

Abstract
Existing mouse models of lethal Ebola virus infection do not reproduce hallmark symptoms of Ebola hemorrhagic fever, neither delayed blood coagulation and disseminated intravascular coagulation, nor death from shock, thus restricting pathogenesis studies to non-human primates. Here we show that mice from the Collaborative Cross exhibit distinct disease phenotypes following mouse-adapted Ebola virus infection. Phenotypes range from complete resistance to lethal disease to severe hemorrhagic fever characterized by prolonged coagulation times and 100% mortality. Inflammatory signaling was associated with vascular permeability and endothelial activation, and resistance to lethal infection arose by induction of lymphocyte differentiation and cellular adhesion, likely mediated by the susceptibility allele Tek. These data indicate that genetic background determines susceptibility to Ebola hemorrhagic fever.
A mouse-adapted strain of Ebola virus (MA-EBOV) does not cause hemorrhagic syndrome despite causing lethal disease in laboratory mice, and cannot be used effectively to study Ebola hemorrhagic fever (EHF) pathogenesis, as the dissimilarity to human disease limits the ability to identify key correlates of viral pathogenesis or accurately assess the effect of vaccines or therapeutics. Pathogenesis studies of EHF have thus been restricted to macaques (1–4), guinea pigs (5, 6), and Syrian hamsters (7). Although these models accurately recapitulate most of the disease features of EHF, practical and ethical concerns limit their use, including non-reproducible genetic backgrounds, cost, animal availability, and reagent availability. Epidemiologic studies of EBOV infection have identified a range of pathogenic phenotypes, which are not linked to specific mutations in the viral genome (8, 9). This suggests that the host response may determine disease severity after EBOV infection.
We tested the role of host genetics in Ebola virus disease (EVD) using the Collaborative Cross (CC) resource, a genetically diverse panel of recombinant inbred (CC-RI) mice obtained through a systematic cross of eight inbred founder mouse strains, five of which are classic laboratory strains (C57BL/6J, A/J, 129S1/SvImJ, NOD/ShiLtJ, NZO/H1LtJ) and three of which are wild-derived inbred strains (CAST/EiJ, PWK/PhJ, and WSB/EiJ) (10). The founders represent 90% of the common genetic variation across the three major Mus musculus subspecies (M. m. musculus, M. m. domesticus, and M. m. castaneus) (11). Different strains can be crossed with one another to generate CC-RI intercrossed (CC-RIX) F1 progeny. We recently observed a spectrum of pathogenic phenotypes in CC mice, and identified genetic loci associated with influenza severity and disease outcome (12, 13). Thus we tested whether a similar range of phenotypes would emerge after infecting CC-RIX animals with MA-EBOV.
To determine phenotypic baseline, we challenged the eight CC founders intraperitoneally with MA-EBOV or the Mayinga strain of wild-type EBOV (WT-EBOV). MA-EBOV differs from the published WT-EBOV sequence by only 13 nucleotide changes, three of which are silent (14). MA-EBOV is pathogenic in guinea pigs and macaques (1), and causes lethal EHF in Syrian hamsters (7). Despite observing 25–100% mortality following MA-EBOV challenge at multiple doses (Figure S1), we found no evidence of hemorrhagic disease or susceptibility to lethal disease after infection with WT-EBOV. We assessed the pathogenic phenotype produced by intraperitoneal infection with 100 focus forming units (FFU) of MA-EBOV in 47 available CC-RIX lines (Table 1). We observed disease phenotypes ranging from complete resistance to lethal disease to severe EHF-associated pathology prior to death, as well as lines that lethal infection without symptoms of EHF, but sometimes with hepatic discoloration.
Table 1.
