

Abstract

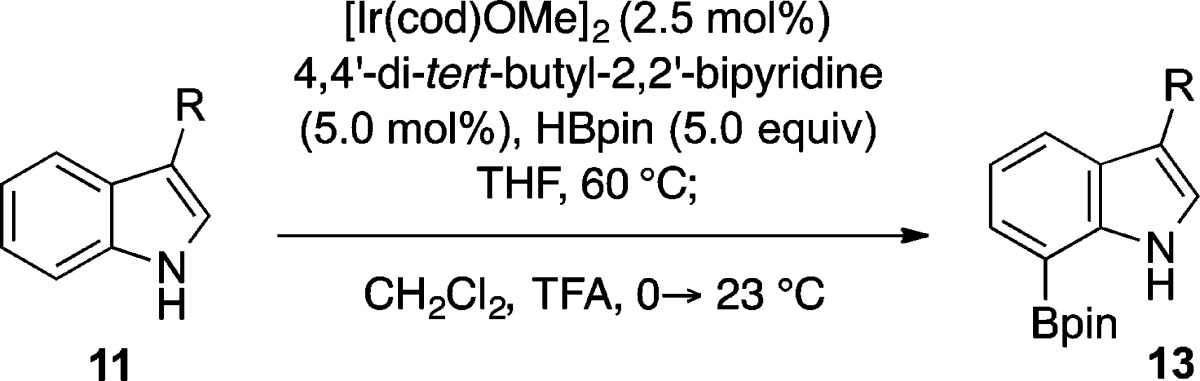
A versatile strategy for C7-selective boronation of tryptophans, tryptamines, and 3-alkylindoles by way of a single-pot C2/C7-diboronation–C2-protodeboronation sequence is described. The combination of a mild iridium-catalyzed C2/C7-diboronation followed by an in situ palladium-catalyzed C2-protodeboronation allows efficient entry to valuable C7-boroindoles that enable further C7-derivatization. The versatility of the chemistry is highlighted by the gram-scale synthesis of C7-boronated N-Boc-L-tryptophan methyl ester and the rapid synthesis of C7-halo, C7-hydroxy, and C7-aryl tryptophan derivatives.
Indole derivatives are prevalent in many natural products and pharmaceutical compounds, often as complex tryptophan- and tryptamine-derived substructures.1 The demand for such diversely substituted indolic structures has led to the development of ever more efficient and direct methods for their synthesis, none more so than in the nascent field of C–H functionalization.2−6 Within this realm, some inventive and practical chemistry has been developed to functionalize different positions around the indole ring, mainly through the innate reactivity of the C2,3 C3,4 and C55 sites and to a lesser extent C4 and C6.6 The growing number of structurally and biologically fascinating natural and unnatural products bearing an indole substituent at C7 (i.e., 1–6, Figure 1) has generated further interest in methods that expedite functionalization at this position.7−11 Many such targets, including chloropeptin I,8 the teleocidins (i.e., 1),9 the indole-glyoxamide class of HIV-1 attachment inhibitors (i.e., 4),10 and the 3-aroylindole isosteres of combretastatin (6),11 have been shown to exhibit potent biological activities.
Figure 1.

Representative biologically active C7-substituted indoles.
Selective C7 indole functionalization has proven particularly challenging in the case of tryptamine and tryptophan derivatives. In the latter case, the most commonly used strategies target 7-bromotryptophan, which requires either enzymatic processes12 or a multistep synthesis from 7-bromoindole.9g,13 Our group’s continuing goal to unearth and expand new avenues for the elaboration of complex indole-derived natural products spurred our interest in addressing this challenge.14 Herein we wish to report a practical and versatile one-pot method for C7 boronation of C3-substituted indoles, including tryptophans and tryptamines, in turn permitting rapid C7 indole functionalization via the corresponding C7-boronated products.
In the past decade, important inroads into C7 indole functionalization have been made through metal-catalyzed arene and heteroarene C–H boronations.15,16 Of particular relevance to us were the reports into iridium-catalyzed indole boronations,17 which Smith17c and later James17e showed would proceed selectively at C7 in the presence of C2 substituents to block the inherently greater azole reactivity. An interesting substrate in Smith’s study was 2-trimethylsilyl indole, which was boronated in 76% yield under their conditions, with subsequent desilylation affording a 7-boroindole in 88% yield.17c Then in 2010, Hartwig’s group utilized a variant of their hydrosilyl-directed ortho-boronation chemistry18,19 as a platform for the first truly C7-selective boronation reaction. Their three-step methodology exploited the N1-appended silane directing group on indole 8 to enable regioselective boronation to 7-boroindole 9 (Scheme 1).18c
Scheme 1. Silane-Directed C7 Boronation of Indoles.

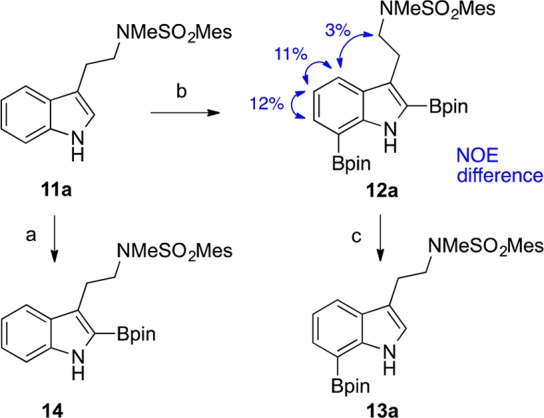
Inspired by these studies, we sought to develop a more streamlined process for direct conversion of tryptamines and tryptophans to the corresponding C7-boronated indoles. By taking advantage of the more nucleophilic/basic C2 position of these C3-substituted indoles (i.e., 11), we have now been able to fashion a two-step, one-pot procedure for C7-selective boronation. This methodology provides an expedient means for C7 derivatization of these compounds on a large scale through direct activation of unfunctionalized tryptamines and tryptophans. The premise behind this diboronation/protodeboronation sequence was our recognition of the high propensity of five-membered heterocycles to readily undergo C2 protodeboronation.20−24 Indeed, this oft-maligned side reaction of Suzuki–Miyaura cross couplings involving C2-boronated indoles,14a,14g,21a−21h furans,22 and thiophenes23 has been extensively studied in acidic media.24 Thus, a synthetic operation was envisioned involving iridium-catalyzed boronation of C3-substituted indole 11, first at C2,17 and then C7, to give diboronated indole 12, followed by acidic workup to induce ipso-protonation, rearomatization, and cleavage of the carbon–boron bond to afford 7-boroindole 13 (Scheme 2).
Scheme 2. Planned Synthesis of C7-Borotryptamines.

During initial investigations into the diboronation of tryptamine, introduction of a mesitylsulfonyl on the amine side chain helped avoid some of the undesired aryl boronations that we first observed when utilizing phthalimido,25 benzenesulfonyl, or p-nitrobenzenesulfonyl protective groups. Furthermore, the use of Nα-methylated mesitylsulfonamide 11a (Scheme 3) would also permit direct mapping of a C7-derivatized tryptamine onto many of the biologically interesting cyclotryptamine natural products that carry an Nα-methyl substituent.26 As confirmation of the order of ring boronation, exposure of tryptamine 11a to 1 equiv of boronating reagent, in conjunction with 2 mol % [Ir(cod)OMe]2 and 4 mol % 4′-di-tert-butyl-2,2′-bipyridine (dtbpy), delivered the C2-boronated product 14 in 68% yield (Scheme 3). Under these conditions, a minor amount (<10%) of the 2,7-diboronated product 12a was also generated, an observation previously noted by James and co-workers in their microwave-assisted C2-boronation of N-Boc-tryptophan methyl ester.17e In our case, inclusion of a second equivalent of bis(pinacolato)diboron under the same conditions (see (a), Scheme 3) led to substantial decomposition. Attempts to remedy this by extending the reaction time to 16 h at ambient temperature however only increased slightly the conversion to 2,7-diborotryptamine 12a, with 2-borotryptamine 14 still the major product. While switching to n-hexane as solvent and using higher catalyst loadings improved substantially the conversion of precursor 11a to the desired product 12a (>75% by NMR), these more forcing conditions led to only 27% yield of the desired product being isolated. After further optimization, tetrahydrofuran (see conditions (b), Scheme 3) proved to be the optimal solvent for full incorporation of the second boron at C7, delivering tryptamine 12a in 80% yield. At slightly elevated reaction temperatures and/or prolonged times, triboronated tryptamine, resulting from further boronation at C5 of diborotryptamine 12a, was also observed in minor amounts. Our regiochemical assignment of the two newly formed carbon–boron bonds in compound 12a was confirmed by 1D NOE difference experiments (Scheme 3).
Scheme 3. Initial Screening of C2/C7 Boronations.
Conditions: (a) [Ir(cod)OMe]2 (2 mol %), 4,4′-di-tert-butyl-2,2′-bipyridine (4 mol %), (Bpin)2 (1 equiv), CH2Cl2, 60 °C, 3 h, 68%; (b) [Ir(cod)OMe]2 (2 mol %), 4,4′-di-tert-butyl-2,2′-bipyridine (4 mol %), (Bpin)2 (2 equiv), THF, 65 °C, 20 h, 80%; (c) TFA (10 equiv), CH2Cl2, 23 °C, 15 min, 81%.
Guided by the wealth of literature on acid-promoted protodemetalations of heteroarenes,21n,22e,23b,24,27,28 we were then able to extend our diboronation procedure to one that also included in situ C2 protodeboronation. After initial success in converting diboroindole 12a into 7-boroindole 13a by simple exposure to trifluoroacetic acid (i.e., conditions (c) in Scheme 3),28 we then explored using the same conditions once the C2/C7 diboronation of tryptamine 11a (i.e., conditions (a) in Scheme 3) had been allowed to run its course. To minimize bis-protodeboronation and recovery of starting material 11a, the reaction mixture was first diluted with dichloromethane prior to addition of trifluoroacetic acid. Gratifyingly, addition of an equal volume (relative to tetrahydrofuran) of trifluoroacetic acid at 0 °C to a 3:2 (v:v) dichloromethane–THF solution afforded 7-borotryptamine 13a in 60% yield (entry 1, Table 1).
Table 1. Rapid Synthesis of C7-Boronated 3-Substituted Indole Derivatives.
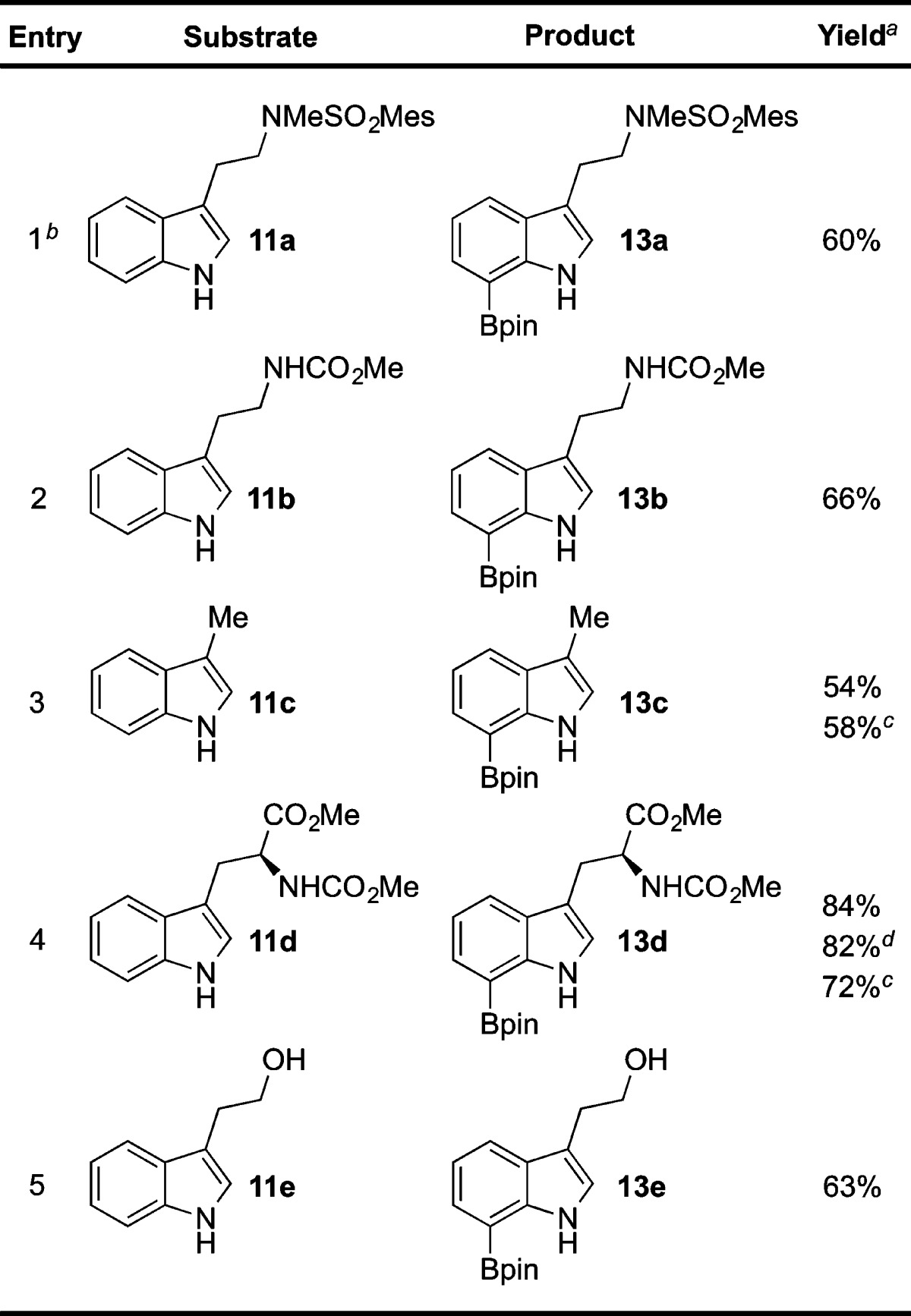
Isolated yield after purification.
