

Abstract
In Parkinson disease (PD), profound putamen dopamine (DA) depletion reflects denervation and a shift from vesicular sequestration to oxidative deamination of cytoplasmic DA in residual terminals. PD also involves cardiac sympathetic denervation. Whether PD entails myocardial norepinephrine (NE) depletion and a sequestration-deamination shift have been unknown. We measured apical myocardial tissue concentrations of NE, DA, and their neuronal metabolites 3,4-dihydroxyphenylglycol (DHPG), and 3,4-dihydroxyphenylacetic acid (DOPAC) from 23 PD patients and 23 controls and ascertained the extent of myocardial NE depletion in PD. We devised, validated in VMAT2-Lo mice, and applied 5 neurochemical indices of the sequestration-deamination shift—concentration ratios of DOPAC:DA, DA:NE, DHPG:NE, DOPAC:NE, and DHPG:DOPAC—and used a kinetic model to estimate the extent of the vesicular storage defect. The PD group had decreased myocardial NE content (p<0.0001). The majority of patients (70%) had severe NE depletion (mean 2% of control), and in this subgroup all 5 indices of a sequestration-deamination shift were increased compared to controls (p<0.001 for each). Vesicular storage in residual nerves was estimated to be decreased by 84-91% in this subgroup. We conclude that most PD patients have severe myocardial NE depletion, due to both sympathetic denervation and decreased vesicular storage in residual nerves.
Keywords: Parkinson's disease, Sympathetic nervous system, Neurochemistry, Catecholamines, Neurodegenerative mechanisms
The landmark discovery by Hornykiewicz and colleagues of striatal dopamine (DA) depletion in Parkinson disease (PD) (Ehringer & Hornykiewicz 1960) helped explain the movement disorder and rationalized development of levodopa therapy, the first successful treatment for a neurodegenerative disease (Cotzias 1971). Since then, many post-mortem neurochemical studies have confirmed DA depletion in PD, especially in the putamen (Kish et al. 1988).
In addition to loss of nigrostriatal dopaminergic innervation, decreased vesicular sequestration of cytosolic DA in residual terminals seems to be a major determinant of the depletion. In a recent study, vesicular storage was estimated to be decreased by 89% in putamen tissue samples from PD patients (Goldstein et al. 2013).
PD has been reported to involve not only loss of nigrostriatal dopaminergic innervation but also loss of cardiac sympathetic noradrenergic innervation, based on decreased post-mortem epicardial nerve content of immunoreactive tyrosine hydroxylase (Amino et al. 2005, Fujishiro et al. 2008). Numerous in vivo studies have fit with cardiac sympathetic denervation (Goldstein 2003); however, decreased vesicular uptake of cytosolic catecholamines in cardiac sympathetic nerves could contribute to the abnormal neuroimaging results. A neuroimaging/neurochemical in vivo study (Goldstein et al. 2011) provided indirect support for a vesicular sequestration-to-oxidative deamination shift in PD, but there was no neuropathologic proof that the diagnosis was correct. In the present post-mortem neurochemical study of tissues from patients with neuropathologically confirmed PD, we asked whether PD involves myocardial depletion of the sympathetic neurotransmitter, norepinephrine (NE), and if so whether decreased vesicular storage in residual nerves contributes to the depletion.
The approach we used was based on assaying simultaneously tissue contents of NE and DA and their respective neuronal metabolites, 3,4-dihydroxyphenylglycol (DHPG) and 3,4-dihydroxyphenylacetic acid (DOPAC). NE is synthesized within vesicles by the action of DA-beta-hydroxylase acting on DA that is taken up from the cytoplasm into vesicles via the type 2 vesicular monoamine transporter (VMAT2), whereas DHPG is synthesized in the cytoplasm by the action of monoamine oxidase-A (MAO-A) on NE that leaks from vesicles into the cytoplasm (Figure 1).
Figure 1. Concept diagrams about sources and metabolic fates of catecholamines in sympathetic nerves.
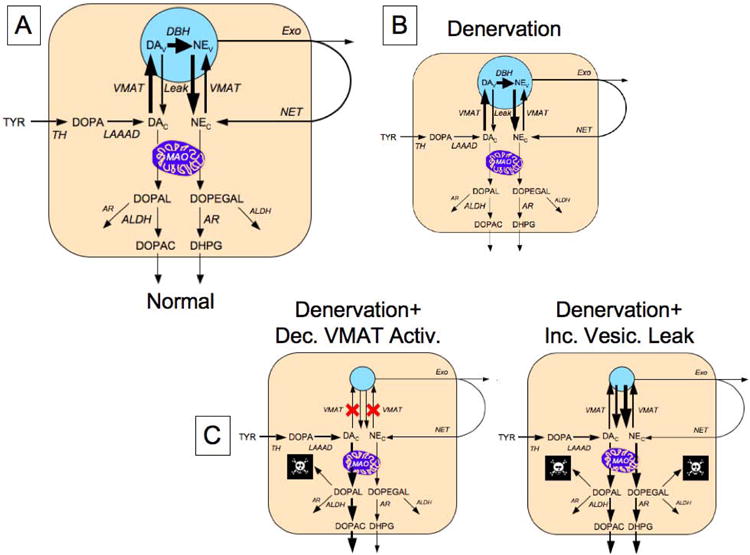
Under resting conditions, loss of norepinephrine (NE) from the neurons is due mainly to passive leakage from the vesicles (NEv) into the cytosol (NEc), followed by enzymatic deamination catalyzed by monoamine oxidase (MAO). Cytosolic NE is taken up into the vesicles via the type 2 vesicular monoamine transporter (VMAT). Release by exocytosis from the vesicles, with escape of reuptake via the cell membrane NE transporter (NET), is a minor determinant of NE turnover. NE loss is balanced by catecholamine biosynthesis from the action of cytosolic L-aromatic-amino-acid decarboxylase (LAAAD) on 3,4-dihydroxyphenylalanine (DOPA) produced from tyrosine (TYR) by tyrosine hydroxylase (TH) and of dopamine-beta-hydroxylase (DBH), which is localized in the vesicles. The action of MAO on cytosolic DA produces 3,4-dihydroxyphenylacetaldehyde (DOPAL) and on NE produces 3,4-dihydroxyphenylglycolaldehyde (DOPEGAL). DOPEGAL is mainly reduced by aldehyde/aldose reductase (AR), to form 3,4-dihydroxyphenylglycol (DHPG), and DOPAL is mainly oxidized by aldehyde dehydrogenase (ALDH) to form 3,4-dihydroxyphenylacetic acid (DOPAC). Red X marks indicate decreased vesicular uptake. When vesicular uptake is attenuated, as in VMAT2-Lo mice, myocardial NE depletion reflects decreased NE synthesis, because less DA is taken up into the vesicles and more is deaminated to form DOPAC. Furthermore, decreased reuptake of NE that leaks from vesicles into the cytoplasm, where the NE is deaminated and is converted to DHPG, accelerates the turnover of NE.