Distribution of Phenotypes Across CC-RIX Lines
| Outcome of Infection | Frequency of phenotype (%) | Phenotypic characteristics | CC-RIX line ID | Mortality (%) |
|---|---|---|---|---|
|
| ||||
| Resistant | 19 (9/47) | 0% mortality | 15156×1566 | 0 |
| 3252×8042 | 0 | |||
| 5119×8018 | 0 | |||
| 3252×8002 | 0 | |||
| 8034×8048 | 0 | |||
| 8048×8026 | 0 | |||
| 8026×5080 | 0 | |||
| 1566×8043 | 0 | |||
| 16012×15119 | 0 | |||
|
| ||||
| Partially resistant | 11 (5/47) | <50% mortality | 18042×3032 | 20 |
| 15156×3252 | 20 | |||
| 477×16912 | 40 | |||
| 13140×16680 | 20 | |||
| 16072×15119 | 20 | |||
|
| ||||
| Lethal | 17 (8/47) | >50% mortality | 3032×16188 | 80 |
| 8004×8043 | 60 | |||
| 8002×3032 | 60 | |||
| 16188×8005 | 100 | |||
| 8008×8016 | 100 | |||
| 16441×8024 | 100 | |||
| 16912×5489 | 100 | |||
| 3415×16012 | 100 | |||
|
| ||||
| Lethal with hepatitis | 19 (9/47) | >50% mortality, hepatic discoloration | 8042×16513 | 60 |
| 16513×15156 | 100 | |||
| 16188×3252 | 75 | |||
| 13067×16912 | 100 | |||
| 5489×16557 | 80 | |||
| 16912×16211 | 60 | |||
| 16211×13140 | 80 | |||
| 8024×8049 | 100 | |||
| 8049×8010 | 100 | |||
|
| ||||
| Lethal with EHF | 34 (16/47) | >50% mortality, severe coagulopathy (discolored blood, prolonged blood clotting) | 3609×5119 | 60 |
| 8018×3154 | 80 | |||
| 13140×3015 | 100 | |||
| 8016×8034 | 100 | |||
| 16441×8005 | 100 | |||
| 8010×16441 | 100 | |||
| 3032×16441 | 60 | |||
| 8005×8002 | 100 | |||
| 3154×3609 | 100 | |||
| 3609×5489 | 100 | |||
| 16557×13067 | 100 | |||
| 16513×16188 | 100 | |||
| 15155×8054 | 100 | |||
| 3393×8052 | 100 | |||
| 8043×8008 | 80 | |||
| 8048×15155 | 80 | |||
Boldface type indicates CC-RIX crosses used in this study
We performed detailed studies on two representative lines, 13140×3015 (susceptible to lethal EHF) and 15156×1566 (resistant to lethal disease). Mice from both lines lost approximately 15% of their body weight over the first five days post-infection (p.i.) (Fig. 1A). However, susceptible mice succumbed to lethal infection on days 5–6 p.i., while resistant mice survived and fully recovered body weight by day 14 (Fig. 1B). At day 5 p.i., susceptible mice presented pathological findings consistent with EHF, including prolonged blood coagulation, internal hemorrhage, coffee-colored blood, splenomegaly, and hepatic discoloration and softened texture (Fig. 1C). The resistant mice, however, had no evident gross pathology at the time of maximum body weight loss and no alteration in the appearance of the liver (Fig. 1D). Neither susceptible nor resistant mice developed observable clinical disease after challenge with WT-EBOV. We detected extremely low titers of virus at day 3 in the liver and spleen of animals following WT-EBOV infection, and these were 100–1000-fold lower than organ titers detected in mice infected with MA-EBOV (Fig. S2). We did not detect virus at day 5 in any organ or any mouse, indicating that WT-EBOV is not able to productively replicate in these mouse strains. In liver and spleen from both mouse lines, equivalent levels of viral RNA were observed (Figs. 2A,B). However, we observed 1–2 logs higher levels of infectious virus in susceptible liver and spleen compared to resistant liver and spleen after virus titration by focus forming assay when infectious virion production became detectable on day 3 (Figs. 2C,D), suggesting that resistance may be associated with a defect in virion assembly, secretion, or other post-transcriptional processes. We confirmed this finding by staining liver sections from susceptible and resistant mice on day 5 p.i. for VP40, the viral matrix protein. We observed substantially less VP40 staining in resistant liver (Fig. 2E,F) compared to susceptible liver (Figs. 2G,H, Fig. S3). Sequence analysis showed no nucleotide changes between virus genomes in either line, indicating that these effects cannot be readily attributed to selection of quasispecies with different viral fitness (Table S1). Despite significant differences in infectious virus titers between the two mouse lines, we observed similar levels of inflammation and apoptosis in spleen and liver, although the two lines displayed distinct histopathology (Figs. S4, S5, S6). Despite similar organ tropism, virus infection occurred in different hepatic cell types in the two mouse lines. Susceptible mice had viral antigen in essentially every hepatocyte (Fig. 2F, Table S2), while resistant mice viral antigen was restricted to cells that lack typical hepatocyte morphology, most likely endothelial cells and Kupffer cells (Fig. 2G), consistent with low-pathogenicity Reston virus infection (15). Possibly in resistant mice, infected hepatic endothelial cell and macrophage responses limit virus production and control systemic inflammation and coagulopathy. Widespread hepatic infection in susceptible mice may explain how they both produce increased amounts of infectious virus and induce dysregulated coagulation pathways.