Boronation conducted at 80 °C.
Second step: Pd(OAc)2 (5 mol %), AcOH, 30 °C.
Gram-scale reaction.
We next focused on applying our one-pot diboronation/protodeboronation regimen to other valuable C3-substituted indole derivatives (Table 1).29 We were pleased to confirm that changing the tryptamine protective group to a methyl carbamate in substrate 11b was in no way detrimental to the outcome of our reaction and improved our yield of 7-borotryptamine to 66% for carbamate 13b (entry 2). We also found that the time and temperature for initial diboronation could be tempered without needing to increase the stoichiometry of reagents. Apart from tryptamine 11a (entry 1, Table 1), a temperature of 60 °C and reaction time of 4–7 h was found to be ideal for most of the diboronations (entries 2–5, Table 1).30,31 The applicability of this protocol to other 3-substituted indoles was also highlighted by the efficient conversion of indoles 11c and 11e to 7-boroindoles 13c and 13e, respectively (entries 3 and 5). In the latter case (entry 5), the presence of a primary alcohol did not impede diboronation of tryptophol 11e from proceeding just as efficiently as was observed with 3-methylindole 13c, as evidenced by the 63% yield for 7-borotryptophol 13e. Importantly, selective boronation of tryptophan derivative 11d to the corresponding 7-borotryptophan 13d was achieved in 84% yield (entry 4).
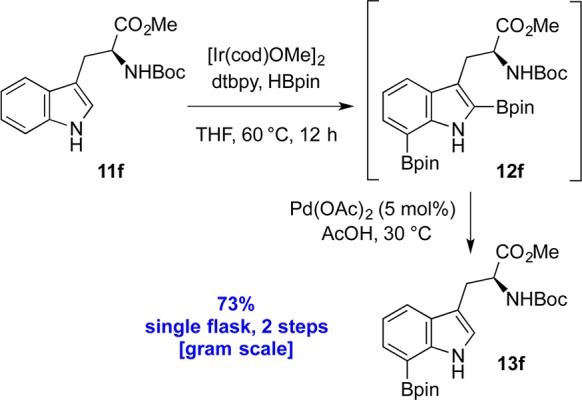
It should be noted that in all these examples the only other side product recovered from the two-step sequence was a minor amount of starting material 11a–e (<5%). The fact that traces of starting material 11a were also observed upon converting pure 2,7-diborotryptamine 12a to 7-borotryptamine 13a suggested that the conditions in the second step may cause unwanted C7 protodeboronation. This led us to focus our efforts on further optimization of the C2 protodeboronation step. More specifically, to make our process more amenable to Nα-Boc-protected tryptophans,17d,17e it was deemed necessary to move toward a milder protodeboronation that obviated the use of trifluoroacetic acid.
The diboronation of N-Boc-tryptophan methyl ester 11f proceeded smoothly under the optimal iridium-catalyzed conditions (Scheme 4). Diborotryptophan 12f could be isolated by concentration of the reaction mixture and purified by flash column chromatography in ca. 88% yield.32 After investigation of a range of basic and acidic conditions, we found that C2 protodeboronation of 12f could be efficiently promoted by a catalytic amount of palladium(II) acetate in acetic acid.21a−21h The optimal temperature for this step of 30 °C delivered 13f in 85% yield (2.0 mmol scale) from tryptophan 12f. Further still, the dried crude mixture from the diboronation of 11f could also be subjected to these same protodeboronation conditions to provide 13f in 73% yield on gram scale, once more rendering this a two-step, one-pot method (Scheme 4). Compared to the conditions used in Table 1, this alternative procedure gave similar yields of 58% and 72%, respectively, for the C7 boronation of indole 11c (entry 3, Table 1) and tryptophan 11d (entry 4, Table 1).
Scheme 4. Optimized C7 Boronation of N-Boc-tryptophan.
The broader utility of this one-pot boronation protocol for the rapid synthesis of 7-substituted tryptamines and tryptophans is highlighted by the facile conversion of 7-boroindole 13 to a variety of derivatives (Scheme 5). The 7-borotryptophan 13d was converted to the corresponding 7-chloro-, 7-bromo-, and 7-iodotryptophan derivatives 15–17, respectively, in high yields using mild copper-mediated conditions.18c,33 Similarly, compound 13d could undergo efficient Suzuki–Miyaura cross coupling to furnish C7-arylated product 18 (Scheme 5).34 Another valuable transformation was the peroxide-mediated oxidation of 7-borotryptophan 13f that delivered the corresponding phenol 19 in 65% yield.35 Interestingly, aqueous hydroxylamine in methanol also proved highly efficient for this oxidation, with these milder conditions providing compound 19 in 86% yield.36
Scheme 5. Derivatization of 7-Tryptophans.

Conditions: (a) 13d, (i) for 15, CuCl2 2H2O, MeOH, H2O, 80 °C, 3 h, 93%; (ii) for 16, CuBr2, MeOH, H2O, 80 °C, 5 h, 76%; (b) 13d, Cu2O, NaI, aq NH3/MeOH, 23 °C, 14 h, 85%; (c) 13f, (i) 30% aq H2O2, 2% aq NaOH, THF, 23 °C, 30 min, 65%; (ii) 50% aq NH2OH, MeOH, 23 °C, 21 h, 86%; (d) 13d, p-bromoanisole, SPhos, Pd2(dba)3, K3PO4, PhMe, 80 °C, 12 h, 76%.
In summary, we have described a direct, practical, and scalable methodology for the one-pot conversion of C3-alkylindoles to the corresponding C7-boronated derivatives. The synthesis and study of a series of C2/C7-diboronated 3-alkylindoles and development of conditions for C2-selective protodeboronation of these C2/C7-diboronated indoles, including tryptophan and tryptamine derivatives, was described (Scheme 3, Table 1). The value of this strategy as an expedient means for rapid C7 derivatization of C3-alkylindoles was underscored by the gram-scale synthesis of C7-boronated N-Boc-L-tryptophan methyl ester (Scheme 4), providing a rapid entry to various unnatural amino acid derivatives (Scheme 5).
Experimental Section
General Methods
All reactions were performed in oven- or flame-dried round-bottomed flasks, reinforced screw-capped pressure tubes, or modified Schlenk (Kjeldahl shape) flasks. The flasks were fitted with rubber septa, and reactions were conducted under a positive pressure of argon. Stainless steel syringes or cannulae were used to transfer air- and moisture-sensitive liquids. Flash column chromatography was performed as described by Still et al. using silica gel (60 Å pore size, 40–63 μm, 4–6% H2O content).37 Analytical thin-layer chromatography was performed using glass plates precoated with 0.25 mm 230–400 mesh silica gel impregnated with a fluorescent indicator (254 nm). Thin layer chromatography plates were visualized by exposure to ultraviolet light and/or an aqueous solution of ceric ammonium molybdate (CAM) followed by heating (<1 min) on a hot plate (∼250 °C). Unless otherwise stated, all chemicals and solvents were obtained from commercial sources and used as received with the following exceptions: dichloromethane, tetrahydrofuran, hexane, toluene, methanol, and triethylamine were purified by the method of Grubbs et al. under positive argon pressure.38 Proton (1H) and carbon (13C) nuclear magnetic resonance spectra were recorded with 600, 500, and 400 MHz spectrometers. Proton nuclear magnetic resonance (1H NMR) spectra are reported in parts per million on the δ scale and are referenced from the residual protium in the NMR solvent (CDCl3, δ 7.24 (CHCl3); CD3OD, δ 3.31 (CH3OH)). Data is reported as follows: chemical shift [multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, st = sextet, sp = septet, m = multiplet, app = apparent, br = broad)], coupling constant(s) in hertz, integration. Carbon-13 nuclear magnetic resonance (13C NMR) spectra are reported in parts per million on the δ scale and are referenced from the carbon resonances of the solvent (CDCl3, δ 77.23; CD3OD, δ 49.15). Assignments of 13C chemical shifts for boron-bound carbons were not made owing to the effect of quadrupolar relaxation on the intensity of these signals.39 Data is reported as follows: chemical shift. Infrared data (IR) were obtained with a FTIR and are reported as follows: [frequency of absorption (cm–1), intensity of absorption (s = strong, m = medium, w = weak, br = broad)]. High-resolution mass spectrometric data (HRMS) were recorded on a FT-ICR-MS spectrometer using electrospray ionization (ESI) source or direct analysis in real-time (DART) ionization source.
N-(2-(1H-Indol-3-yl)ethyl)-N,2,4,6-tetramethylbenzenesulfonamide (11a)
Triethylamine (1.80 mL, 12.9 mmol, 1.04 equiv) was added via syringe to a solution of tryptamine (2.0 g, 12.4 mmol, 1 equiv) in dichloromethane (60 mL) at 23 °C under an argon atmosphere. A sample of 2-mesitylenesulfonyl chloride (2.6 g, 11.9 mmol, 0.96 equiv) was added cautiously, resulting in a slight exotherm. After 1 h, the reaction mixture was diluted with diethyl ether (500 mL) and washed with distilled water (300 mL). The aqueous layer was removed, and the organic layer was acidified to pH 1–2 by slow addition of aqueous hydrochloric acid (1N, 600 mL). The organic layer was washed with saturated aqueous sodium bicarbonate solution (2 × 300 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was dissolved in toluene and concentrated (2 × 100 mL) to give N-(2-(1H-indol-3-yl)ethyl)-2,4,6-trimethylbenzenesulfonamide S1 as a yellow solid. This material was sufficiently clean to be used in the next step without further purification; mp 92–95 °C. 1H NMR (500 MHz, CDCl3, 20 °C): δ 8.15 (br s, 1H), 7.44–7.27 (m, 2H), 7.17 (dd, J = 7.5, 8.5 Hz, 1H), 7.02 (t, J = 7.5 Hz, 1H), 6.94 (d, J = 2.5 Hz, 1H), 6.83 (s, 2H), 4.51 (t, J = 6.0 Hz, 1H), 3.20 (q, J = 6.5 Hz, 2H), 2.92 (t, J = 6.5 Hz, 2H), 2.45 (s, 6H), 2.26 (s, 3H). 13C NMR (125 MHz, CDCl3, 20 °C): δ 142.3, 139.2, 136.7, 133.5, 132.1, 127.0, 122.8, 122.5, 119.7, 118.6, 111.7, 111.5, 42.6, 25.5, 22.9, 21.1. FTIR (thin film, cm–1): 3397 (br s), 1604 (m), 1458 (s), 1317 (s), 1153 (s), 1076 (m), 744 (s), 656 (s). HRMS (ESI, TOF) (m/z): calcd for C19H23N2O2S [M + H+], 343.1475; found, 343.1466. TLC (30% ethyl acetate in hexanes), Rf = 0.36 (CAM, UV). The sulfonamide S1 was dissolved in dimethylformamide (60 mL), and 1,8-diazabicyclo[5.4.0]undec-7-ene (7.18 mL, 48.0 mmol, 3.87 equiv) was added. A sample of dimethyl sulfate (2.88 mL, 30.0 mmol, 2.42 equiv) was added, and the reaction mixture was stirred at 23 °C. After 3 h, another sample of 1,8-diazabicyclo[5.4.0]undec-7-ene (7.18 mL, 48.0 mmol, 3.87 equiv) and dimethyl sulfate (2.88 mL, 30.0 mmol, 2.42 equiv) was added. After 3 h, the reaction mixture was diluted with diethyl ether (200 mL) and washed with aqueous hydrochloric acid (1N, 500 mL). The organic layer was washed with saturated aqueous sodium bicarbonate solution (2 × 300 mL) and brine (300 mL) and was dried over anhydrous sodium sulfate. The organic layer was filtered and concentrated under reduced pressure, and the resulting brown residue was purified by flash column chromatography (silica gel, 4 cm × 8 cm; eluent, 50% ethyl acetate in hexanes) to afford N-methyl sulfamide 11a as an orange powder (3.30 g, 77.3% over two steps); mp 104–105 °C. 1H NMR (500 MHz, CDCl3, 20 °C): δ 8.03 (br s, 1H), 7.36 (d, J = 7.5 Hz, 1H), 7.32 (app dd, J = 0.5, 8.0 Hz, 1H), 7.15 (t, J = 8.0 Hz, 1H), 7.03 (t, J = 7.0 Hz, 1H), 6.94 (d, J = 2.0 Hz, 1H), 6.87 (s, 2H), 3.43–3.38 (m, 2H), 3.32–2.97 (m, 2H), 2.84 (s, 3H), 2.55 (s, 6H), 2.27 (s, 3H). 13C NMR (125 MHz, CDCl3, 20 °C): δ 142.5, 140.5, 136.4, 132.4, 132.0, 127.3, 122.2, 122.1, 119.5, 118.6, 112.6, 111.3, 49.6, 33.1, 23.9, 22.9, 21.2. FTIR (thin film, cm–1): 3403 (br s), 1604 (m), 1457 (s), 1309 (s), 1148 (s), 949 (s), 742 (s), 727 (s). HRMS (ESI, TOF) (m/z): calcd for C20H25N2O2S [M + H]+, 357.1631; found, 357.1617. TLC (30% ethyl acetate in hexanes), Rf = 0.49 (CAM, UV).