We devised 5 neurochemical indices of a vesicular sequestration-to-oxidative deamination shift, by the following reasoning. A decrease in vesicular sequestration would be expected to result in increased NE turnover, reflected by an elevated tissue concentration ratio of DHPG:NE (see Appendix). The DA:NE ratio would be increased, because of greater NE turnover than DA turnover. Since cytoplasmic DA is converted to DOPAC via MAO-A, a shift from vesicular sequestration to oxidative deamination would also be expected to increase the DOPAC:DA ratio. Both vesicular reuptake of NE and NE synthesis depend on vesicular sequestration of cytoplasmic catecholamines, and so attenuation of vesicular uptake would be expected to result in especially high DOPAC:NE ratios. Finally, if there were decreased vesicular uptake, the differential decrease in intra-vesicular NE synthesis with respect to cytoplasmic DA synthesis would be expected to increase the DOPAC:DHPG ratio. Thus, we used a total of 5 neurochemical indices of a sequestration-to-deamination shift—DOPAC:DA, DA:NE, DHPG:NE, DOPAC:NE, and DOPAC:DHPG.
As explained in the Appendix, by applying a kinetic model (Figure 2) to previously published data about rates of catecholamine synthesis, release, reuptake, and metabolism (Eisenhofer et al. 1996a, Goldstein et al. 1991) and to data about tissue contents of NE, DA, DHPG, and DOPAC from the present study, we estimated the relative contributions of denervation and decreased vesicular storage in residual nerves to myocardial NE depletion.
Figure 2. Detailed and condensed kinetic models for the fate of catecholamines in sympathetic nerves.
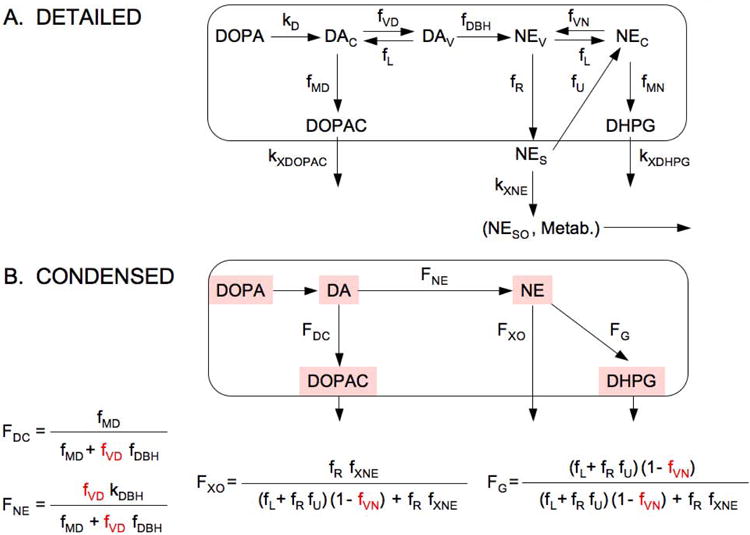
Abbreviations: FDC = fraction of DA that is metabolized to DOPAC; FNE = fraction of DA that is converted to NE; FG = fraction of vesicular NE that is lost by oxidative deamination; FXO = fraction of NE that is lost by release; kMD = rate constant for cytosolic DA being deaminated to form DOPAC; kVD = rate constant for uptake of cytosolic DA into the vesicles; kL = rate constant for leakage of vesicular DA or NE into the cytosol; kR = rate constant for NE release into the interstitial fluid; kU = rate constant for uptake of NE from the interstitial fluid into the cytosol via the cell membrane NE transporter; kXNE = rate constant for loss of NE from the interstitial fluid by metabolism or spillover (NESO) into the bloodstream; kXDOPAC = rate constant for DOPAC entry into the interstitial fluid; and kXDHPG = rate constant for DHPG entry into the interstitial fluid.
We validated the 5 indices of the sequestration-deamination shift by assaying myocardial tissue contents of catechols from mice with genetically determined very low VMAT2 activity (Taylor et al. 2014). Such mice have depletion of myocardial NE and DA (Taylor et al. 2014); however, whether they also have increased myocardial DOPAC:DA, DA:NE, DHPG:NE, DOPAC:NE, and DOPAC:DHPG ratios has not been reported.
Methods
Sources of Samples
Human Myocardial Tissue
All the assayed samples were from human myocardial tissue of the left ventricular apex obtained at autopsy from 23 neuropathologically confirmed cases of end-stage, Lewy body positive PD and 23 control subjects. One patient had PD in the setting of Gaucher disease.
Most of the tissues (18 PD patients, 18 controls) were obtained from Banner Sun Health Research Institute (Sun City, AZ). The study also included 3 PD patients who during life had been evaluated under IRB-approved protocols of the Division of Intramural Research, NINDS (2 autopsied at the NIH Clinical Center and 1 off-site). Another PD patient had not been evaluated at the NIH during life but was autopsied at the NIH Clinical Center.
Of the 23 control subjects, 18 were autopsied under the auspices of Banner Sun and 5 at the NIH Clinical Center. Research use of tissues harvested at autopsy at the NIH Clinical Center was in accordance with guidance by the NCI/CCR/Laboratory of Pathology Tissue Resource Committee. One of the control subjects had Lewy bodies noted incidentally.