Figure 1. Distinct Morbidity and Mortality Following MA-EBOV Infection in CC-RIX Mouse Lines.
A. Percent of starting body weight over course of infection in susceptible (red squares) and resistant mice (blue circles). Data shown are mean ± SEM from five mice per CC-RIX line. B. Kaplan-Meier survival curve for susceptible (red) and resistant (blue) mice. Five mice were used for each CC-RIX line. C,D,E,F. Gross appearance of liver at necropsy in uninfected susceptible (C) and resistant (E) mice, and on day 5 post-infection in susceptible (D) and resistant (F) mice.
Figure 2. MA-EBOV Replication in CC-RIX Mouse Lines.
A,B. Quantitative real-time PCR showing expression of MA-EBOV genomes relative to mouse 18S rRNA in spleen (A) and liver (B). Data shown are mean ± SEM for three mice per time point per RIX line. C,D. Titration of infectious MA-EBOV in organ homogenates from spleen (C) and liver (D) quantified as focus forming units per milliliter. No infectious virus was detected prior to day 3 p.i. Data shown are mean ± SEM from two experiments using 2–3 mice per time point per CC-RIX line. E,F,G,H. Immunohistochemical staining for VP40 in resistant liver (E,F) and susceptible liver (G,H). Arrow indicates representative hepatocyte morphology. (t-test, *p<0.05)
We quantified the extent of coagulopathy by measuring blood clotting times. On days 5–6 p.i., susceptible mice showed significantly prolonged thrombin time (TT), prothrombin time (PTT), and activated partial thromboplastin time (aPTT) compared to resistant and C57BL/6J mice (Fig. 3A–3C). An initial spike in serum fibrinogen levels in susceptible mice on day 3 p.i. was followed by a precipitous drop (Fig. 3D) prior to death. This increase may be due to compensatory fibrinogen production in response to hepatic cell death and consequent clotting factor depletion, consistent with observations in other EHF models in which severe hemorrhage and coagulopathy typically peaks within 48 hours preceding death (3, 7).
Figure 3. Quantification of Coagulopathy and Hemorrhage in CC-RIX Mouse Lines.
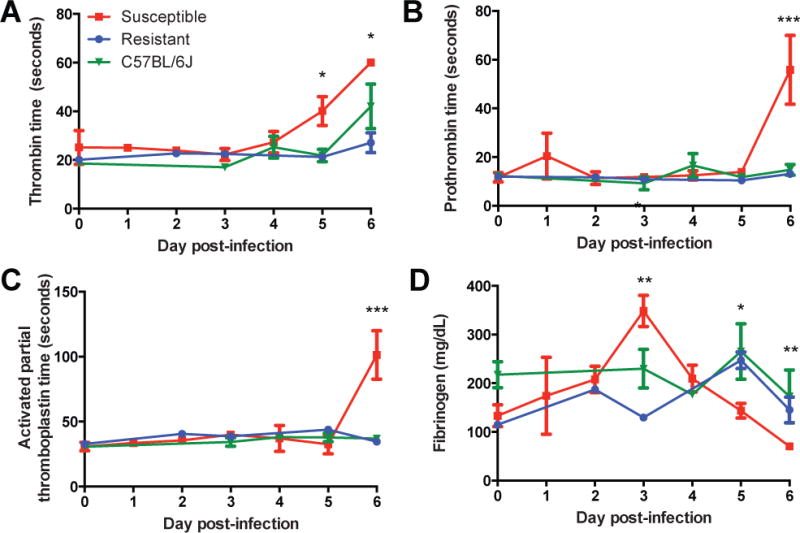
A,B,C. Coagulation times in seconds for thrombin (A), prothrombin (B), and activated partial thromboplastin (C) over course of MA-EBOV infection. D. Serum fibrinogen levels in CC-RIX mice over course of MA-EBOV infection. All data shown are the mean ± SEM for 2 experiments including 2–5 animals per time point. (ANOVA with Tukey’s HSD post-hoc. *p<0.05, **p<0.05, ***p<0.0000001).