N-(2-(2,7-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indol-3-yl)ethyl)-N,2,4,6-tetra-methylbenzenesulfonamide (12a)
A round-bottomed flask equipped with a stir bar was charged sequentially with bis(pinacolato)diboron (142 mg, 560 μmol, 2.00 equiv), tryptamine 11a (100 mg, 281 μmol, 1 equiv), (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (3.7 mg, 5.6 μmol, 2.0 mol %), and 4,4′-di-tert-butyl-2,2′-bipyridine (3.0 mg, 11 μmol, 4.0 mol %). The flask was placed under an argon atmosphere, anhydrous tetrahydrofuran (1.4 mL) was added to the flask, and the resulting red–brown solution was stirred at 65 °C. After 20 h, the volatiles were then removed under reduced pressure, and the resulting black residue was purified by flash column chromatography (eluent: 25% ethyl acetate in hexanes) to afford 12a as a white foamy solid (136 mg, 80.1%); mp 163.5–165 °C. 1H NMR (400 MHz, CDCl3, 20 °C): δ 9.06 (br s, 1H), 7.62 (dd, J = 1.0, 7.0 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 6.96 (t, J = 7.0 Hz, 1H), 6.72 (s, 2H), 3.27–3.20 (m, 2H), 3.19–3.12 (m, 2H), 2.97 (s, 3H), 2.46 (s, 6H), 2.21 (s, 3H), 1.39 (s, 12H), 1.33 (s, 12H). 13C NMR (100 MHz, CDCl3, 20 °C): δ 143.1, 142.2, 140.4, 132.4, 131.8, 131.5, 127.0, 125.0, 122.9, 119.0, 84.0, 83.9, 50.6, 32.5, 25.2, 25.1, 23.4, 22.9, 21.2. FTIR (thin film, cm–1): 3455 (m), 2978 (m), 2930 (m), 1598 (m), 1551 (m), 1372 (s), 1320 (s), 1292 (s), 1141 (s), 683 (m). HRMS (ESI, TOF) (m/z): calcd for C32H47B2N2O6S [M + H]+, 609.3360; found, 609.3371. TLC (20% ethyl acetate in hexanes), Rf = 0.47 (CAM, UV).
N,2,4,6-Tetramethyl-N-(2-(7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indol-3-yl)ethyl)-benzenesulfonamide (13a)
A sample of diborotryptamine 12a (500.0 mg, 822 μmol, 1 equiv) in a 25 mL round-bottomed flask was flushed three times with argon, then was dissolved in anhydrous dichloromethane (8.0 mL) and cooled to 0 °C. Trifluoroacetic acid (0.620 mL, 8.34 mmol, 10.2 equiv) was then added dropwise via a gastight syringe, and the colorless reaction solution was stirred at 0 °C for 5 min before warming to 23 °C over 10 min. After a further 5 min, the resulting light-green solution was concentrated under reduced pressure to leave a pale-green residue, which was purified by flash column chromatography on silica gel (eluent: 20% ethyl acetate in hexanes) to provide the boronic ester 13a as a white foamy solid (320 mg, 80.7%). Structural assignments were made with additional information from gCOSY, HSQC, and gHMBC data; mp 44–44.5 °C. 1H NMR (600 MHz, CDCl3, 20 °C): δ 9.03 (br s, 1H), 7.62 (d, J = 7.0 Hz, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.05 (app t, J = 7.3 Hz, 1H), 7.01 (d, J = 1.7 Hz, 1H), 6.86 (s, 2H), 3.39 (t, J = 7.7 Hz, 2H), 3.00 (t, J = 8.0 Hz, 2H), 2.85 (s, 3H), 2.55 (s, 6H), 2.27 (s, 3H), 1.38 (s, 12H). 13C NMR (150 MHz, CDCl3, 20 °C): δ 142.4, 141.5, 140.3, 132.4, 131.9, 129.3, 126.0, 122.1, 122.0, 118.9, 112.0, 83.9, 49.7, 33.0, 25.1, 23.8, 22.9, 21.1. FTIR (thin film, cm–1): 3457 (s), 2978 (s), 2936 (s), 1605 (m), 1592 (m), 1372 (s), 1328 (s), 1151 (s), 729 (w). HRMS (ESI, TOF) (m/z): calcd for C26H36BN2O4S [M + H]+, 483.2498; found, 483.2488. TLC (5% acetone, 15% dichloromethane, 80% hexanes), Rf = 0.11 (CAM, UV).
N,2,4,6-Tetramethyl-N-(2-(2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indol-3-yl)ethyl)-benzenesulfonamide (14)
A round-bottomed flask equipped with a stir bar was charged sequentially with bis(pinacolato)diboron (71.1 mg, 280 μmol, 1.00 equiv), tryptamine 11a (100 mg, 280 μmol, 1 equiv), (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (3.7 mg, 5.6 μmol, 2.0 mol %), and 4,4′-di-tert-butyl-2,2′-bipyridine (3.0 mg, 11 mmol, 4.0 mol %). After flushing with argon for 5 min, anhydrous dichloromethane (1.4 mL) was added to the flask and the resulting red solution was stirred at 60 °C. After 3 h, the volatiles were removed under reduced pressure, and the resulting black residue was purified by flash column chromatography (eluent: 25% ethyl acetate in hexanes) to afford 14 as a white foamy solid (91.6 mg, 67.7%); mp 42–43 °C. 1H NMR (500 MHz, CDCl3, 20 °C): δ 8.34 (br s, 1H), 7.26 (t, J = 8.0 Hz, 2H), 7.16 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 6.95 (ddd, J = 1.0, 7.0, 8.0 Hz, 1H), 6.74 (s, 2H), 3.32–3.27 (m, 2H), 3.21–3.17 (m, 2H), 2.98 (s, 3H), 2.49 (s, 6H), 2.21 (s, 3H), 1.33 (s, 12H). 13C NMR (125 MHz, CDCl3, 20 °C): δ 142.2, 140.3, 138.2, 132.3, 132.0, 131.8, 127.9, 125.2, 123.8, 119.4, 111.4, 84.0, 50.4, 32.5, 25.1, 23.4, 22.9, 21.2. FTIR (thin film, cm–1): 3395 (br m), 2975 (s), 1550 (s), 1315 (s), 1144 (s), 730 (s). HRMS (ESI, TOF) (m/z): calcd for C26H36BN2O4S [M + H]+, 483.2498; found, 483.2490. TLC (30% ethyl acetate in hexanes), Rf = 0.41 (CAM, UV).
N,2,4,6-Tetramethyl-N-(2-(7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indol-3-yl)ethyl)-benzenesulfonamide (13a)
A pressure tube was charged sequentially with (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (5.0 mg, 7.5 μmol, 2.5 mol %), 4,4′-di-tert-butyl-2,2′-bipyridine (4.0 mg, 15 μmol, 5.0 mol %), and tryptamine 11a (107 mg, 300 μmol, 1 equiv). The contents of the reaction vessel were kept under an argon atmosphere. Freshly distilled anhydrous tetrahydrofuran (2 mL) was then introduced to the flask via a gastight syringe to afford a dark-brown solution. Pinacolborane (218 μL, 1.50 mmol, 5.00 equiv) was added in a single portion via a gastight syringe to afford a red solution. The pressure tube was sealed and was stirred at 80 °C for 21 h and 15 min. The tube was subsequently cooled to 0 °C, and anhydrous dichloromethane (3 mL) was added under an argon atmosphere. Trifluoroacetic acid (2 mL) was then added dropwise via a gastight syringe to afford a clear-orange solution. The solution was stirred at 0 °C for 10 min and was then warmed to 23 °C and was stirred for 5 h and 15 min. The resulting solution was diluted with dichloromethane (50 mL) and was washed with saturated aqueous sodium bicarbonate solution (50 mL). The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting brown residue was purified by flash column chromatography on silica gel (eluent: 5% acetone, 15% dichloromethane, 80% hexanes) to provide the boronic ester 13a (86.4 mg, 59.6%). All data was in accordance with that obtained from the aforementioned protodeboronation of 12a.
Methyl (2-(7-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indol-3-yl)ethyl)carbamate (13b)
A pressure tube was charged sequentially with (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (5.0 mg, 7.5 μmol, 2.5 mol %), 4,4′-di-tert-butyl-2,2′-bipyridine (4.0 mg, 15 μmol, 5.0 mol %), and tryptamine methyl carbamate4011b (67.0 mg, 307 μmol, 1 equiv). The contents of the reaction vessel were kept under an argon atmosphere. Freshly distilled anhydrous tetrahydrofuran (2 mL) was introduced to the flask via a gastight syringe to afford a dark-brown solution. Pinacolborane (218 μL, 1.50 mmol, 4.86 equiv) was added in a single portion via a gastight syringe, at which point the solution turned bright red. The pressure tube was sealed and the reaction mixture stirred at 60 °C for 6 h and 15 min and then cooled to 0 °C, whereupon anhydrous dichloromethane (3 mL) was added under an argon atmosphere. Trifluoroacetic acid (2 mL) was then added dropwise via a gastight syringe to afford an orange solution. The solution was stirred at 0 °C for 10 min and then warmed to 23 °C and stirred for 2 h and 30 min. The solution was diluted with dichloromethane (50 mL) and washed with saturated aqueous sodium bicarbonate solution (50 mL). The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting brown residue was purified by flash column chromatography on silica gel (eluent: 10% acetone, 20% dichloromethane, 70% hexanes) to provide the boronic ester 13b (70.0 mg, 66.2%) as a white waxy solid. Structural assignments were made with additional information from gCOSY, HSQC, and gHMBC data; mp 138–140 °C. 1H NMR (600 MHz, CDCl3, 20 °C): δ 9.07 (br s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.64 (d, J = 6.9 Hz, 1H), 7.13 (app t, J = 7.3 Hz, 1H), 7.09 (br s, 1H), 4.71 (br s, 1H), 3.63 (s, 3H), 3.49 (app q, 2H), 2.97 (app t, 2H), 1.37 (s, 12H). 13C NMR (150 MHz, CDCl3, 20 °C): δ 157.2, 141.7, 129.6, 126.3, 122.4, 122.2, 119.1, 112.4, 84.0, 52.2, 41.4, 25.9, 25.1. FTIR (thin film, cm–1): 3451 (s), 2978 (s), 2939 (s), 1713 (s), 1522 (m), 1373 (m), 1135 (s), 966 (w), 684 (m). HRMS (ESI, TOF) (m/z): calcd for C18H26BN2O4 [M + H]+, 345.1994; found, 345.1996. TLC (10% acetone, 20% dichloromethane, 70% hexanes), Rf = 0.21 (CAM, UV).