VMAT2-Lo Mice
Hearts from 8 mice with very low activity of the type 2 vesicular monoamine transporter (VMAT2-Lo) and from 8 control wild-type mice were used for this study. To create VMAT2-Lo mice, the VMAT2 locus (SLC18A2) was cloned from the 129/Sv genomic library and a 2.2 kb PvuII fragment from the third intron of the VMAT2 gene, and cloned into the blunt-ended NotI site of the construct (Caudle et al. 2007, Taylor et al. 2009). The targeting vector was introduced into 129/Ola CGR 8.8 embryonic stem cells and injected into blastocysts of C57BL/6 mice. The construct was designed to generate a full knockout of the gene, but due to a rare recombination event, a low level of transcription remained (Mooslehner et al. 2001). Chimeric males (genotype confirmed by Southern blot analysis) were bred with C57BL/6 females. The procedures were approved by the Institutional Animal Care and Use Committee at Emory University. Frozen myocardial tissue was sent from the Emory laboratory to the Clinical Neurocardiology Section at the NIH in Bethesda, MD, where the neurochemical assays were done.
Assays of Tissue Catechols
The same person (P.S.) conducted the assays in the laboratory of the Clinical Neurocardiology Section in intramural NINDS.
The assays were conducted by methods developed and published by our group (Holmes et al. 1994, Holmes et al. 2010). Briefly, frozen tissue samples were homogenized in a mixture of 20:80 of 0.2 M phosphoric:0.2 M acetic acid and the supernate transferred to plastic cryotubes and stored at -80 °C until assayed by batch alumina extraction followed by liquid chromatography with series electrochemical detection. Concentrations of catechols in cell lysates were expressed in units of pmoles per mg wet weight.
Data Analysis and Statistics
Neurochemical data were graphed and analyzed using KaleidaGraph 4.01 (Synergy Software, Reading, PA). Differences between controls and patients groups were assessed by two-tailed, independent-means t-tests conducted on log-transformed data. Since log-transformed data were used, and some of the data were ratios, all data with zero values were excluded from statistical testing. Mean values were expressed ± SEM. A p value of less than 0.05 defined statistical significance.
Results
Cardiac Tissue Catechols in PD
The PD and control groups had similar ages and gender makeup (Table 1). The PD group had highly significantly decreased myocardial tissue NE, DHPG, and DA contents compared to the controls (p<0.0001 each; Table 2). The groups did not differ in mean DOPAC or DOPA content.
Table 1. Clinical Characteristics.
| Control | Parkinson Disease | |
|---|---|---|
| Age (years) | 70 ± 4 | 78 ± 1 |
| Gender (M/F) | 14/9 | 14/9 |
| Post-mortem Interval (hours) | 5 ± 1 | 6 ± 1 |
Table 2. Mean (± SEM) Left Ventricular Apical Tissue Concentrations (pmol per mg wet weight) of Catechols in Wild-Type and VMAT2-Lo Mice and in Control Subjects and PD Patients.
Numbers in parentheses are the numbers of subjects.
| Catechol | Wild-Type | VMAT2-Lo | VMAT2-Lo/Wild-Type | p |
|---|---|---|---|---|
| DOPA | 0.188 ± 0.022 | 0.173 ± 0.025 | 92% | n.s. |
| DA | 0.088 ± 0.007 | 0.031 ± 0.003 | 35% | 1.0 × 10-6 |
| NE | 2.52 ± 0.096 | 0.099 ± 0.010 | 3.9% | 4.0 × 10-14 |
| DOPAC | 0.018 ± 0.003 | 0.035 ± 0.004 | 194% | 0.001 |
| DHPG | 0.088 ± 0.008 | 0.016 ± 0.001 | 17.5% | 1.4 × 10-10 |
| Catechol | Control (23) | PD | PD/Control | p |
| DOPA | 0.219 ± 0.028 | 1.66 ± 0.74 (23) | 757% | n.s. |
| DA | 0.073 ± 0.016 | 0.021 ± 0.008 (21) | 29% | 0.00005 |
| NE | 1.54 ± 0.25 | 0.77 ± 0.56 (23) | 50.2% | 3 × 10-6 |
| DOPAC | 0.036 ± 0.007 | 0.058 ± 0.024 (23) | 164% | n.s. |
| DHPG | 0.091 ± 0.019 | 0.020 ± 0.007 (21) | 21.6% | 0.00002 |
| Catechol | PD No Depl. (7) | PD NE Depl. | PD NE Depl./No Depl. | p |
| DOPA | 0.47 ± 0.23 | 2.17 ± 1.04 (16) | 458% | n.s. |
| DA | 0.038 ± 0.022 | 0.013 ± 0.006 (14) | 32.5% | n.s. |
| NE | 0.643 ± 0.242 | 0.030 ± 0.008 (16) | 4.7% | 1.7 × 10-7 |
| DOPAC | 0.027 ± 0.008 | 0.072 ± 0.035 (16) | 271% | n.s. |
| Catechol | PD No Depl. | PD NE Depl. | PD NE Depl./No Depl. | p |
| DHPG | 0.041 ± 0.020 (7) | 0.011 ± 0.005 (15) | 26.9% | 0.07 |
The majority of PD patients had markedly decreased tissue NE (Figure 3). Among a subgroup of 16 (70%) PD patients, tissue NE was decreased by 98% from the controls (Figure 4A). Tissue DA and DHPG were also decreased in this subgroup but to a lesser extent than was tissue NE.
Figure 3. Individual values for myocardial DHPG and DHPG:NE as a function of NE in patients with PD (red circles) and controls (gray circles).
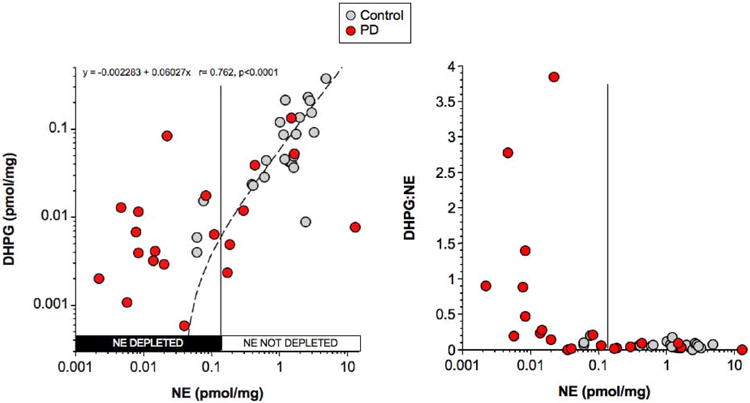
Vertical line separates PD subgroups with or without NE depletion. Dashed line shows line of best fit for the relationship between DHPG and NE in controls. Equation is for the line of best fit in controls.