We investigated transcriptional host responses linked to disease outcome in the CC-RIX lines. Significant differentially expressed genes relative to time-matched mock-infected samples (FDR-adjusted p-value <0.05; fold change > 1.5) in both spleen and liver were 10–100 fold higher number of DEG in susceptible mice than resistant mice (Fig 4A–B; Supplementary Data 2 and 3). These data suggest that EHF is characterized by earlier induction of a larger magnitude transcriptional response. In susceptible mice relative to resistant mice, genes associated with EBOV infection were differentially induced. Early in infection in the spleens of susceptible mice at day 1 p.i., we observed enrichment of p38 MAPK and ERK signaling, processes that stimulate productive EBOV infection (16, 17). Additionally we observed increased NFkB expression and induction of proinflammatory processes, which may reflect early targets of infection in the secondary lymphoid organs. By day 3 p.i. in both liver and spleen, inflammatory pathways became increasingly enriched in susceptible mice, as did pathways associated with cell death, including those associated with cytotoxicity and apoptosis in macrophages and endothelial cells. Both resistant and susceptible lines induced multiple immune pathways in the spleen. By day 5, although differential gene expression peaked in both lines, the gene sets involved were distinct and probably reflect different courses of disease.
Figure 4. Distinct Host Responses Associated with Disease Phenotype.
A,B. Number of differentially expressed genes (DEG) either up-regulated (positive y-axis) or down-regulated (negative y-axis) relative to time-matched mock-infected samples in spleen (A) and liver (B).
We identified differentially expressed genes unique to susceptible mice in liver and observed enrichment in genes related to vascular integrity at days 3 and 5, including the endothelial tyrosine kinases Tie1 and Tek (Tie2). Tie1 and Tek expression was depressed compared with levels in mock-infected animals at day 5, concurrent with the onset of coagulopathy. We used Ingenuity Pathway Analysis (IPA) software to generate networks predicting molecular activity (18), and predicted activation of processes associated with vascular differentiation and endothelial activation, IL-6-mediated inflammation, and bleeding, and inhibition of pathways associated with vascular integrity and inflammatory regulation in susceptible livers (Fig. S7). TIE1 and TEK signaling promote activation of coagulation factors, such as thrombin (F2), tissue factor (F3), and protease activated receptors 1, 3, and 4 (PAR1/F2R, PAR3/F2RL2, PAR4/F2RL3) (19), which have been mechanistically implicated in coagulopathies mediated by EBOV and other viruses (4, 20), and are differentially regulated in these mice (Fig. S8). Tie1 and Tek expression was consistently elevated in resistant mouse spleens, implying that endothelial signaling regulation and vascular leakage contributes to disease resistance in susceptible mice. In livers from resistant mice at day 5, gene expression associated with vascular density and angiogenesis increased, suggesting that this line effectively controls vascular leakage, potentially through repair or structural maintenance of blood vessels. It seems likely that restriction of MA-EBOV infection to endothelial and Kupffer cells in resistant mice prevents induction of hepatocyte-specific molecules that enhance systemic inflammation, thrombocytopenia, and coagulopathy.
We investigated the genomes and found that the Tie1 alleles across the eight CC founders are from all three Mus musculus subspecies, and are highly divergent from one another (21), which prevented us from identifying significant relationships between Tie1 alleles and phenotype. In contrast, Tek alleles in the CC-RIX are derived from only two subspecies: M. m. domesticus and M.m. musculus, and are very different from one another. Distinct Tek alleles were previously associated with inflammatory coagulopathies and vascular dysfunction (22–26). In our preliminary analysis, we identified statistically significant relationships between subspecific Tek alleles and initial onset of weight loss (ANOVA, F2,31=5.581, p=0.0085), average day of death (ANOVA F2,34=10.519, p=0.00028), and mortality (ANOVA F2,37=8.5553, p=0.0008) (Fig. S9). Here, we reproduced EHF in a mouse model that will allow linkage of specific genetic polymorphisms to tropism, infectious virus production, cell type-specific responses, and phenotypic outcome. The CC model provides a unique platform to map susceptibility alleles in the context of EHF pathogenesis, and rapidly apply these findings to the development of candidate therapeutics and vaccines. Ongoing screening activities in CC-RIX mice will identify additional genetic loci that contribute to hemorrhagic disease, lethality, or resistance to severe disease.