3-Methyl-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (13c)
A pressure tube was charged sequentially with (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (5.0 mg, 7.5 μmol, 2.5 mol %), 4,4′-di-tert-butyl-2,2′-bipyridine (4.0 mg, 15 μmol, 5.0 mol %), and 3-methylindole 11c (39.0 mg, 0.297 mmol, 1 equiv). The contents of the reaction vessel were kept under an argon atmosphere. Anhydrous tetrahydrofuran (2 mL) was introduced to the flask under an argon atmosphere to afford a clear - brown solution. Pinacolborane (218 μL, 1.50 mmol, 5.05 equiv) was added in a single portion via a gastight syringe, the pressure tube was sealed, and the resulting red solution was stirred at 60 °C. After 5 h and 30 min, the reaction contents were cooled to 0 °C, and anhydrous dichloromethane (3 mL) was added under an argon atmosphere. Trifluoroacetic acid (2 mL) was then added dropwise via a gastight syringe to afford an orange solution that was stirred at 0 °C for 10 min and then warmed to 23 °C and stirred for a further 3 h. The resulting solution was diluted with dichloromethane (50 mL) and washed with saturated aqueous sodium bicarbonate solution (50 mL). The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give a brown residue that was purified by flash column chromatography on silica gel (eluent: 1.5% acetone in hexanes) to provide the boronic ester 13c (41.0 mg, 53.6%) as a white waxy solid. Structural assignments were made with additional information from gCOSY, HSQC, and gHMBC data; mp 46–47 °C. 1H NMR (600 MHz, CDCl3, 20 °C): δ 8.94 (br s, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.63 (d, J = 7.0 Hz, 1H), 7.11 (t, J = 7.2 Hz, 1H), 7.00 (d, J = 0.6 Hz, 1H), 2.33 (s, 3H), 1.37 (s, 12H). 13C NMR (150 MHz, CDCl3, 20 °C): δ 141.5, 129.2, 127.3, 122.5, 121.6, 118.7, 111.2, 83.9, 25.1, 9.8. FTIR (thin film, cm–1): 3462 (s), 2977 (s), 2923 (m), 1607 (m), 1592 (m), 1437 (m), 1371 (s), 1136 (s), 848 (m). HRMS (ESI, TOF) (m/z): calcd for C15H21BNO2 [M + H]+, 258.1673; found, 258.1668. TLC (5% diethyl ether in hexanes), Rf = 0.26 (CAM, UV).
Alternative Procedure, 3-Methyl-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (13c)
Anhydrous tetrahydrofuran (11 mL) was added via syringe to 3-methylindole 11c (201 mg, 1.53 mmol, 1 equiv), (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (25.4 mg, 38.3 μmol, 2.50 mol %), and 4,4′-di-tert-butyl-2,2′-bipyridine (20.6 mg, 76.6 μmol, 5.00 mol %) sealed in a flame-dried round-bottomed flask under an argon atmosphere to result in a dark-brown solution. Pinacolborane (1.10 mL, 7.66 mmol, 5.00 equiv) was introduced in a single portion via a gastight syringe, and the resulting red solution was stirred at 60 °C for 8 h. After cooling to 23 °C and removal of volatiles under reduced pressure, the subsequent brown residue was dissolved in acetic acid (4.0 mL) and palladium(II) acetate (17.2 mg, 76.6 μmol, 5.00 mol %) was added and the resulting mixture was stirred under argon at 30 °C. After 8 h, the mixture was cooled to 23 °C and was filtered through Celite with ethyl acetate (150 mL) as eluent. The filtrate was washed with saturated aqueous sodium bicarbonate solution (200 mL), and the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting brown residue was purified by flash column chromatography on silica gel (eluent: 1.5% acetone in hexanes) to provide the boronic ester 13c (228 mg, 57.9%) as a white waxy solid. All data for 13c was in accordance with that obtained from the previous procedure.
(S)-Methyl 2-((methoxycarbonyl)amino)-3-(7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl))-1H-indol-3-yl)propanoate (13d)
A pressure tube was charged sequentially with (1,5-cyclooctadiene)(methoxy)-iridium(I) dimer (5.0 mg, 7.5 μmol, 2.5 mol %), 4,4′-di-tert-butyl-2,2′-bipyridine (4.0 mg, 15 μmol, 5.0 mol %), and L-tryptophan methyl ester 11d (83.0 mg, 300 μmol, 1 equiv). The contents of the reaction vessel were kept under an argon atmosphere. Anhydrous tetrahydrofuran (2 mL) was introduced to the flask via a gastight syringe to afford a clear dark-brown solution. Pinacolborane (218 μL, 1.50 mmol, 5.00 equiv) was then added in a single portion via a gastight syringe, the pressure tube was sealed, and the resulting red solution was stirred at 60 °C. After 4 h, the tube was subsequently cooled to 0 °C and anhydrous dichloromethane (3 mL) was added via a gastight syringe. Trifluoroacetic acid (2 mL) was added dropwise via a gastight syringe to afford an orange solution that was stirred at 0 °C for 10 min, then was warmed to 23 °C and was stirred for a further 5 h and 15 min. The resulting solution was diluted with dichloromethane (50 mL) and washed with saturated aqueous sodium bicarbonate solution (50 mL). The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting brown residue was purified by flash column chromatography on silica gel (eluent: 8% acetone, 30% dichloromethane, 62% hexanes) to provide the boronic ester 13d (101 mg, 83.5%) as a white powder. Structural assignments were made with additional information from gCOSY, HSQC, and gHMBC data; mp 151–152 °C. 1H NMR (600 MHz, CDCl3, 20 °C): δ 9.16 (br s, 1H), 7.65 (d, J = 8.4 Hz, 1H), 7.62 (d, J = 7.2 Hz, 1H), 7.13 (app t, J = 7.3 Hz, 1H), 7.05 (d, J = 1.7 Hz, 1H), 5.28 (d, J = 8.1 Hz, 1H), 4.68 (q, J = 2.8 Hz, 1H), 3.66 (s, 3H), 3.64 (s, 3H), 3.30 (d, J = 4.7 Hz, 2H), 1.37 (s, 12H). 13C NMR (150 MHz, CDCl3, 20 °C): δ 172.6, 156.5, 141.3, 129.5, 126.5, 122.9, 122.2, 119.2, 109.3, 83.9, 54.5, 52.4, 52.3, 28.0, 25.0. FTIR (thin film, cm–1): 3448 (s), 2979 (s), 2953 (m), 1722 (s), 1591 (m), 1516 (s), 1374 (m), 1329 (m), 1134 (s), 684 (w). HRMS (ESI, TOF) (m/z): calcd for C20H28BN2O6 [M + H]+, 403.2050; found, 403.2030. TLC (10% acetone, 20% dichloromethane, 70% hexanes), Rf = 0.19 (CAM, UV).
Alternative Procedure, (S)-Methyl 2-((methoxycarbonyl)amino)-3-(7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl))-1H-indol-3-yl)propanoate (13d)
Anhydrous tetrahydrofuran (6 mL) was added to L-tryptophan methyl ester 11d (203 mg, 735 μmol, 1 equiv), (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (12.2 mg, 18.4 μmol, 2.50 mol %), and 4,4′-di-tert-butyl-2,2′-bipyridine (9.8 mg, 37 μmol, 5.0 mol %) sealed in a flame-dried round-bottomed flask under an argon atmosphere. Pinacolborane (530 μL, 3.68 mmol, 5.00 equiv) was added to the resulting dark-brown solution in a single portion via a gastight syringe, and the resulting red solution was stirred at 60 °C. After 8 h, the mixture was allowed to cool to 23 °C and the volatiles were removed under reduced pressure to give a brown residue. Acetic acid (1.8 mL) and palladium(II) acetate (8.3 mg, 37 μmol, 5.0 mol %) were added, and the resulting mixture was stirred under argon at 30 °C. After 18 h, the mixture was allowed to cool to 23 °C and filtered through Celite with ethyl acetate (100 mL) as eluent. The filtrate was washed with saturated aqueous sodium bicarbonate solution (150 mL), and the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting brown residue was purified by flash column chromatography on silica gel (eluent: 8% acetone, 30% dichloromethane, 62% hexanes) to provide the boronic ester 13d (213 mg, 72.3%) as a white powder. All data for 13d was in accordance with that obtained from the previous procedure.
2-(7-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indol-3-yl)ethan-1-ol (13e)
A pressure tube was charged sequentially with (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (20.4 mg, 30.7 μmol, 2.42 mol %), 4,4′-di-tert-butyl-2,2′-bipyridine (16.5 mg, 61.4 μmol, 4.84 mol %), and tryptophol 11e (205 mg, 1.27 mmol, 1 equiv). The contents of the reaction vessel were kept under an argon atmosphere. Freshly distilled anhydrous tetrahydrofuran (10 mL) was introduced to the flask via a gastight syringe to afford a dark-brown solution. Pinacolborane (895 mL, 6.17 mmol, 4.87 equiv) was added in a single portion via a gastight syringe, at which point the solution turned bright red. The pressure tube was sealed and the reaction mixture was stirred at 60 °C for 7 h, then cooled to 0 °C, whereupon anhydrous dichloromethane (15 mL) was added under an argon atmosphere. Trifluoroacetic acid (10 mL) was then added dropwise via a gastight syringe to afford an orange solution. The solution was stirred at 0 °C for 10 min and was then warmed to 23 °C and was stirred for 4 h. The reaction solution was diluted with dichloromethane (100 mL) and was washed with saturated aqueous sodium bicarbonate solution (2 × 200 mL). The organic layer was concentrated under reduced pressure, dissolved in pentane/diethyl ether (2:1, 300 mL), and washed with water (5 × 300 mL). The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to leave a yellow residue that was purified by flash column chromatography on silica gel (eluent: 15% ethyl acetate in hexanes) to give a white solid. This material was vacuum-dried (6–8 mTorr) for 48 h at 70 °C to remove remaining pinacol, and 7-borotryptophol 13e was obtained as a white solid (227 mg, 62.7%); mp 78–78.5 °C. 1H NMR (500 MHz, CDCl3, 20 °C): δ 9.12 (br s, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.67 (d, J = 7.0 Hz, 1H), 7.15 (t, J = 7.4 Hz, 1H), 7.13 (s, 1H), 3.92–3.86 (m, 2H), 3.05 (t, J = 6.2 Hz, 2H), 1.73 (br s, 1H), 1.40 (s, 12H). 13C NMR (125 MHz, CDCl3, 20 °C): δ 142.3, 130.2, 127.0, 123.2, 123.1, 119.7, 112.3, 84.5, 63.4, 29.5, 25.7. FTIR (thin film, cm–1): 3457 (br), 2978 (s), 1684 (s), 1559 (m), 1437 (m), 1374 (w), 1329 (w), 1135 (w), 1048 (s), 668 (m). HRMS (ESI, TOF) (m/z): calcd for C16H23BNO3 [M + H]+, 288.1766; found, 288.1763. TLC (30% ethyl acetate in hexanes), Rf = 0.24 (CAM, UV).
(S)-Methyl 2-((tert-Butoxycarbonyl)amino)-3-(7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indol-3-yl)propanoate (13f)
N-Boc-L-tryptophan methyl ester 11f (1.01 g, 3.18 mmol, 1 equiv), (1,5-cyclooctadiene)(methoxy)iridium(I) dimer (52.8 mg, 79.6 μmol, 2.50 mol %), and 4,4′-di-tert-butyl-2,2′-bipyridine (42.7 mg, 159 μmol, 5.00 mol %) were sealed in a flame-dried round-bottomed flask under an argon atmosphere and anhydrous tetrahydrofuran (32 mL) was added. To the resulting dark-brown solution was added pinacolborane (2.31 mL, 15.9 mmol, 5.00 equiv) in a single portion via a gastight syringe, and the resulting red solution was stirred at 60 °C for 12 h. After cooling to 23 °C and removal of volatiles under reduced pressure, the subsequent brown residue was dissolved in acetic acid (3.5 mL) and palladium(II) acetate (35.7 mg, 0.159 mmol, 5.00 mol %) was added. The mixture was stirred under argon at 30 °C for 10 h, then cooled to 23 °C and filtered through Celite with ethyl acetate (50 mL) as eluent. The filtrate was washed with saturated aqueous sodium bicarbonate solution (50 mL), and the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting brown residue was purified by flash column chromatography on silica gel (eluent: 5% acetone, 15% dichloromethane, 80% hexanes) to provide the 7-borotryptophan derivative 13f as a white powder (1.03 g, 73.1%); mp 152–153 °C. 1H NMR (500 MHz, CDCl3, 20 °C): δ 9.11 (br s, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.62 (d, J = 7.0 Hz, 1H), 7.11 (t, J = 7.5 Hz, 1H), 7.04 (s, 1H), 5.03 (d, J = 8.0 Hz, 1H), 4.65–4.57 (m, 1H), 3.65 (s, 3H), 3.29 (d, J = 5.0 Hz, 2H), 1.41 (s, 9H), 1.37 (s, 12H). 13C NMR (125 MHz, CDCl3, 20 °C): δ 172.9, 155.4, 141.5, 129.7, 126.8, 122.9, 122.5, 119.3, 109.8, 84.0, 79.9, 54.4, 52.4, 28.5, 28.1, 25.2. FTIR (thin film, cm–1): 3451 (br s), 2978 (m), 1745 (s), 1714 (s), 1592 (m), 1493 (s), 1436 (m), 1373 (m), 1329 (m), 1167 (m), 1134 (s), 1050 (m), 966 (s), 914 (s), 849 (m). HRMS (ESI, TOF) (m/z): calcd for C23H34BN2O6 [M + H]+, 445.2520; found, 445.2515. Anal. Calcd for C23H33BN2O6: C, 62.17; H, 7.49; N, 6.30. Found: C, 62.24; H, 7.42; N, 6.24. TLC (5% acetone, 15% dichloromethane, 80% hexanes), Rf = 0.28 (CAM, UV).