Figure 4. Mean (± SEM) values for catechols in PD subgroups with NE depletion (PD NE Depl.) or no NE depletion (PD No Depl.) and in controls (Control).
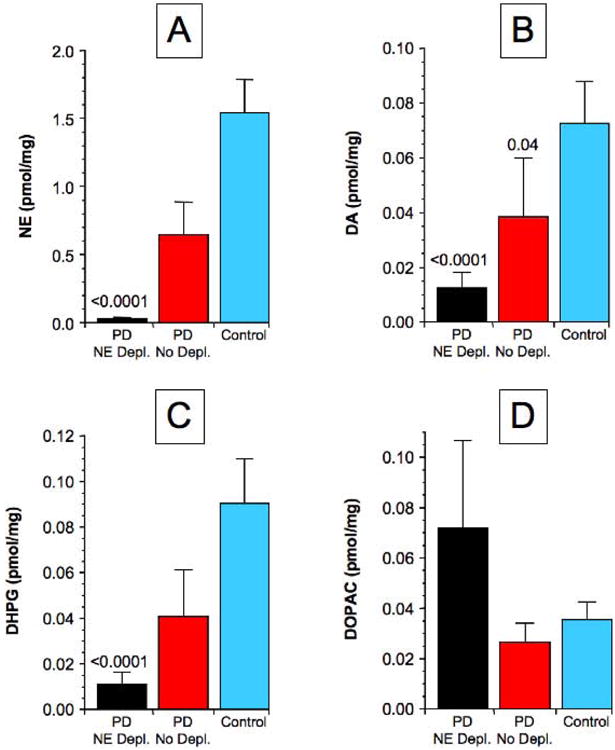
(A) NE, (B) DA, (C) DHPG, (D) DOPAC. P values indicate significant differences from Control.
Indices of a Vesicular Sequestration-to-Oxidative Deamination Shift
In the PD subgroup with NE depletion, tissue DHPG:NE averaged 11.2 times control (p<0.0001), DOPAC:DA 27.5 times control (p<0.0001), DA:NE 10.5 times control (p=0.0004), DOPAC:NE 163 times control (p<0.0001), and DOPAC:DHPG 49.7 times control (p=0.0002) (Figure 5).
Figure 5. Myocardial mean (± SEM) ratios of DHPG:NE, DOPAC:DA, DA:NE, DOPAC:NE in control subjects (CON, light colors) and patients with Parkinson disease (PD, dark colors) and in wild-type (WT) mice (light colors) and mice with very low activity of the type 2 vesicular monoamine transporter (VMAT2-Lo, dark colors).
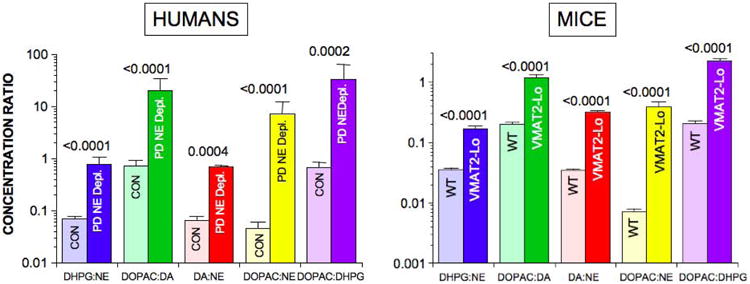
P values are for PD vs. CON and WT vs. VMAT2-Lo groups for each ratio.
Denervation and Decreased Vesicular Storage as Determinants of NE Depletion
We used two methods to estimate the contributions of denervation and decreased vesicular storage to myocardial NE depletion in PD. First, since the previously published mean extraction fraction of 3H-NE across the heart was 79.30% in controls (Eisenhofer et al. 1996b) and 12.24% in PD patients with severely decreased myocardial 18F-DA-derived radioactivity (Goldstein et al. 2000), the extent of innervation in the PD group was 12.24%/79.3% = 15.4% of control, corresponding to an 84.6% decrease. Given a decrease in innervation to 15.4% of control and a decrease in NE content to 1.98% of control in the PD subgroup with NE depletion, vesicular sequestration was 1.98%/15.4% = 12.9% of control, corresponding to an 87.1% decrease.
A second method was based on the NE turnover rate in the NE-depleted subgroup being several times that in controls. Using mean DHPG: mean NE as the measure of the NE turnover rate (see Appendix), kT in the NE Depl. subgroup was 6.14 times that in the controls, meaning that vesicular storage in the No Depl. subgroup was 1/6.14 = 16.3% of control (83.7% decrease). Given a decrease in vesicular sequestration to 16.3% of control and a decrease in NE content to 1.98% of control in the NE-depleted subgroup, innervation in the subgroup was then 1.98%/16.3% = 12.3% (87.7% decrease in innervation). Using mean DHPG:NE ratios, kT in the NE-depleted subgroup was 11.23 times control, which would correspond to a 91.1% decrease in vesicular sequestration and 82% decrease in innervation. Vesicular sequestration was therefore estimated to be decreased by 84-91% and innervation decreased by 82-88% among the PD patients with myocardial NE depletion.
Relationships of Catechol Ratios to Post-Mortem Intervals and Tissue DOPA
Post-mortem intervals averaged 6 hours in PD patients and 5 hours in controls and were less than 24 hours in all subjects.
To address the issue of possible confounding effects of post-mortem autolysis on the obtained results, we examined relationships of catechol levels and ratios with postmortem intervals. In the control and PD groups, individual values for myocardial tissue contents of DHPG, NE, DA, and DOPAC were unrelated to post-mortem intervals (r=0.12, -0.09; r=0.19, -0.03; r=-0.23, -0.03; r=-0.38, 0.19). Individual values for all 5 indices of the vesicular sequestration-to-oxidative deamination shift were also unrelated to post-mortem intervals (Figure 6, Table 3). There were 12 PD patients with a postmortem interval ≤ 6 hours who had severe NE depletion (mean 2% of control). In this subgroup, DOPAC:DA averaged 8.2 times that in control subjects with post-mortem intervals ≤ 6 hours (p=0.0005), DA:NE 12.4 times control (p=0.0001), DHPG:NE 13.2 times control (p<0.0001), DOPAC:NE 56 times control (p<0.0001), and DOPAC:DHPG 4.7 times control (p<0.0001).
