetzel SK, Settanni G, Plückthun A, Caflisch A. Characterization and further stabilization of designed ankyrin repeat proteins by combining molecular dynamics simulations and experiments. J Mol Biol. 2008;375:837–854. doi: 10.1016/j.jmb.2007.09.042. [DOI] [PubMed] [Google Scholar]
- Wetzel SK, Settanni G, Kenig M, Binz HK, Plückthun A. Folding and unfolding mechanism of highly stable full-consensus ankyrin repeat proteins. J Mol Biol. 2008;376:241–257. doi: 10.1016/j.jmb.2007.11.046. [DOI] [PubMed] [Google Scholar]
- Kramer MA, Wetzel SK, Plückthun A, Mittl PR, Grütter MG. Structural determinants for improved stability of designed ankyrin repeat proteins with a redesigned C-capping module. J Mol Biol. 2010;404:381–391. doi: 10.1016/j.jmb.2010.09.023. [DOI] [PubMed] [Google Scholar]
- Wetzel SK, Ewald C, Settanni G, Jurt S, Plückthun A, Zerbe O. Residue-resolved stability of full-consensus ankyrin repeat proteins probed by NMR. J Mol Biol. 2010;402:241–258. doi: 10.1016/j.jmb.2010.07.031. [DOI] [PubMed] [Google Scholar]
- Watson RP, Christen MT, Ewald C, Bumbak F, Reichen C, Mihajlovic M, Schmidt E, Güntert P, Caflisch A, Plückthun A, Zerbe O. Spontaneous self-assembly of engineered armadillo repeat protein fragments into a folded structure. Structure. 2014;22:985–995. doi: 10.1016/j.str.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Leslie AGW. Joint CCP4 + ESF-EAMCB. Newsletter Protein Crystallogr. 1992:26. Available at: http://www.ccp4.ac.uk/newsletters/No26.pdf. [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- DeLano WL. 2002. PyMOL. Available at: www.pymol.org.
- DiMaio F, Leaver-Fay A, Bradley P, Baker D, Andre I. Modeling symmetric macromolecular structures in Rosetta3. PloS One. 2011;6:e20450. doi: 10.1371/journal.pone.0020450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.