(S)-Methyl 2-((Methoxycarbonyl)amino)-3-(7-chloro-1H-indol-3-yl)propanoate (15)
To a methanol solution (2.4 mL) of 7-borotryptophan 13d (80.5 mg, 200 μmol, 1 equiv) was added an aqueous solution (2.4 mL) of copper(II) chloride dihydrate (102 mg, 670 μmol, 3.35 equiv). The resulting mixture was stirred for 3 h at 80 °C, then cooled to 23 °C and diluted with ethyl acetate (10 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (2 × 20 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 25% ethyl acetate in hexanes) to provide tryptophan 15 as white needles (57.7 mg, 92.7% yield); mp 118.5–119 °C. 1H NMR (500 MHz, CDCl3, 20 °C): δ 8.33 (br s, 1H), 7.42 (d, J = 8.0 Hz, 1H), 7.17 (d, J = 7.0 Hz, 1H), 7.08–7.01 (m, 2H), 5.22 (d, J = 7.5 Hz, 1H), 4.72–4.64 (m, 1H), 3.66 (s, 3H), 3.65 (s, 3H), 3.27 (d, J = 5.0 Hz, 2H). 13C NMR (125 MHz, CDCl3, 20 °C): δ 172.5, 156.6, 133.6, 129.2, 123.6, 121.9, 120.7, 117.5, 117.0, 111.4, 54.6, 52.63, 52.57, 28.3. FTIR (thin film, cm–1): 3341 (br s), 2952 (s), 1709 (s), 1521 (m), 1439 (s), 1341 (m), 1205 (m), 1082 (m), 895 (s), 843 (s), 779 (s), 738 (s). HRMS (ESI, TOF) (m/z): calcd for C14H16ClN2O4 [M + H]+, 311.0793; found, 311.0808. TLC (25% ethyl acetate in hexanes), Rf = 0.17 (CAM, UV).
(S)-Methyl 2-((Methoxycarbonyl)amino)-3-(7-bromo-1H-indol-3-yl)propanoate (16)
To a methanol solution (2.4 mL) of 7-borotryptophan 13d (80.5 mg, 200 μmol, 1 equiv) was added an aqueous solution (2.4 mL) of copper(II) bromide (134 mg, 600 μmol, 3.00 equiv). The resulting mixture was heated at 80 °C for 4 h and 30 min. The reaction mixture was cooled to 23 °C and was diluted with ethyl acetate (10 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 25% ethyl acetate in hexanes) to provide tryptophan 16 as a light-yellow powder (54.2 mg, 76.2% yield); mp 123–124 °C. 1H NMR (500 MHz, CDCl3, 20 °C): δ 8.30 (br s, 1H), 7.46 (d, J = 7.5 Hz, 1H), 7.32 (d, J = 7.5 Hz, 1H), 7.04 (d, J = 1.0 Hz, 1H), 6.99 (t, J = 8.0 Hz, 1H), 5.23 (d, J = 8.0 Hz, 1H), 4.72–4.64 (m, 1H), 3.66 (s, 3H), 3.65 (s, 3H), 3.26 (d, J = 5.0 Hz, 2H). 13C NMR (125 MHz, CDCl3, 20 °C): δ 172.5, 156.6, 135.0, 128.9, 124.8, 123.6, 121.1, 118.1, 111.5, 105.1, 54.6, 52.64, 52.57, 28.4. FTIR (thin film, cm–1): 3335 (br s), 2952 (s), 1707 (s), 1521 (m), 1437 (s), 1337 (m), 1203 (m), 1077 (m), 882 (s), 827 (s), 777 (s), 738 (s). HRMS (ESI, TOF) (m/z): calcd for C14H16BrN2O4 [M + H]+, 355.0288; found, 355.0281. TLC (25% ethyl acetate in hexanes), Rf = 0.17 (CAM, UV).
(S)-Methyl 2-((Methoxycarbonyl)amino)-3-(7-iodo-1H-indol-3-yl)propanoate (17)
A round bottomed flask was charged with copper(I) oxide (2.9 mg, 20 μmol, 10 mol %) and 28% aqueous ammonia (62 μL, 500 μmol, 2.5 equiv), and the resulting mixture was stirred under air at 23 °C for 15 min. 7-Borotryptophan 13d (80.5 mg, 200 μmol, 1 equiv), sodium iodide (150 mg, 1.00 mmol, 5.00 equiv), and methanol (0.6 mL) were then added to the reaction solution, and stirring under air was continued at 23 °C for 14 h. The resulting solution was concentrated under reduced pressure, and the residue was dissolved in water (5 mL). This aqueous solution was then extracted with ethyl acetate (3 × 20 mL), and the combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 25% ethyl acetate in hexanes) to provide tryptophan 17 as a white solid (68.5 mg, 85.1% yield); mp 119–119.5 °C. 1H NMR (500 MHz, CDCl3, 20 °C): δ 8.13 (br s, 1H), 7.53 (d, J = 7.5 Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.05 (d, J = 2.0 Hz, 1H), 6.88 (t, J = 8.0 Hz, 1H), 5.19 (d, J = 8.0 Hz, 1H), 4.71–4.65 (m, 1H), 3.66 (s, 3H), 3.65 (s, 3H), 3.25 (d, J = 5.5 Hz, 2H). 13C NMR (125 MHz, CDCl3, 20 °C): δ 172.5, 156.6, 138.1, 131.1, 127.8, 123.3, 121.6, 119.0, 111.8, 54.5, 52.64, 52.57, 28.5. FTIR (thin film, cm–1): 3464 (br s), 1708 (s), 1517 (m), 1432 (s), 1333 (m), 1202 (m), 1075 (m), 776 (s), 666 (s). HRMS (ESI, TOF) (m/z): calcd for C14H16IN2O4 [M + H]+, 403.0149; found, 403.0151. TLC (25% ethyl acetate in hexanes), Rf = 0.14 (CAM, UV).
(S)-Methyl 2-((Methoxycarbonyl)amino)-3-(7-(4-methoxyphenyl)-1H-indol-3-yl)propanoate (18)
A dry round-bottomed flask was charged sequentially with 7-borotryptophan 13d (80.5 mg, 200 μmol, 1 equiv), tris(dibenzylideneacetone)dipalladium(0) (9.1 mg, 10 μmol, 5.0 mol %), SPhos (8.2 mg, 20 μmol, 10 mol %), and tribasic potassium phosphate (84.9 mg, 400 μmol, 2.00 equiv), sealed, and placed under an argon atmosphere. Toluene (0.8 mL) was added, and to the resulting red–brown slurry p-bromoanisole (30.0 μL, 239 μmol, 1.20 equiv) was added via syringe in a single portion. The resulting dark-green reaction mixture was stirred at 80 °C for 12 h, then was cooled to 23 °C and filtered through Celite with ethyl acetate as eluent. The filtrate was concentrated under reduced pressure to yield a residue that was purified by flash column chromatography on silica gel (eluent: 40% ethyl acetate in hexanes) to provide tryptophan 18 as a white waxy solid (58.5 mg, 76.4% yield); mp 165 °C (decomp.). 1H NMR (400 MHz, CDCl3, 20 °C): δ 8.26 (br s, 1H), 7.52 (d, J = 8.5 Hz, 2H), 7.49 (dd, J = 7.3 Hz, 1.3 Hz, 1H), 7.20–7.14 (m, 2H), 7.02 (d, J = 9.0 Hz, 2H), 6.99 (s, 1H), 5.24 (d, J = 7.5 Hz, 1H), 4.74–4.67 (m, 1H), 3.86 (s, 3H), 3.69 (s, 3H), 3.65 (s, 3H), 3.31 (d, J = 5.5 Hz, 2H). 13C NMR (125 MHz, CDCl3, 20 °C): δ 172.7, 159.3, 156.7, 134.3, 131.5, 129.5, 128.1, 125.7, 123.1, 122.2, 120.5, 117.6, 114.8, 110.6, 55.6, 54.7, 52.6, 52.5, 28.2. FTIR (thin film, cm–1): 3374 (br s), 2952 (s), 1710 (s), 1610 (s), 1513 (s), 1438 (m), 1355 (m), 1247 (m), 1056 (s), 1029 (s), 836 (s), 799 (m), 748 (s). HRMS (ESI, TOF) (m/z): calcd for C21H23N2O5 [M + H]+, 383.1601; found, 383.1617. TLC (40% ethyl acetate in hexanes), Rf = 0.29 (CAM, UV).
(S)-Methyl 2-((tert-Butoxycarbonyl)amino)-3-(7-hydroxy-1H-indol-3-yl)propanoate (19)
Conditions: (i) A sample of N-Boc tryptophan boronic ester 13f (20.2 mg, 45.5 μmol, 1 equiv) was dissolved in anhydrous tetrahydrofuran (0.5 mL) and a solution of 2% aqueous sodium hydroxide was added (0.5 mL), followed by slow addition of 30% (wt) aqueous hydrogen peroxide (51.5 μL, 455 μmol, 10.0 equiv). The resulting purple reaction solution was stirred for 30 min at 23 °C and then quenched by addition of a solution of saturated ammonium hydroxide/saturated ammonium chloride (3:1, 5 mL). After extracting with ethyl acetate (3 × 5 mL), the organic layers were combined and washed with brine (2 × 10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting brown residue was purified by flash column chromatography on silica gel (eluent: 15% acetone, 15% dichloromethane, 70% hexanes) to afford phenol 19 as an off-white solid (9.8 mg, 64.6%). (ii) To a sample of N-Boc tryptophan boronic ester 13f (25.3 mg, 56.9 μmol, 1 equiv) in methanol (1.0 mL) was added 50% aqueous hydroxylamine (18.8 μL, 285 μmol, 5.00 equiv), and the resulting white slurry was stirred at 23 °C. After 3 h, another dose of 50% aqueous hydroxylamine (18.8 mg, 285 μmol, 5.00 equiv) was added and the reaction was stirred at 23 °C. After 15 h, the reaction mixture had turned into a light-brown solution, at which time TLC analysis indicated complete disappearance of starting material. The reaction was diluted with water (5 mL) and was extracted with ethyl acetate (3 × 5 mL). The organic layers were combined and were washed sequentially with saturated aqueous ammonium chloride (2 × 10 mL), saturated aqueous sodium bicarbonate (10 mL), and brine (10 mL) before being dried over anhydrous sodium sulfate. After filtration and concentration under reduced pressure, the resulting brown residue was purified by flash column chromatography on silica gel (eluent: 15% acetone, 15% dichloromethane, 70% hexanes) to afford phenol 19 as an off-white powder (16.8 mg, 86.3%); mp 185 °C (decomp.). 1H NMR (500 MHz, CD3OD, 20 °C): δ 7.02 (d, J = 7.0 Hz, 1H), 7.01 (s, 1H), 6.83 (t, J = 7.6 Hz, 1H), 6.51 (d, J = 7.6 Hz, 1H), 4.40–4.37 (m, 1H), 3.65 (s, 3H), 3.20 (d, J = 14.5, 5.5 Hz, 1H), 3.08 (d, J = 14.5, 8.2 Hz, 1H), 1.39 (s, 9H). 13C NMR (125 MHz, CDCl3, 20 °C): δ 174.9, 157.9, 144.8, 130.9, 128.1, 124.1, 120.7, 111.3, 111.0, 106.8, 80.8, 56.3, 52.7, 29.0, 28.8. FTIR (thin film, cm–1): 3384 (br), 2899 (m), 1684 (s), 1583 (m), 1506 (m), 1437 (w), 1368 (m), 1164 (m). HRMS (ESI, TOF) (m/z): calcd for C17H23N2O5 [M + H]+, 335.1601; found, 335.1614. TLC (15% acetone, 15% dichloromethane, 70% hexanes), Rf = 0.17 (CAM, UV).
Acknowledgments
We acknowledge financial support by NIH-NIGMS (GM089732 and GM074825) and the NSF under CCI Center for selective C–H functionalization (CHE-1205646). R.P.L. thanks the Fonds de Recherche du Québec—Nature et Technologies for a postdoctoral fellowship. K.A. acknowledges support from the Institute of Transformative Bio-Molecules, Nagoya University, and the NSF program for Science Across Virtual Institutes for a summer fellowship. E.O. thanks MECYD for a FPU fellowship.