Figure 6. Individual values for myocardial NE content and DHPG:NE ratios expressed as functions of post-mortem intervals (PMIs, A, B) and DOPA content (C, D) in patients with PD (red circles) and controls (gray circles).
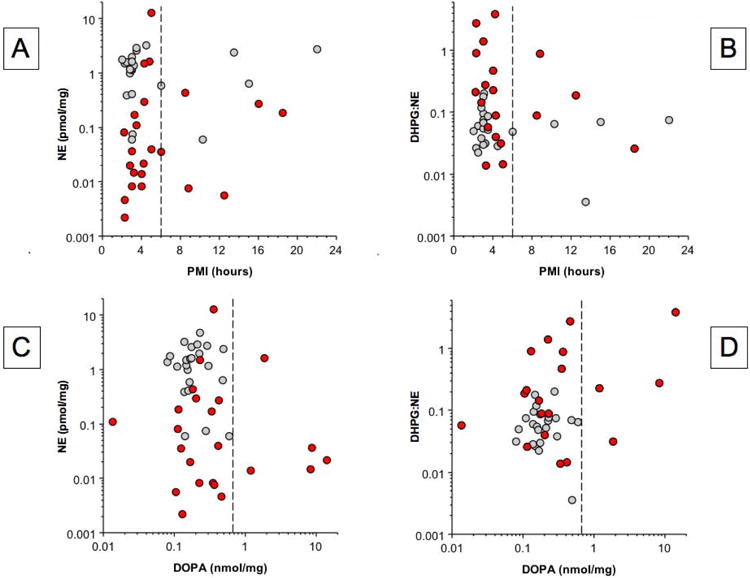
Vertical dashed lines in A and B indicate PMI of 6 hours and in C and D indicate the upper range of control values.
Table 3. Correlation Coefficients for Individual Values of Catechol Ratios as a Function of Post-Mortem Intervals and Tissue DOPA Levels in Control Subjects (CON) and Parkinson Disease Patients (PD).
| Ratio | Correlation with PMI (CON) | Correlation with PMI (PD) |
|---|---|---|
| DOPAC:DA | -0.26 | 0.32 |
| DA:NE | -0.09 | -0.15 |
| DHPG:NE | -0.10 | -0.22 |
| DOPAC:NE | -0.19 | 0.29 |
| DOPAC:DHPG | -0.21 | 0.42 |
| Ratio | Correlation with DOPA (CON) | Correlation with DOPA (PD) |
| DOPAC:DA | -0.22 | -0.11 |
| DA:NE | 0.14 | 0.73* |
| DHPG:NE | -0.05 | 0.63* |
| DOPAC:NE | -0.18 | 0.08 |
| DOPAC:DHPG | -0.25 | -0.07 |
p=0.0002
We used data about myocardial DOPA to take into account possible artifactual effects of levodopa treatment before death on tissue contents of catechols and catechol ratios. In the PD group, individual values for DOPAC:DA, DOPAC:NE, and DOPAC:DHPG ratios were unrelated to DOPA content; however, DA:NE and DHPG:NE ratios were positively correlated with DOPA content (Table 3). From inspection of Figure 4C, there were 5 PD patients with myocardial DOPA above the control range. When all data from these 5 patients were excluded, then there were 12 PD patients with NE depletion (mean 2% of control). In this subgroup, DOPAC:DA averaged 37 times control (p=0.0002), DA:NE 2.8 times control (p=0.02), DHPG:NE 8.7 times control (p=0.0003), DOPAC:NE 172 times control (p<0.0001), and DOPAC:DHPG 60 times control (p=0.003).
Thus, after taking into account post-mortem intervals and myocardial DOPA content, all 5 indices of a sequestration-deamination shift were found to remain elevated in PD patients with myocardial NE depletion.
Validation of Neurochemical Indices of a Sequestration-to-Deamination Shift in VMAT2-Lo Mice
In VMAT2-Lo mice, myocardial tissue concentrations of NE were decreased by 96.1% compared to wild-type mice (p=5.0 ×10-13; Table 2). Myocardial DHPG in VMAT2-Lo mice averaged 17.5% that in wild-type mice. The decrease in DHPG content was consistently less than the decrease in NE content, so that ratios of DHPG:NE concentrations in VMAT2-Lo mice averaged 4.8 times those in wild-type mice (p=1.9 × 10-8; Figure 5). DA was decreased by 64.7%, and the mean ratio of DA:NE was 9.0 times that in wild-type mice (p=3.6 ×10-13). Myocardial tissue DOPAC averaged 1.9 times that in wild-type mice, with the mean DOPAC:DA ratio 5.9 times and the mean DOPAC:NE ratio 49.5 times the corresponding mean ratios in the wild-type mice. DOPAC:DHPG ratios were markedly increased by about 11-fold compared to wild-type mice (2.23 ± 0.20 vs. 0.205 ± 0.002, p=9.7 × 10-11).
The results in the VMAT2-Lo mice therefore validated high DHPG:NE, DOPAC:DA, DA:NE, DOPAC:NE, and DOPAC:DHPG ratios as indices of a vesicular sequestration-to-oxidative deamination shift in the fate of cytoplasmic catecholamines.
Discussion
This study shows that the majority (70%) of PD patients have drastic myocardial NE depletion (mean 2% of control). The magnitude of the depletion in this subgroup was similar to that reported previously for DA in the caudal putamen (Wilson et al. 1996), the site of the most severe catecholaminergic lesion in the brain in PD. The results reinforce the view that most PD patients have a profound cardiac sympathetic lesion (Amino et al. 2005).
One might presume that myocardial NE depletion in PD directly reflects catecholamine neuron loss; however, we obtained evidence for marked attenuation of vesicular storage in residual myocardial sympathetic nerves, after taking denervation into account. From applying a kinetic model to previously published data and the present results, we estimated an 84-91% decrease in vesicular storage in residual nerves of PD patients in the NE-depleted subgroup.