The frequency of different pathological manifestations across the 47 CC-RIX lines screened so far are similar in variety and proportion to the spectrum of clinical disease observed in patients with Ebola virus disease in the 2014 West Africa outbreak, with hemorrhagic symptoms appearing in 30–50% of patients (27, 28). Although we cannot rule out the possibility that human survivors have pre-existing immunity to EBOV or a related virus, our data suggest that genetic factors play a significant role in determining disease outcome in naïve individuals without prior exposure or immunologic priming.
While we have not yet screened CC-RIX mice for susceptibility to other ebolavirus species, we anticipate that we would observe a similar distribution of pathogenic phenotypes following infection with viruses that are capable of replicating in mice. The current 2014 West Africa outbreak is caused by the same species of ebolavirus as the MA-EBOV used in this screen. There are also similarities in the spectrum of disease observed in CC-RIX mice infected with MA-EBOV and in clinical cases in the current outbreak. The model described in this paper can be implemented promptly to identify genetic markers, conduct meticulous pathogenesis studies, and evaluate therapeutic strategies that have broad-spectrum antiviral activity against all Zaire ebolaviruses, including the virus responsible for the current West Africa outbreak.
Supplementary Material
Acknowledgments
This study was supported in part by awards U54 AI081680, U19 AI109761, and U19 AI100625 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, P51 OD010425 from the Office of the Director, National Institutes of Health, and by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. Microarray data has been deposited with the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo) (accession number GSE57214), and raw data can be obtained at https://www.ccebola.org/project/Supplemental/begin.view?.
Footnotes
Author Contributions. A.L.R. designed the study, performed functional analysis of microarray data, and wrote the manuscript, A.O. performed infections, veterinary examinations, necropsies, assessed phenotype, collected and processed samples, and titrated virus from organs by focus forming assay, M.T.F., M.T.H., F.P.M.V. and R.S.B. established systems for designing and breeding CC-RIX mouse populations and utilizing them for virus pathogenesis studies and contributed to strain selection and data analysis, R.G. performed microarray data normalization, batch correction, and differential expression analysis, S.M.K. and J.M.W. performed target preparation and hybridization of microarrays, R.L. coordinated veterinary care for experimental animals, D.P.S. performed histopathological staining and analyzed the histopathology data, F.F., D.S., and E.H. assisted with mouse procedures in high biocontainment, M.J.T. and R.G. performed sequencing and subsequent analysis of viral RNA, A.F. performed functional analysis of microarray data, P.S. quantified viral RNA by quantitative PCR, M.J.K. edited the manuscript, H.F. and M.G.K. contributed significantly to study design, provided space and infrastructure for the experiments and analysis, assisted in data analysis, and edited the manuscript.
References and Notes
- 1.Bray M, Hatfill S, Hensley L, Huggins JW. Haematological, biochemical and coagulation changes in mice, guinea-pigs and monkeys infected with a mouse-adapted variant of Ebola Zaire virus. Journal of comparative pathology. 2001 Nov;125:243. doi: 10.1053/jcpa.2001.0503. [DOI] [PubMed] [Google Scholar]
- 2.Yen JY, et al. Therapeutics of Ebola hemorrhagic fever: whole-genome transcriptional analysis of successful disease mitigation. The Journal of infectious diseases. 2011 Nov;204(Suppl 3):S1043. doi: 10.1093/infdis/jir345. [DOI] [PubMed] [Google Scholar]
- 3.Ebihara H, et al. Host response dynamics following lethal infection of rhesus macaques with Zaire ebolavirus. The Journal of infectious diseases. 2011 Nov;204(Suppl 3):S991. doi: 10.1093/infdis/jir336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geisbert TW, et al. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. The Journal of infectious diseases. 2003 Dec 1;188:1618. doi: 10.1086/379724. [DOI] [PubMed] [Google Scholar]
- 5.Connolly BM, et al. Pathogenesis of experimental Ebola virus infection in guinea pigs. The Journal of infectious diseases. 1999 Feb;179(Suppl 1):S203. doi: 10.1086/514305. [DOI] [PubMed] [Google Scholar]
- 6.Ryabchikova E, et al. Ebola virus infection in guinea pigs: presumable role of granulomatous inflammation in pathogenesis. Archives of virology. 1996;141:909. doi: 10.1007/BF01718165. [DOI] [PubMed] [Google Scholar]