Supporting Information Available
1H and 13C NMR spectra of all products described in the Experimental Section. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For indole reviews, see:; a Sundberg R. J.Indoles; Academic Press: London, 1996. [Google Scholar]; b Sumpter W. G.; Miller F. M., Eds. In Natural Products Containing the Indole Nucleus; Heterocyclic Compounds with Indole and Carbazole Systems, Vol. 8; John Wiley & Sons, Inc.: Hoboken, NJ, 2008. [Google Scholar]; c Gribble G. W. In Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Topics in Heterocyclic Chemistry, Vol. 26; Springer-Verlag: Berlin, Heidelberg, 2010. [Google Scholar]; d Vicente R. Org. Biomol. Chem. 2011, 9, 6469. [DOI] [PubMed] [Google Scholar]; e Shiri M. Chem. Rev. 2012, 112, 3508. [DOI] [PubMed] [Google Scholar]; f Kaushik N. K.; Kaushik N.; Attri P.; Kumar N.; Kim C. H.; Verma A. K.; Choi E. H. Molecules 2013, 19, 6620. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Ishikura M.; Abe T.; Choshi T.; Hibino S. Nat. Prod. Rep. 2013, 30, 694. [DOI] [PubMed] [Google Scholar]
- a Cacchi S.; Fabrizi G. Chem. Rev. 2005, 105, 2873. [DOI] [PubMed] [Google Scholar]; and references therein.; b Bandini M.; Eichholzer A. Angew. Chem., Int. Ed. 2009, 48, 9608. [DOI] [PubMed] [Google Scholar]; c Xie Y.; Zhao Y.; Qian B.; Yang L.; Xia C.; Huang H. Angew. Chem., Int. Ed. 2011, 50, 5682. [DOI] [PubMed] [Google Scholar]; d Davies H. M. L.; Du Bois J.; Yu J.-Q. Chem. Soc. Rev. 2011, 40, 1855. [DOI] [PubMed] [Google Scholar]; e Broggini G.; Beccalli E. M.; Fasana A.; Gazzola S. Beilstein J. Org. Chem. 2012, 8, 1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recent examples include:; a Ferreira E. M.; Stoltz B. M. J. Am. Chem. Soc. 2003, 125, 9578. [DOI] [PubMed] [Google Scholar]; b Ferrer C.; Echavarren A. M. Angew. Chem., Int. Ed. 2006, 45, 1105. [DOI] [PubMed] [Google Scholar]; c Campeau L.-C.; Parisien M.; Jean A.; Fagnou K. J. Am. Chem. Soc. 2006, 128, 581. [DOI] [PubMed] [Google Scholar]; d Bajtos B.; Yu M.; Zhao H.; Pagenkopf B. L. J. Am. Chem. Soc. 2007, 129, 9631. [DOI] [PubMed] [Google Scholar]; e Zhao J.; Zhang Y.; Cheng K. J. Org. Chem. 2008, 73, 7428. [DOI] [PubMed] [Google Scholar]; f Lebrasseur N.; Larrosa I. J. Am. Chem. Soc. 2008, 130, 2926. [DOI] [PubMed] [Google Scholar]; g Yang S.-D.; Sun C.-L.; Fang Z.; Li B.-J.; Li Y.-Z.; Shi Z.-Y. Angew. Chem., Int. Ed. 2008, 47, 1473. [DOI] [PubMed] [Google Scholar]; h Bandini M.; Eichholzer A. Angew. Chem., Int. Ed. 2009, 48, 9533. [DOI] [PubMed] [Google Scholar]; i Thornton A. R.; Martin V. I.; Blakey S. B. J. Am. Chem. Soc. 2009, 131, 2434. [DOI] [PubMed] [Google Scholar]; j Suarez L. L.; Greaney M. F. Chem. Commun. 2011, 47, 7992. [DOI] [PubMed] [Google Scholar]; k Zhu Y.; Rawal V. J. Am. Chem. Soc. 2012, 134, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Shi J.; Zhou B.; Yang Y.; Li Y. Org. Biomol. Chem. 2012, 10, 8953. [DOI] [PubMed] [Google Scholar]; m Ding Z.; Yoshikai N. Angew. Chem., Int. Ed. 2012, 51, 4698. [DOI] [PubMed] [Google Scholar]; n Jiao L.; Herdtweck E.; Bach T. J. Am. Chem. Soc. 2012, 134, 14563. [DOI] [PubMed] [Google Scholar]; o Gregory A. W.; Jakubec P.; Turner P.; Dixon D. J. Org. Lett. 2013, 15, 4330. [DOI] [PMC free article] [PubMed] [Google Scholar]; p Potukuchi H. K.; Bach T. J. Org. Chem. 2013, 78, 12263. [DOI] [PubMed] [Google Scholar]; q Islam S.; Larrosa I. Chem.—Eur. J. 2013, 19, 15093. [DOI] [PubMed] [Google Scholar]; r Williams T. J.; Reay A. J.; Whitwood A. C.; Fairlamb I. J. S. Chem. Commun. 2014, 50, 3052. [DOI] [PubMed] [Google Scholar]
- Recent examples include:; a Grimster N. P.; Gauntlett C.; Godfrey C. R. A.; Gaunt M. J. Angew. Chem., Int. Ed. 2005, 44, 3125. [DOI] [PubMed] [Google Scholar]; b Phipps R. J.; Grimster N. P.; Gaunt M. J. J. Am. Chem. Soc. 2008, 130, 8172. [DOI] [PubMed] [Google Scholar]; c Bellina F.; Benelli F.; Rossi R. J. Org. Chem. 2008, 73, 5529. [DOI] [PubMed] [Google Scholar]; d Wolf C.; Zhang P. Adv. Synth. Catal. 2011, 353, 760. [Google Scholar]; e McKeon S. C.; Mueller-Bunz H.; Guiry P. J. Eur. J. Org. Chem. 2011, 7107. [Google Scholar]; f Peng J.; Liu L.; Hu Z.; Huang J.; Zhu Q. Chem. Commun. 2012, 48, 3772. [DOI] [PubMed] [Google Scholar]; g Zhang Y.; Stephens D.; Hernandez G.; Mendoza R.; Larionov O. V. Chem.—Eur. J. 2012, 18, 16612. [DOI] [PubMed] [Google Scholar]; h Stahl T.; Müther K.; Ohki Y.; Tatsumi K.; Oestreich M. J. Am. Chem. Soc. 2013, 135, 10978. [DOI] [PubMed] [Google Scholar]; i Tsyshchuk I. E.; Vorobyeva D. V.; Peregudov A. S.; Osipov S. N. Eur. J. Org. Chem. 2014, 2480. [Google Scholar]; j Chu X.-Q.; Zi Y.; Lu X.-M.; Wang S.-Y.; Ji S.-J. Tetrahedron 2014, 70, 232. [Google Scholar]; k Johansson Seechurn C. C. C.; Sivakumar V.; Satoskar D.; Colacot T. J. Organometallics 2014, 33, 3514. [Google Scholar]; l Zhou A.-X.; Mao L.-L.; Wang G.-W.; Yang S.-D. Chem. Commun. 2014, 50, 8529. [DOI] [PubMed] [Google Scholar]
- a Paris D.; Cottin M.; Demonchaux P.; Augert G.; Dupassieux P.; Lenoir P.; Peck M. J.; Jasserand D. J. Med. Chem. 1995, 38, 669. [DOI] [PubMed] [Google Scholar]; b Somei M.; Iwaki T.; Yamada F.; Tanaka Y.; Shigenobu K.; Koike K.; Suzuki N.; Hattori A. Heterocycles 2006, 68, 1565. [Google Scholar]; c Gotoh H.; Duncan K. K.; Robertson W. M.; Boger D. L. ACS Med. Chem. Lett. 2011, 2, 948. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Sun D.; Zhao Q.; Li C. Org. Lett. 2011, 13, 5302. [DOI] [PubMed] [Google Scholar]; e Patureau F. W.; Besset T.; Glorius F. Angew. Chem., Int. Ed. 2011, 50, 1064. [DOI] [PubMed] [Google Scholar]; f Liebhold M.; Xie X.; Li S.-M. Org. Lett. 2012, 14, 4882. [DOI] [PubMed] [Google Scholar]; g Wood I.; Martini M. F.; Pickholz M. J. Mol. Struct. 2013, 1045, 124. [Google Scholar]; h Xu Q.-L.; Dai L.-X.; You S.-L. Chem. Sci. 2013, 4, 97. [Google Scholar]; i Liebhold M.; Li S.-M. Org. Lett. 2013, 15, 5834. [DOI] [PubMed] [Google Scholar]; j Fan A.; Li S.-M. Adv. Synth. Catal. 2013, 355, 2659. [Google Scholar]; k Rudolf J. D.; Wang H.; Poulter C. D. J. Am. Chem. Soc. 2013, 135, 1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Davies H. M. L.; Manning J. R. J. Am. Chem. Soc. 2006, 128, 1060. [DOI] [PubMed] [Google Scholar]; b Liu Q.; Li Q.; Ma Y.; Jia Y. Org. Lett. 2013, 15, 4528. [DOI] [PubMed] [Google Scholar]; c Lanke V.; Prabhu K. R. Org. Lett. 2013, 15, 6262. [DOI] [PubMed] [Google Scholar]; d Liu H.; Zheng C.; You S.-L. J. Org. Chem. 2014, 79, 1047. [DOI] [PubMed] [Google Scholar]; e Gritsch P. J.; Leitner C.; Pfaffenbach M.; Gaich T. Angew. Chem., Int. Ed. 2014, 53, 1208. [DOI] [PubMed] [Google Scholar]; f Shi Z.; Boultadakis-Arapinis M.; Koester D. C.; Glorius F. Chem. Commun. 2014, 50, 2650. [DOI] [PubMed] [Google Scholar]; g Yang G.; Lindovska P.; Zhu D.; Kim J.; Wang P.; Tang R.-Y.; Movassaghi M.; Yu J.-Q. J. Am. Chem. Soc. 2014, 136, 10807. [DOI] [PubMed] [Google Scholar]
- a Zhang M.; Wang W.-L.; Fang Y.-C.; Zhu T.-J.; Gu Q.-Q.; Zhu W.-M. J. Nat. Prod. 2008, 71, 985. [DOI] [PubMed] [Google Scholar]; b Roach S. L.; Hudson A. R.; Valdez L. J.; Higuchi R. I.; Zhi L.; Vassar A. C.; Landry-Bayle A.; Adams M. E.; Rowley C. V.; Lamer R. B.; Grant V.; Heather S. PCT Int. Appl.WO 2009103007, 2009. (CA: 2009, 146, 408201).; c Kikuchi H.; Ohtsuki T.; Koyano T.; Kowithayakorn T.; Sakai T.; Ishibashi M. J. Nat. Prod. 2010, 73, 452. [DOI] [PubMed] [Google Scholar]; d Berlin M. Expert Opin. Ther. Pat. 2010, 20, 855. [DOI] [PubMed] [Google Scholar]; e Chen M.; Gan L.; Lin S.; Wang X.; Li L.; Li Y.; Zhu C.; Wang Y.; Jiang B.; Jiang J.; Yang Y.; Shi Y. J. Nat. Prod. 2012, 75, 1167. [DOI] [PubMed] [Google Scholar]
- Garfunkle J.; Kimball F. S.; Trzupek J. D.; Takizawa S.; Shimamura H.; Tomishima M.; Boger D. L. J. Am. Chem. Soc. 2009, 131, 16036.and references therein.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Takashima M.; Sakai H. Bull. Agr. Chem. Soc. Jpn. 1960, 24, 647. [Google Scholar]; b Fujiki H.; Suganuma M.; Matsukura N.; Sugimura T.; Takayama S. Carcinogenesis 1982, 3, 895. [DOI] [PubMed] [Google Scholar]; c Hitotsuyanagi Y.; Fujiki H.; Suganuma N.; Aimi S.; Sakai S.; Endo Y.; Shudo K.; Sugimura T. Chem. Pharm. Bull. 1984, 32, 4233. [DOI] [PubMed] [Google Scholar]; d Muratake H.; Natsume M. Tetrahedron Lett. 1987, 28, 2265. [Google Scholar]; e Yamashita T.; Imoto M.; Isshiki K.; Sawa T.; Naganawa H.; Kurasawa S.; Zhu B.-Q.; Umezawa K. J. Nat. Prod. 1988, 51, 1184. [Google Scholar]; f Muratake H.; Okabe K.; Natsume M. Tetrahedron 1991, 47, 8545. [Google Scholar]; g Fine Nathel N. F.; Shah T. K.; Bronner S. M.; Garg N. K. Chem. Sci. 2014, 5, 2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yeung K. S.; Qiu Z.; Xue Q.; Fang H.; Yang Z.; Zadjura L.; D’Arienzo C. J.; Eggers B. J.; Riccardi K.; Shi P. Y.; Gong Y. F.; Browning M. R.; Gao Q.; Hansel S.; Santone K.; Lin P. F.; Meanwell N. A.; Kadow J. F. Bioorg. Med. Chem. Lett. 2013, 23, 198. [DOI] [PubMed] [Google Scholar]; b Yeung K. S.; Qiu Z.; Yin Z.; Trehan A.; Fang H.; Pearce B.; Yang Z.; Zadjura L.; D’Arienzo C. J.; Riccardi K.; Shi P. Y.; Spicer T. P.; Gong Y. F.; Browning M. R.; Hansel S.; Santone K.; Barker J.; Coulter T.; Lin P. F.; Meanwell N. A.; Kadow J. F. Bioorg. Med. Chem. Lett. 2013, 23, 203. [DOI] [PubMed] [Google Scholar]