By all of 5 indices of a shift from vesicular sequestration to oxidative deamination of cytoplasmic catecholamines—tissue concentration ratios of DOPAC:DA, DA:NE, DHPG:NE, DOPAC:NE, and DOPAC:DHPG—PD patients with NE depletion had neurochemical evidence for a sequestration-to-deamination shift. The present results therefore agreed with and extended on those from our previous in vivo study based on myocardial 18F-DA-derived radioactivity and arterial plasma 6F-DOPAC levels (Goldstein et al. 2011). A potential limitation of the in vivo study was that there was no proof that the diagnostic assignment was correct. The present results based on neuropathologically confirmed PD therefore provide important support for the concept of a sequestration-deamination shift in PD.
There are also potential limitations of a post-mortem neurochemical study of PD patients, including autolytic changes after death and artifactual influences of levodopa treatment before death. In the present study, the control and PD groups had similar, quite short post-mortem intervals, and in both the control and PD groups individual values for catechols and DOPAC:DA, DA:NE, DHPG:NE, DOPAC:NE, and DOPAC:DHPG ratios were unrelated to post-mortem intervals. Moreover, after exclusion of data from all subjects with post-mortem intervals exceeding 6 hours, mean values for the 5 indices of a sequestration-to-deamination shift remained highly significantly increased in the PD patients with NE depletion. We were able to assess whether the indices of the sequestration-deamination shift were related to tissue DOPA content in the PD group, because we assayed myocardial DOPA simultaneously with other catechols. After exclusion of data from PD patients with myocardial DOPA above the control range, mean values for all 5 indices of a sequestration-to-deamination shift remained increased in the PD patients with NE depletion.
There are two means by which vesicular storage might be decreased in PD–decreased vesicular uptake, such as from interference with VMAT2 or from decreased vesicle populations (decreased fVN and fVD in the kinetic model in Figure 2), and increased vesicular leakage (increased fL in the kinetic model). It is impossible to distinguish between the two without a recognizable change in the amine taken up into the vesicles. Efficient conversion of DA to NE in vesicles provides such a marker (Kopin & Weise 1968). In the present study, NE formation, reflected by tissue DHPG content (see Appendix) could be used to test whether decreased VMAT2 activity is a determinant of tissue catecholamine depletion in PD. Substantially increased DOPAC:DHPG ratios in the NE Depl. subgroup fit with impaired vesicular uptake. The vesicular storage lesion might involve attenuation of VMAT2 expression or function or decreased numbers of vesicles in residual nerve terminals; however, the present results do not exclude increased vesicle permeability as a contributing factor. Alpha-synucleinopathy can produce all these abnormalities (Lotharius & Brundin 2002, Mosharov et al. 2006, Volles & Lansbury 2002, Gaugler et al. 2012, Janezic et al. 2013).
In putamen tissue, the ratio of tissue DA:DOPA provides an index of vesicular sequestration of DA (Goldstein et al. 2013). By this measure PD entails about a 90% decrease in vesicular sequestration in the residual dopaminergic terminals. In the heart, however, DOPA content is unrelated to sympathetic innervation, since chemical sympathectomy with 6-hydroxydopamine DA does not alter myocardial tissue DOPA content (Eldrup et al. 1989), and so the DA:DOPA ratio in myocardial tissue may not validly indicate vesicular uptake.
In keeping with the well established medical concept that cellular dysfunction precedes cellular death, decreased vesicular storage might indicate a functional abnormality before the loss noradrenergic terminals. Thus, a recent study found evidence for aging-related myocardial sympathetic denervation in VMAT2-Lo mice (Taylor et al. 2014).
Braak's concept about pathogenetic stages in the development of PD includes early involvement of autonomic nerves (Braak et al. 2004, Del Tredici & Braak 2012), and patients with incidental Lewy body disease, thought to be a precursor of PD, have neurohistochemical evidence of cardiac sympathetic denervation (Orimo et al. 2008, Fujishiro et al. 2008, Del Tredici et al. 2010). The fact that about 30% of PD patients did not have severe NE depletion does not fit well with Braak's theory, since patients with symptomatic PD would all be expected to have myocardial NE depletion. In vivo studies have indicated a degree of independence between the nigrostriatal dopaminergic and cardiac noradrenergic lesions in PD. For instance, individual values for neuroimaging indices of striatal dopaminergic innervation are unrelated to indices of myocardial noradrenergic innervation (Raffel et al. 2006, Goldstein et al. 2008). Braak's concept may be enhanced by incorporating differential susceptibility to damaging effects of alpha-synucleinopathy, depending on the neurotransmitter phenotype.
Conclusions and Implications
About 70% of patients with autopsy-proven PD have profound myocardial NE depletion. The depletion is due to both sympathetic denervation and a shift from vesicular sequestration to oxidative deamination of cytoplasmic catecholamines in the residual nerves. The vesicular storage lesion seems to involve decreased vesicular uptake, such as by attenuation of VMAT2 expression or function or decreased numbers of vesicles.
The sequestration-deamination shift might be relevant to treatment, as follows. The immediate products of MAO-A acting on cytoplasmic catecholamines are aldehydes and hydrogen peroxide, both of which challenge the integrity of catecholaminergic neurons and cells (Li et al. 2001, Panneton et al. 2010, Su et al. 2013, Mattammal et al. 1995, Goldstein et al. 2012). Decreased vesicular storage would be expected to bias toward cytoplasmic buildup of the catecholaldehyde 3,4-dihydroxyphenylacetaldehyde (DOPAL), which is cytotoxic (Panneton et al. 2010), via generation of reactive oxygen species (Li et al. 2001), covalent binding to proteins (Rees et al. 2009), and oligomerization of alpha-synuclein (Burke et al. 2008), especially in the setting of divalent metal cations (Jinsmaa et al. 2014). DOPAL and alpha-synuclein may be nodes in a complex nexus of interacting homeostatic systems, in which decreased vesicular sequestration of cytoplasmic catecholamines, decreased aldehyde dehydrogenase activity (Goldstein et al. 2013), and oligomerization of alpha-synuclein lead to conversion from the stability afforded by negative feedback regulation to the instability, degeneration, and system failure caused by induction of positive feedback loops (Goldstein 2013). Since catecholaldehydes and hydrogen peroxide are the immediate products of metabolism of DA by MAO, the catecholaldehyde hypothesis predicts that treatment with an MAO inhibitor should slow neuronal loss by attenuating DOPAL production.