- 7.Ebihara H, et al. A Syrian golden hamster model recapitulating ebola hemorrhagic fever. The Journal of infectious diseases. 2013 Jan 15;207:306. doi: 10.1093/infdis/jis626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leroy EM, Baize S, Mavoungou E, Apetrei C. Sequence analysis of the GP, NP, VP40 and VP24 genes of Ebola virus isolated from deceased, surviving and asymptomatically infected individuals during the 1996 outbreak in Gabon: comparative studies and phylogenetic characterization. The Journal of general virology. 2002 Jan;83:67. doi: 10.1099/0022-1317-83-1-67. [DOI] [PubMed] [Google Scholar]
- 9.Leroy EM, et al. Human asymptomatic Ebola infection and strong inflammatory response. Lancet. 2000 Jun 24;355:2210. doi: 10.1016/s0140-6736(00)02405-3. [DOI] [PubMed] [Google Scholar]
- 10.C. Collaborative Cross. The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics. 2012 Feb;190:389. doi: 10.1534/genetics.111.132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts A, Pardo-Manuel de Villena F, Wang W, McMillan L, Threadgill DW. The polymorphism architecture of mouse genetic resources elucidated using genome-wide resequencing data: implications for QTL discovery and systems genetics. Mammalian genome : official journal of the International Mammalian Genome Society. 2007 Jul;18:473. doi: 10.1007/s00335-007-9045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bottomly D, et al. Expression quantitative trait Loci for extreme host response to influenza a in pre-collaborative cross mice. G3. 2012 Feb;2:213. doi: 10.1534/g3.111.001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferris MT, et al. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS pathogens. 2013 Feb;9:e1003196. doi: 10.1371/journal.ppat.1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebihara H, et al. Molecular determinants of Ebola virus virulence in mice. PLoS pathogens. 2006 Jul;2:e73. doi: 10.1371/journal.ppat.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groseth A, et al. The Ebola virus glycoprotein contributes to but is not sufficient for virulence in vivo. PLoS pathogens. 2012;8:e1002847. doi: 10.1371/journal.ppat.1002847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson JC, et al. Pyridinyl imidazole inhibitors of p38 MAP kinase impair viral entry and reduce cytokine induction by Zaire ebolavirus in human dendritic cells. Antiviral research. 2014 Jul;107:102. doi: 10.1016/j.antiviral.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strong JE, et al. Stimulation of Ebola virus production from persistent infection through activation of the Ras/MAPK pathway. Proceedings of the National Academy of Sciences of the United States of America. 2008 Nov 18;105:17982. doi: 10.1073/pnas.0809698105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.S. Text.
- 19.Sato TN, et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995 Jul 6;376:70. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- 20.Antoniak S, Mackman N. Coagulation, protease-activated receptors, and viral myocarditis. Journal of cardiovascular translational research. 2014 Mar;7:203. doi: 10.1007/s12265-013-9515-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang H, et al. Subspecific origin and haplotype diversity in the laboratory mouse. Nature genetics. 2011 Jul;43:648. doi: 10.1038/ng.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brouillard P, Olsen BR, Vikkula M. High-resolution physical and transcript map of the locus for venous malformations with glomus cells (VMGLOM) on chromosome 1p21–p22. Genomics. 2000 Jul 1;67:96. doi: 10.1006/geno.2000.6232. [DOI] [PubMed] [Google Scholar]
- 23.Flanagan JM, et al. Genetic predictors for stroke in children with sickle cell anemia. Blood. 2011 Jun 16;117:6681. doi: 10.1182/blood-2011-01-332205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nolan VG, et al. Sickle cell leg ulcers: associations with haemolysis and SNPs in Klotho, TEK and genes of the TGF-beta/BMP pathway. British journal of haematology. 2006 Jun;133:570. doi: 10.1111/j.1365-2141.2006.06074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vikkula M, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996 Dec 27;87:1181. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- 26.Zheng Q, et al. Association study between of Tie2/angiopoietin-2 and VEGF/KDR pathway gene polymorphisms and vascular malformations. Gene. 2013 Jul 10;523:195. doi: 10.1016/j.gene.2013.02.053. [DOI] [PubMed] [Google Scholar]
- 27.Baize S, et al. Emergence of Zaire Ebola virus disease in Guinea. The New England journal of medicine. 2014 Oct 9;371:1418. doi: 10.1056/NEJMoa1404505. [DOI] [PubMed] [Google Scholar]
- 28.Dixon MG, Schafer IJ, C. D. C. Eis officer Ebola viral disease outbreak – west Africa, 2014. MMWR Morbidity and mortality weekly report. 2014 Jun 27;63:548. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.