- Wu Y. S.; Coumar M. S.; Chang J. Y.; Sun H. Y.; Kuo F. M.; Kuo C. C.; Chen Y. J.; Chang C. Y.; Hsiao C. L.; Liou J. P.; Chen C.-P.; Yao H.-T.; Chiang Y.-K.; Tan U.-K.; Chen C.-T.; Chu C.-Y.; Wu S.-Y.; Yeh T.-K.; Lin C.-Y.; Hsieh H.-P. J. Med. Chem. 2009, 52, 4941. [DOI] [PubMed] [Google Scholar]
- a Allen M. C.; Brundish D. E.; Wade R. J. Chem. Soc., Perkin Trans. 1 1980, 1928. [Google Scholar]; b Groll M.; Kaiser M.; Milbradt A. G.; Moroder L.; Siciliano C.; Assfalg-Machleidt I. Org. Lett. 2003, 5, 3435. [DOI] [PubMed] [Google Scholar]; c Unversucht S.; Hollmann F.; Schmid A.; van Pée K.-H. Adv. Synth. Catal. 2005, 347, 1163. [Google Scholar]; d Baran P. S.; Foo K.; Newhouse T.; Mori I.; Takayama H. Angew. Chem., Int. Ed. 2011, 50, 2716. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Andorfer M. C.; Lewis J. C.; Payne J. T. Angew. Chem., Int. Ed. 2013, 52, 5271. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Smith D. R. M.; Willemse T.; Gkotsi D. S.; Schepens W.; Maes U. W.; Ballet S.; Goss R. J. M. Org. Lett. 2014, 16, 2622. [DOI] [PubMed] [Google Scholar]; g Frese M.; Guzowska P. H.; Voß H.; Sewald N. ChemCatChem 2014, 6, 1270. [Google Scholar]
- a Bartoli G.; Palmieri G. Tetrahedron Lett. 1989, 30, 2129. [Google Scholar]; b Arima S.; Harigaya Y.; Kai T.; Konda-Yamada Y.; Okada C.; Sato N.; Takayanagi H.; Umeda Y.; Yoshida K. Tetrahedron 2002, 58, 7851. [Google Scholar]; c Dalpozzo R.; Bartoli G. Curr. Org. Chem. 2005, 9, 163. [Google Scholar]; d Mentzel U. V.; Tanner D.; Tønder J. E. J. Org. Chem. 2006, 71, 5807. [DOI] [PubMed] [Google Scholar]; e Berthelot A.; Piguel S.; Le Dour G.; Vidal J. J. Org. Chem. 2003, 68, 9835. [DOI] [PubMed] [Google Scholar]; f Teng X.; Degterev A.; Jagtap P.; Xing X.; Choi S.; Denu R.; Yuan J.; Cuny G. D. Bioorg. Med. Chem. Lett. 2005, 15, 5039. [DOI] [PubMed] [Google Scholar]
- a Movassaghi M.; Schmidt M. A.; Ashenhurst J. A. Org. Lett. 2008, 10, 4009. [DOI] [PubMed] [Google Scholar]; b Movassaghi M.; Ahmad O. K.; Lathrop S. P. J. Am. Chem. Soc. 2011, 133, 13002. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kim J.; Movassaghi M. J. Am. Chem. Soc. 2011, 133, 14940. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Medley J. W.; Movassaghi M. Org. Lett. 2013, 15, 3614. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Coste A.; Kim J.; Adams T. C.; Movassaghi M. Chem. Sci. 2013, 4, 3191. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lathrop S. P.; Movassaghi M. Chem. Sci. 2014, 5, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Han S.; Morrison K. C.; Hergenrother P. J.; Movassaghi M. J. Org. Chem. 2014, 79, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For comprehensive reviews and mechanistic insights:; a Cho J. Y.; Tse M. K.; Holmes D.; Maleczka R. E. Jr.; Smith M. R. III. Science 2002, 295, 305. [DOI] [PubMed] [Google Scholar]; b Ishiyama T.; Takagi J.; Ishida K.; Miyaura N.; Anastasi N. R.; Hartwig J. F. J. Am. Chem. Soc. 2002, 124, 390. [DOI] [PubMed] [Google Scholar]; c Tamura H.; Yamazaki H.; Sato H.; Sakaki S. J. Am. Chem. Soc. 2003, 125, 16114. [DOI] [PubMed] [Google Scholar]; d Boller T. M.; Murphy J. M.; Hapke M.; Ishiyama T.; Miyaura N.; Hartwig J. F. J. Am. Chem. Soc. 2005, 127, 14263. [DOI] [PubMed] [Google Scholar]; e Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. Chem. Rev. 2010, 110, 890. [DOI] [PubMed] [Google Scholar]; f Hartwig J. F. Chem. Soc. Rev. 2011, 40, 1992. [DOI] [PubMed] [Google Scholar]; g Hartwig J. F. Acc. Chem. Res. 2012, 45, 864. [DOI] [PubMed] [Google Scholar]; h Preshlock S. M.; Ghaffari B.; Maligres P. E.; Krska S. W.; Maleczka R. E. Jr.; Smith M. R. III. J. Am. Chem. Soc. 2013, 135, 7572. [DOI] [PubMed] [Google Scholar]; i Larsen M. A.; Hartwig J. F. J. Am. Chem. Soc. 2014, 136, 4287. [DOI] [PubMed] [Google Scholar]; j Green A. G.; Liu P.; Merlic C. A.; Houk K. N. J. Am. Chem. Soc. 2014, 136, 4575. [DOI] [PubMed] [Google Scholar]
- For representative reports:; a Cho J. Y.; Iverson C. N.; Smith M. R. III. J. Am. Chem. Soc. 2000, 122, 12868. [Google Scholar]; b Chen H.; Schlecht S.; Semple T. C.; Hartwig J. F. Science 2000, 287, 1995. [DOI] [PubMed] [Google Scholar]; c Shimada S.; Batsanov A. S.; Howard J. A. K.; Marder T. B. Angew. Chem., Int. Ed. 2001, 40, 2168. [DOI] [PubMed] [Google Scholar]; d Ishiyama T.; Takagi J.; Hartwig J. F.; Miyaura N. Angew. Chem., Int. Ed. 2002, 41, 3056. [DOI] [PubMed] [Google Scholar]; e Chotana G. A.; Rak M. A.; Smith M. R. III. J. Am. Chem. Soc. 2005, 127, 10539. [DOI] [PubMed] [Google Scholar]; f Shi F.; Smith M. R. III; Maleczka R. E. Jr. Org. Lett. 2006, 8, 1411. [DOI] [PubMed] [Google Scholar]; g Mkhalid I. A. I.; Coventry D. N.; Albesa-Jove D.; Batsanov A. S.; Howard J. A. K.; Perutz R. N.; Marder T. B. Angew. Chem., Int. Ed. 2006, 45, 489. [DOI] [PubMed] [Google Scholar]; h Chotana G. A.; Kallepalli V. A.; Maleczka R. E. Jr.; Smith M. R. III. Tetrahedron 2008, 64, 6103. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Finke A. D.; Moore J. S. Org. Lett. 2008, 10, 4851. [DOI] [PubMed] [Google Scholar]; j Beck E. M.; Hatley R.; Gaunt M. J. Angew. Chem., Int. Ed. 2008, 47, 3004. [DOI] [PubMed] [Google Scholar]; k Harrison P.; Morris J.; Marder T. B.; Steel P. G. Org. Lett. 2009, 11, 3586. [DOI] [PubMed] [Google Scholar]; l Fischer D. F.; Sarpong R. J. Am. Chem. Soc. 2010, 132, 5926. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Tajuddin H.; Harrisson P.; Bitterlich B.; Collings J. C.; Sim N.; Batsanov A. S.; Cheung M. S.; Kawamorita S.; Maxwell A. C.; Shukla L.; Morris J.; Lin Z.; Marder T. B.; Steel P. G. Chem. Sci. 2012, 3, 3505. [Google Scholar]; n Robbins D. W.; Hartwig J. F. Angew. Chem., Int. Ed. 2013, 52, 933. [DOI] [PubMed] [Google Scholar]; o Liskey C. W.; Hartwig J. F. J. Am. Chem. Soc. 2013, 135, 3375. [DOI] [PubMed] [Google Scholar]; p Obligacion J. V.; Semproni S. P.; Chirik P. J. J. Am. Chem. Soc. 2014, 136, 4133. [DOI] [PubMed] [Google Scholar]
- a Takagi J.; Sato K.; Hartwig J. F.; Ishiyama T.; Miyaura N. Tetrahedron Lett. 2002, 43, 5649. [Google Scholar]; b Ishiyama T.; Takagi J.; Yonekawa Y.; Hartwig J. F.; Miyaura N. Adv. Synth. Catal. 2003, 345, 1103. [Google Scholar]; c Paul S.; Chotana G. A.; Holmes D.; Reichle R. C.; Maleczka R. E. Jr.; Smith M. R. III. J. Am. Chem. Soc. 2006, 128, 15552. [DOI] [PubMed] [Google Scholar]; d Kallepalli V. A.; Shi F.; Paul S.; Onyeozili E. N.; Maleczka R. E. Jr.; Smith M. R. III. J. Org. Chem. 2009, 74, 9199. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Meyer F. M.; Liras S.; Guzman-Perez A.; Perreault C.; Bian J.; James K. Org. Lett. 2010, 12, 3870. [DOI] [PubMed] [Google Scholar]; f Liskey C. W.; Hartwig J. F. Synthesis 2013, 45, 1837. [Google Scholar]; g Zhou S.; Jia Y. Org. Lett. 2014, 16, 3416. [DOI] [PubMed] [Google Scholar]; h Sadler S. A.; Tajuddin H.; Mkhalid I. A. I.; Batsanov A. S.; Albesa-Jove D.; Cheung M. S.; Maxwell A. C.; Shukla L.; Roberts B.; Blakemore D. C.; Lin Z.; Marder T. B.; Steel P. G. Org. Biomol. Chem. 2014, 12, 7318. [DOI] [PubMed] [Google Scholar]; i Homer J. A.; Sperry J. Tetrahedron Lett. 2014, 55, 5798. [Google Scholar]
- a Boebel T. A.; Hartwig J. F. J. Am. Chem. Soc. 2008, 130, 7534. [DOI] [PubMed] [Google Scholar]; b Boebel T. A.; Hartwig J. F. Organometallics 2008, 27, 6013. [Google Scholar]; c Robbins D. W.; Boebel T. A.; Hartwig J. F. J. Am. Chem. Soc. 2010, 132, 4068. [DOI] [PubMed] [Google Scholar]; d Cho S. H.; Hartwig J. F. J. Am. Chem. Soc. 2013, 135, 8157. [DOI] [PubMed] [Google Scholar]; e Cho S. H.; Hartwig J. F. Chem. Sci. 2014, 5, 694. [Google Scholar]
- For other examples of ortho-directed boronations:; a Kawamorita S.; Ohmiya H.; Hara K.; Fukuoka A.; Sawamura M. J. Am. Chem. Soc. 2009, 131, 5058. [DOI] [PubMed] [Google Scholar]; b Yamazaki K.; Kawamorita S.; Ohmiya H.; Sawamura M. Org. Lett. 2010, 12, 3978. [DOI] [PubMed] [Google Scholar]; c Ishiyama T.; Isou H.; Kikuchi T.; Miyaura N. Chem. Commun. 2010, 46, 159. [DOI] [PubMed] [Google Scholar]; d Kawamorita S.; Ohmiya H.; Sawamura M. J. Org. Chem. 2010, 75, 3855. [DOI] [PubMed] [Google Scholar]; e Ros A.; Estepa B.; López-Rodríguez R.; Álvarez E.; Fernández R.; Lassaletta J. M. Angew. Chem., Int. Ed. 2011, 50, 11724. [DOI] [PubMed] [Google Scholar]; f Roosen P. C.; Kallepalli V. A.; Chattopadhyay B.; Singleton D. A.; Maleczka R. E. Jr.; Smith M. R. III. J. Am. Chem. Soc. 2012, 134, 11350. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Sasaki I.; Doi H.; Hashimoto T.; Kikuchi T.; Ito H.; Ishiyama T. Chem. Commun. 2013, 49, 7546. [DOI] [PubMed] [Google Scholar]; h Ros A.; Fernández R.; Lassaletta J. M. Chem. Soc. Rev. 2014, 43, 3229. [DOI] [PubMed] [Google Scholar]; i Sasaki I.; Amou T.; Ito H.; Ishiyama T. Org. Biomol. Chem. 2014, 12, 2041. [DOI] [PubMed] [Google Scholar]; j Konishi S.; Kawamorita S.; Iwai T.; Steel P. G.; Marder T. B.; Sawamura M. Chem.—Asian J. 2014, 9, 434. [DOI] [PubMed] [Google Scholar]