Supplementary Material
Acknowledgments
This work was funded by Division of Intramural Research, NINDS, NIH (grant number): This information is usually included already, but please add to the Acknowledgments if not.
Funding: We thank Dr. Thomas G. Beach and the Banner Sun Health Research Institute for providing most of the myocardial tissue samples used in this report.
Mr. Shawn Alter provided the mouse heart tissue used in this study.
The research reported here was supported by the intramural research program of the National Institute of Neurological Disorders and Stroke.
Dr. Miller is supported by grant P50 NS071669.
Abbreviations
- ALDH
aldehyde dehydrogenase
- COMT
catechol-O-methyltransferase
- DA
dopamine
- DHPG
3,4-dihydroxyphenylglycol
- DOPAC
3,4-dihydroxyphenylacetic acid
- DOPAL
3,4-dihydroxyphenylacetaldehyde
- DOPEGAL
3,4-dihydroxyphenylglycolaldehyde
- MAO
monoamine oxidase
- MSA
multiple system atrophy
- PAF
pure autonomic failure
- PD
Parkinson disease
- VMAT
vesicular monoamine transporter
Footnotes
Disclosures: The Authors have no conflicts of interest to disclose.
ARRIVE guidelines have been followed:
No
⇒ if No, skip complete sentence
⇒ if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.”
Conflicts of interest: none
⇒ if ‘none’, insert “The authors have no conflict of interest to declare.”
⇒ otherwise insert info unless it is already included
References
- Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Path. 2005;15:29–34. doi: 10.1111/j.1750-3639.2005.tb00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Kumar VB, Pandey N, et al. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008;115:193–203. doi: 10.1007/s00401-007-0303-9. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, Wang MZ, et al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27:8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotzias GC. Levodopa in the treatment of Parkinsonism. JAMA. 1971;218:1903–1908. [PubMed] [Google Scholar]
- Del Tredici K, Braak H. Lewy pathology and neurodegeneration in premotor Parkinson's disease. Movement Dis. 2012;27:597–607. doi: 10.1002/mds.24921. [DOI] [PubMed] [Google Scholar]
- Del Tredici K, Hawkes CH, Ghebremedhin E, Braak H. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson's disease. Acta Neuropathol. 2010;119:703–713. doi: 10.1007/s00401-010-0665-2. [DOI] [PubMed] [Google Scholar]
- Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Wien Klin Wochenschr. 1960;38:1236–1239. doi: 10.1007/BF01485901. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Friberg P, Rundqvist B, Quyyumi AA, Lambert G, Kaye DM, Kopin IJ, Goldstein DS, Esler MD. Cardiac sympathetic nerve function in congestive heart failure. Circulation. 1996a;93:1667–1676. doi: 10.1161/01.cir.93.9.1667. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004;56:331–349. doi: 10.1124/pr.56.3.1. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Pacak K, Goldstein DS, McCarty R. Sympathetic nervous system activity is increased in aged 344 Fischer rats. In: McCarty R, Aguilera G, Sabban E, Kvetnansky R, editors. Stress: Molecular, Genetic and Neurobiological Advances. Gordon and Breach; New York: 1996b. pp. 949–965. [Google Scholar]
- Eldrup E, Richter AE, Christensen NJ. Dopa, norepinephrine, and dopamine in rat tissues: no effect of sympathectomy on muscle dopa. Am J Physiol. 1989;256:E284–E287. doi: 10.1152/ajpendo.1989.256.2.E284. [DOI] [PubMed] [Google Scholar]
- Fujishiro H, Frigerio R, Burnett M, Klos KJ, Josephs KA, Delledonne A, Parisi JE, Ahlskog JE, Dickson DW. Cardiac sympathetic denervation correlates with clinical and pathologic stages of Parkinson's disease. Mov Disord. 2008;23:1085–1092. doi: 10.1002/mds.21989. [DOI] [PubMed] [Google Scholar]
- Gaugler MN, Genc O, Bobela W, et al. Nigrostriatal overabundance of alpha-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 2012;123:653–669. doi: 10.1007/s00401-012-0963-y. [DOI] [PubMed] [Google Scholar]
- Goldstein DS. Dysautonomia in Parkinson's disease: neurocardiological abnormalities. Lancet Neurol. 2003;2:669–676. doi: 10.1016/s1474-4422(03)00555-6. [DOI] [PubMed] [Google Scholar]
- Goldstein DS. Concepts of scientific integrative medicine applied to the physiology and pathophysiology of catecholamine systems. Comp Physiol. 2013;3:1569–1610. doi: 10.1002/cphy.c130006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Cannon RO, Quyyumi A, Chang P, Duncan M, Brush JE, Jr, Eisenhofer G. Regional extraction of circulating norepinephrine, DOPA, and dihydroxyphenylglycol in humans. J Auton Nerv Sys. 1991;34:17–35. doi: 10.1016/0165-1838(91)90005-n. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Bentho O, Sato T, Moak J, Sharabi Y, Imrich R, Conant S, Eldadah BA. Biomarkers to detect central dopamine deficiency and distinguish Parkinson disease from multiple system atrophy. Parkinsonism Relat Disord. 2008;14:600–607. doi: 10.1016/j.parkreldis.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Kopin IJ, Sharabi Y. Intra-neuronal vesicular uptake of catecholamines is decreased in patients with Lewy body diseases. J Clin Inv. 2011;121:3320–3330. doi: 10.1172/JCI45803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO., 3rd Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med. 2000;133:338–347. doi: 10.7326/0003-4819-133-5-200009050-00009. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Sullivan P, Cooney A, Jinsmaa Y, Sullivan R, Gross DJ, Holmes C, Kopin IJ, Sharabi Y. Vesicular uptake blockade generates the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde in PC12 Cells: Relevance to the pathogenesis of Parkinson disease. J Neurochem. 2012;123:932–943. doi: 10.1111/j.1471-4159.2012.07924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Sullivan P, Holmes C, Miller GW, Alter S, Strong R, Mash DC, Kopin IJ, Sharabi Y. Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson's disease. J Neurochem. 2013;126:591–603. doi: 10.1111/jnc.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Zimlichman R, Stull R, Keiser HR, Kopin IJ. Estimation of intrasynaptic norepinephrine concentrations in humans. Hypertension. 1986;8:471–475. doi: 10.1161/01.hyp.8.6.471. [DOI] [PubMed] [Google Scholar]