- For general discussions, see:; a Miyaura N. Top. Curr. Chem. 2002, 219, 11. [Google Scholar]; b Molander G. A.; Canturk B.; Kennedy L. E. J. Org. Chem. 2009, 74, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kinzel T.; Zhang Y.; Buchwald S. L. J. Am. Chem. Soc. 2010, 132, 14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C2 indole protodeboronation under Pd catalysis conditions:; a Ishikura M.; Terashima M. Heterocycles 1988, 27, 203. [Google Scholar]; b Sutherland A.; Gallagher T. J. Org. Chem. 2003, 68, 3352. [DOI] [PubMed] [Google Scholar]; c Duncton M. A. J.; Estiarte M. A.; Tan D.; Kaub C.; O’Mahony D. J. R.; Johnson R. J.; Cox M.; Edwards W. T.; Wan M.; Kincaid J.; Kelly M. G. Org. Lett. 2008, 10, 3259. [DOI] [PubMed] [Google Scholar]; d Kassis P.; Bénéteau V.; Mérour J.-Y.; Routier S. Synthesis 2009, 2447. [Google Scholar]; e Beaumard F.; Dauban P.; Dodd R. H. Synthesis 2010, 4033. [Google Scholar]; f Thakur A.; Zhang K.; Louie J. Chem. Commun. 2012, 48, 203. [DOI] [PubMed] [Google Scholar]; g Nguyen T. T. B.; Lomberget T.; Tran N. C.; Barret R. Tetrahedron 2013, 69, 2336. [Google Scholar]; h Edwankar C. R.; Edwankar R. V.; Namjoshi O. A.; Liao X.; Cook J. M. J. Org. Chem. 2013, 78, 6471. [DOI] [PMC free article] [PubMed] [Google Scholar]; Other C2 indole protodeboronations:; i Vazquez E.; Davies I. W.; Payack J. F. J. Org. Chem. 2002, 67, 7551. [DOI] [PubMed] [Google Scholar]; j Cai X.; Snieckus V. Org. Lett. 2004, 6, 2293. [DOI] [PubMed] [Google Scholar]; k Zheng S. L.; Reid S.; Lin N.; Wang B. Tetrahedron Lett. 2006, 47, 2331. [Google Scholar]; l Tseng N.-W.; Lautens M. J. Org. Chem. 2009, 74, 1809. [DOI] [PubMed] [Google Scholar]; m Berionni G.; Morozova V.; Heininger M.; Mayer P.; Knochel P.; Mayr H. J. Am. Chem. Soc. 2013, 135, 6317. [DOI] [PubMed] [Google Scholar]; n Zhang N.; Zhang X.; Zhu J.; Turpoff A.; Chen G.; Morrill C.; Huang S.; Lennox W.; Kakarla R.; Liu R.; Li C.; Ren H.; Almstead N.; Venkatraman S.; Njoroge F. G.; Gu Z.; Clausen V.; Graci J.; Jung S. P.; Zheng Y.; Colacino J. M.; Lahser F.; Sheedy J.; Mollin A.; Weetall M.; Nomeir A.; Karp G. M. J. Med. Chem. 2014, 57, 2121. [DOI] [PubMed] [Google Scholar]
- C2-furan protodeboronations:; a Gooßen L. J.; Ghosh K. Chem. Commun. 2001, 2084. [PubMed] [Google Scholar]; b Alfonsi M.; Arcadi A.; Chiarini M.; Marinelli F. J. Org. Chem. 2007, 72, 9510. [DOI] [PubMed] [Google Scholar]; c Chartoire A.; Comoy C.; Fort Y. Tetrahedron 2008, 64, 10867. [Google Scholar]; d De M. Muñoz J.; Alcázar J.; De La Hoz A.; Díaz-Ortiz A. Adv. Synth. Catal. 2012, 354, 3456. [Google Scholar]; e Audi H.; Rémond E.; Eymin M.-J.; Tessier A.; Malacea-Kabbara R.; Jugé S. Eur. J. Org. Chem. 2013, 35, 7960. [Google Scholar]; f Zhao Y.; Snieckus V. Adv. Synth. Catal. 2014, 356, 1527. [Google Scholar]
- C2-thiophene protodeboronations:; a Yang C.-G.; Liu G.; Jiang B. J. Org. Chem. 2002, 67, 9392. [DOI] [PubMed] [Google Scholar]; b Klingensmith L. M.; Bio M. M.; Moniz G. A. Tetrahedron Lett. 2007, 48, 8242. [Google Scholar]; c Billingsley K.; Buchwald S. L. J. Am. Chem. Soc. 2007, 129, 3358. [DOI] [PubMed] [Google Scholar]; d Fleckenstein C. A.; Plenio H. J. Org. Chem. 2008, 73, 3236. [DOI] [PubMed] [Google Scholar]; e Steel P. G.; Woods T. M. Synthesis 2009, 3897. [Google Scholar]; f Gwynne E. A.; Holt J. C.; Dwan J. R.; Appoh F. E.; Vogels C. M.; Decken A.; Westcott S. A. Helv. Chim. Acta 2010, 93, 1093. [Google Scholar]; g Del Grosso A.; Singleton P. J.; Muryn C. A.; Ingleson M. A. Angew. Chem., Int. Ed. 2011, 50, 2102. [DOI] [PubMed] [Google Scholar]; h Migliorini A.; Oliviero C.; Gasperi T.; Loreto M. A. Molecules 2012, 17, 4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kuivila H. G.; Nahabedian K. V. J. Am. Chem. Soc. 1961, 83, 2159. [Google Scholar]; b Kuivila H. G.; Nahabedian K. V. J. Am. Chem. Soc. 1961, 83, 2164. [Google Scholar]; c Nahabedian K. V.; Kuivila H. G. J. Am. Chem. Soc. 1961, 83, 2167. [Google Scholar]; d Kuivila H. G.; Reuwer J. F.; Mangravite J. A. J. Am. Chem. Soc. 1964, 86, 2666. [Google Scholar]; e Brown R. D.; Buchanan A. S.; Humffray A. A. Aust. J. Chem. 1965, 18, 1521. [Google Scholar]; f Roques B. P.; Florentin D. M.; Callanquin M. J. Heterocycl. Chem. 1975, 12, 195. [Google Scholar]; g Florentin D.; Fournié-Zaluski M. C.; Callanquin M.; Roques B. P. J. Heterocycl. Chem. 1976, 13, 1265. [Google Scholar]
- Initial trials revealed that diboronation was actually occurring first at C2, then on the phthalimide ring itself. Longer exposure to the boronation conditions eventually led to the triboronated product that included C7 indole boronation. Attempts with a perchlorophthalimido protecting group were also unsuccessful, leading primarily to reduction products.
- a Cordell G. A.; Saxton J. E. In The Alkaloids: Chemistry and Physiology; Manske R. H. F., Rodrigo R. G. A., Eds.; Academic Press: New York, 1981; Vol. 20, pp 3–294. [Google Scholar]; b Hino T.; Nakagawa M. In The Alkaloids: Chemistry and Pharmacology; Brossi A., Ed.; Academic Press: New York, 1989; Vol. 34, pp 1–75. [Google Scholar]; c Crich D.; Banerjee A. Acc. Chem. Res. 2007, 40, 151. [DOI] [PubMed] [Google Scholar]; d Steven A.; Overman L. E. Angew. Chem., Int. Ed. 2007, 46, 5488. [DOI] [PubMed] [Google Scholar]
- Acid-promoted C2 indole protodestannylations:; a Labadie S. S.; Teng E. J. Org. Chem. 1994, 59, 4250. [Google Scholar]; b Tius M. A.; Kawakami J. K. Tetrahedron 1995, 51, 3997. [Google Scholar]; c Basarić N.; Baruah M.; Qin W.; Metten B.; Smet M.; Dehaen W.; Boens N. Org. Biomol. Chem. 2005, 3, 2755. [DOI] [PubMed] [Google Scholar]
- Acid-promoted C2 indole protodesilylations involving trifluoroacetic acid or acetic acid:; a Masters N. F.; Mathews N.; Nechvatal G.; Widdowson D. A. Tetrahedron 1989, 45, 5955. [Google Scholar]; b Zhang X.; Li X.; Lanter J. C.; Sui Z. Org. Lett. 2005, 7, 2043. [DOI] [PubMed] [Google Scholar]; c Sutou N.; Kato K.; Akita H. Tetrahedron Asymmetry 2008, 19, 1833. [Google Scholar]; d Chung J. Y. L.; Steinhuebel D.; Krska S. W.; Hartner F. W.; Cai C.; Rosen J.; Mancheno D. E.; Pei T.; Dimichele L.; Ball R. G.; Chen C.-Y.; Tan L.; Alorati A. D.; Brewer S. E.; Scott J. P. Org. Process Res. Dev. 2012, 16, 1832. [Google Scholar]; e Gavara L.; Suchaud V.; Nauton L.; Théry V.; Anizon F.; Moreau P. Bioorg. Med. Chem. Lett. 2013, 23, 2298. [DOI] [PubMed] [Google Scholar]; f Shan D.; Gao Y.; Jia Y. Angew. Chem., Int. Ed. 2013, 52, 4902. [DOI] [PubMed] [Google Scholar]; g Breazzano S. P.; Poudel Y. B.; Boger D. L. J. Am. Chem. Soc. 2013, 135, 1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton O. S. M.Sc. Thesis. Massachusetts Institute of Technology, Cambridge, MA, 2012. [Google Scholar]
- See the detailed Experimental Section for the precise reaction conditions for the direct conversion of indoles 11a–e to 7-boroindoles 13a–e, respectively.
- 3-Cyanoindole failed to undergo boronation under the conditions used in Table 1.
- The 2,7-diborotryptophan 12f contained trace impurities and was not subjected to further chromatographic purification due to sensitivity toward C2 protodeboronation.
- a Murphy J. M.; Liao X.; Hartwig J. F. J. Am. Chem. Soc. 2007, 129, 15434. [DOI] [PubMed] [Google Scholar]; b Hartwig J. F.; Partridge B. M. Org. Lett. 2013, 15, 140. [DOI] [PubMed] [Google Scholar]
- a Kikuchi T.; Nobuta Y.; Yamamoto Y.; Ishiyama T.; Miyaura N. Tetrahedron 2008, 64, 4967. [Google Scholar]; b Robbins D. W.; Hartwig J. F. Org. Lett. 2012, 14, 4266. [DOI] [PubMed] [Google Scholar]
- Caruso A.; Voisin-Chiret A. S.; Lancelot J.-C.; Sinicropi M. S.; Garofalo A.; Rault S. Heterocycles 2007, 71, 2203. [Google Scholar]
- a Minutolo F.; Antonello M.; Bertini S.; Rapposelli S.; Rossello A.; Sheng S.; Carlson K. E.; Katzenellenbogen J. A.; Macchia M. Bioorg. Med. Chem. 2003, 11, 1247. [DOI] [PubMed] [Google Scholar]; b Yang C.; Edsall R. Jr.; Harris H. A.; Zhang X.; Manas E. S.; Mewshaw R. E. Bioorg. Med. Chem. 2004, 12, 2553. [DOI] [PubMed] [Google Scholar]; c Kianmehr E.; Yahyaee M.; Tabatabai K. Tetrahedron Lett. 2007, 48, 2713. [Google Scholar]; d Tomita D.; Yamatsugu K.; Kanai M.; Shibasaki M. J. Am. Chem. Soc. 2009, 131, 6946. [DOI] [PubMed] [Google Scholar]; e Liger F.; Pellet-Rostaing S.; Popowycz F.; Lemaire M. Tetrahedron Lett. 2011, 52, 3736. [Google Scholar]; f Zhu C.; Wang R.; Falck J. R. Org. Lett. 2012, 14, 3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Still W. C.; Kahn M.; Mitra A. J. Org. Chem. 1978, 43, 2923. [Google Scholar]
- Pangborn A. B.; Giardello M. A.; Grubbs R. H.; Rosen R. K.; Timmers F. J. Organometallics 1996, 15, 1518. [Google Scholar]
- Aggarwal V. A.; Ball L. T.; Carobene S.; Connelly R. L.; Hesse M. J.; Partridge B. M.; Roth P.; Thomas S. P.; Webster M. P. Chem. Commun. 2012, 48, 9230. [DOI] [PubMed] [Google Scholar]
- Ishikawa H.; Takayama H.; Aimi N. Tetrahedron Lett. 2002, 43, 5637. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.