- Holmes C, Eisenhofer G, Goldstein DS. Improved assay for plasma dihydroxyphenylacetic acid and other catechols using high-performance liquid chromatography with electrochemical detection. J Chromatogr B Biomed Appl. 1994;653:131–138. doi: 10.1016/0378-4347(93)e0430-x. [DOI] [PubMed] [Google Scholar]
- Holmes C, Whittaker N, Heredia-Moya J, Goldstein DS. Contamination of the norepinephrine prodrug droxidopa by dihydroxyphenylacetaldehyde. Clin Chem. 2010;56:832–838. doi: 10.1373/clinchem.2009.139709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janezic S, Threlfell S, Dodson PD, et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A. 2013;110:E4016–4025. doi: 10.1073/pnas.1309143110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinsmaa Y, Sullivan P, Gross D, Cooney A, Sharabi Y, Goldstein DS. Divalent metal ions enhance DOPAL-induced oligomerization of alpha-synuclein. Neurosci Lett. 2014 doi: 10.1016/j.neulet.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura M, Kopin IJ, Kador PF, Sato S, Tjurmina O, Eisenhofer G. Effects of aldehyde/aldose reductase inhibition on neuronal metabolism of norepinephrine. J Auton Nerv Syst. 1997;66:145–148. doi: 10.1016/s0165-1838(97)00086-6. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson's disease. Pathophysiologic and clinical implications. N Engl J Med. 1988;318:876–880. doi: 10.1056/NEJM198804073181402. [DOI] [PubMed] [Google Scholar]
- Kopin IJ. Catecholamine metabolism: basic aspects and clinical significance. Pharmacol Rev. 1985;37:333–364. [PubMed] [Google Scholar]
- Kopin IJ, Weise VK. Effect of reserpine and metaraminol on excretion of homovanillic acid and 3-methoxy-4-hydroxyphenylglycol in the rat. Biochem Pharmacol. 1968;17:1461–1464. doi: 10.1016/0006-2952(68)90083-x. [DOI] [PubMed] [Google Scholar]
- Kopin IJ, Zukowska-Grojec Z, Bayorh MA, Goldstein DS. Estimation of intrasynaptic norepinephrine concentrations at vascular neuroeffector junctions in vivo. Naunyn Schmiedebergs Arch Pharmacol. 1984;325:298–305. doi: 10.1007/BF00504372. [DOI] [PubMed] [Google Scholar]
- Li SW, Lin TS, Minteer S, Burke WJ. 3,4-Dihydroxyphenylacetaldehyde and hydrogen peroxide generate a hydroxyl radical: possible role in Parkinson's disease pathogenesis. Brain Res Mol Brain Res. 2001;93:1–7. doi: 10.1016/s0169-328x(01)00120-6. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P. Pathogenesis of Parkinson's diease: Dopamine, vesicles, and [alpha]-synuclein. Nature Reviews: Neuroscience. 2002;3:932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- Mattammal MB, Haring JH, Chung HD, Raghu G, Strong R. An endogenous dopaminergic neurotoxin: implication for Parkinson's disease. Neurodegeneration. 1995;4:271–281. doi: 10.1016/1055-8330(95)90016-0. [DOI] [PubMed] [Google Scholar]
- Mooslehner KA, Chan PM, Xu W, Liu L, Smadja C, Humby T, Allen ND, Wilkinson LS, Emson PC. Mice with very low expression of the vesicular monoamine transporter 2 gene survive into adulthood: potential mouse model for parkinsonism. Mol Cell Biol. 2001;21:5321–5331. doi: 10.1128/MCB.21.16.5321-5331.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Staal RG, Bove J, et al. Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci. 2006;26:9304–9311. doi: 10.1523/JNEUROSCI.0519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo S, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, Takahashi H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson's disease. Brain. 2008;131:642–650. doi: 10.1093/brain/awm302. [DOI] [PubMed] [Google Scholar]
- Panneton WM, Kumar VB, Gan Q, Burke WJ, Galvin JE. The neurotoxicity of DOPAL: behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS One. 2010;5:e15251. doi: 10.1371/journal.pone.0015251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffel DM, Koeppe RA, Little R, Wang CN, Liu S, Junck L, Heumann M, Gilman S. PET measurement of cardiac and nigrostriatal denervation in parkinsonian syndromes. J Nucl Med. 2006;47:1769–1777. [PubMed] [Google Scholar]
- Rees JN, Florang VR, Eckert LL, Doorn JA. Protein reactivity of 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite, is dependent on both the aldehyde and the catechol. Chem Res Toxicol. 2009;22:1256–1263. doi: 10.1021/tx9000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Duan J, Ying Z, Hou Y, Zhang Y, Wang R, Deng Y. Increased vulnerability of parkin knock down PC12 cells to hydrogen peroxide toxicity: the role of salsolinol and NM-salsolinol. Neuroscience. 2013;233:72–85. doi: 10.1016/j.neuroscience.2012.12.045. [DOI] [PubMed] [Google Scholar]
- Taylor TN, Alter SP, Wang M, Goldstein DS, Miller GW. Reduced vesicular storage of catecholamines causes progressive degeneration in the locus ceruleus. Neuropharmacology. 2014;76:A97–A105. doi: 10.1016/j.neuropharm.2013.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TN, Caudle WM, Shepherd KR, Noorian A, Jackson CR, Iuvone PM, Weinshenker D, Greene JG, Miller GW. Nonmotor symptoms of Parkinson's disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci. 2009;29:8103–8113. doi: 10.1523/JNEUROSCI.1495-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson's disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. 2002;41:4595–4602. doi: 10.1021/bi0121353. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Levey AI, Rajput A, et al. Differential changes in neurochemical markers of striatal dopamine nerve terminals in idiopathic Parkinson's disease. Neurology. 1996;47:718–726. doi: 10.1212/wnl.47.3.718. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.