

Abstract
We report the case of an 8-year-old boy with pyridoxamine 5′-phosphate oxidase (PNPO) deficiency. He developed seizures at 24 h of age that were refractory to standard anticonvulsant therapy and a trial of pyridoxine but responded to pyridoxal phosphate (PLP) at 28 days of life. Genetic testing identified compound heterozygous mutations in the PNPO gene. Management of encephalopathic episodes required escalation of PLP dose to 100 mg/kg/day by 2 years of age. Routine blood tests at this time showed significantly deranged liver function tests (LFTs). A wedge liver biopsy showed early cirrhosis with marked elevation of pyridoxal and pyridoxic acid levels in the liver sample. Despite extensive investigation, no cause other than PLP therapy could be identified for the cirrhosis. The PLP dose was weaned to 50 mg/kg/day before episodes of encephalopathy recurred. Concurrent with the reduction of his PLP dose, LFTs showed improvement. However, at 8 years of age, there is persistent evidence of hepatic fibrosis and early portal hypertension. We hypothesise that hepatic toxicity due to PLP or its degradation products is the cause of cirrhosis in this boy. Until further evidence becomes available, we would suggest that people with PNPO deficiency are treated with the minimum dose of PLP required to prevent episodes of encephalopathy.
Background
Pyridoxal 5′-phosphate (PLP) is the active member of the pyridoxine vitamer family. The first case of neonatal epilepsy responsive to PLP but not pyridoxine was reported in 2002 (Kuo and Wang 2002). Mills and colleagues subsequently described the genotype and phenotype of neonatal epileptic encephalopathy due to pyridoxamine 5′-phosphate oxidase (PNPO) deficiency (Mills et al. 2005). The treatment dose of PLP has largely been empirical, guided by clinical response with reported doses varying from 30 to 60 mg/kg/day (Mills et al. 2005; Hoffmann et al. 2007; Bagci et al. 2008). PLP is not licensed as a drug for the treatment of epilepsy outside of Asia.
Case
We report an eight-year-old boy with PNPO deficiency who developed cirrhosis while being treated with high-dose PLP.
The initial clinical details have been previously reported (Hoffmann et al. 2007; Schmitt et al. 2010). In summary, this boy was born at term and developed seizures at 24 h of age. Despite multiple anticonvulsant medications and a trial of pyridoxine, his seizures proved refractory. At 28 days he was trialled on PLP 50 mg four times a day. His seizures ceased immediately. All antiepileptic medications were successfully withdrawn. He was maintained on PLP and made good developmental progress. His liver function tests (LFTs) were normal at 1 month of age.
A trial of withdrawal of PLP at 7 months resulted in encephalopathy and EEG changes within 7 h which resolved within 20 min of administration of 200 mg of PLP. Genetic studies revealed he was compound heterozygous for a missense mutation, D33V (c.98A>T), and a single base pair deletion, c.246delT, in the PNPO gene.
Over the following 18 months, there were recurrent episodes of PLP-responsive encephalopathy, especially during periods of illness, requiring escalation of the dose of PLP to 1,500 mg/day (~100 mg/kg/day) given in four divided doses. Routine LFTs at 2 years of age were markedly elevated aspartate transaminase (AST): 292 U/L (normal: 10–50 U/L), alanine transaminase (ALT): 169 U/L (normal: 0–45 U/L), gamma-glutamyl transaminase (GGT): 131 U/L (normal: 0–45 U/L) and alpha fetoprotein (AFP): 635 IU/mL (normal: 0–6 IU/mL). The clotting profile was mildly deranged – prothrombin time (PT) 19 s (normal: 11–15 s). There was palpable hepatosplenomegaly (confirmed on ultrasound). The trough whole blood PLP level (6 h post-dose) measured using high-performance liquid chromatography (HPLC) was markedly elevated – 6,090 nmol/L (normal: 35–110 nmol/L).
A wedge liver biopsy (age 2 years 2 months) showed extensive fibrosis and focal nodules of hepatocytes surrounded by fibrous tissue consistent with early cirrhosis (Fig. 1). All investigations for viral and metabolic causes of cirrhosis were negative. Respiratory chain enzyme analyses in the muscle and liver were normal.
Fig. 1.
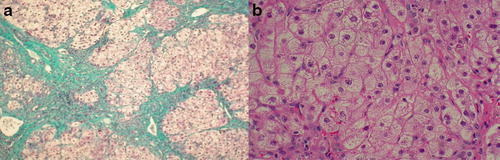
Liver biopsy specimens (2006) age 2 years and 2 months. (a) Wedge liver biopsy (2006): Masson’s trichrome stain shows marked disruption of architecture with extensive fibrosis, portal to portal and central bridging with focal nodules of hepatocyte surrounded by fibrous tissue consistent with early cirrhosis. (b) Wedge liver biopsy: Haematoxylin and eosin stain shows swollen hepatocytes with ballooning degenerative changes in the cytoplasm which have a granular appearance. Stains for glycogen and fat were negative
Assays of hepatic levels of B6 vitamers were performed on the liver samples and samples of two control patients. Liver samples were weighed, immediately homogenised with 25% trichloroacetic acid and centrifuged and the supernatant analysed by HPLC/fluorescence to measure non-phosphorylated forms of the B6 vitamers. Because this process was rapid, we believe that there was no significant hydrolysis of phosphorylated vitamers. Levels of pyridoxal and pyridoxic acid in the liver were approximately 40 times greater than two control specimens (Table 1). We feel it is likely that this technique measured non-phosphorylated forms of the vitamers and pyridoxic acid.
Table 1.
Levels of pyridoxine and its metabolites (non-phosphorylated forms) in the liver tissue of two controls and our patient
| Control liver 1 (μg/g) | Control liver 2 (μg/g) | Patient (μg/g) | |
|---|---|---|---|
| Pyridoxamine | 0.1 | 0.3 | 0.4 |
| Pyridoxine | 0.2 | 0.3 | 0.8 |
| Pyridoxal | 0.03 | 0.02 | 1.3 |
| Pyridoxic acid | 0.02 | 0.02 | 1.0 |
We hypothesised that high doses of PLP accounted for his liver dysfunction and weaned the dose of PLP to ~50 mg/kg/day. Dose frequency was changed from 4 to 6 hourly to smaller 3 hourly doses in the hope that lower peak levels might protect against hepatotoxicity. Vitamin C was added to help with potential oxidative stress. His LFTs improved substantially but did not entirely normalise (Fig. 2).
Fig. 2.
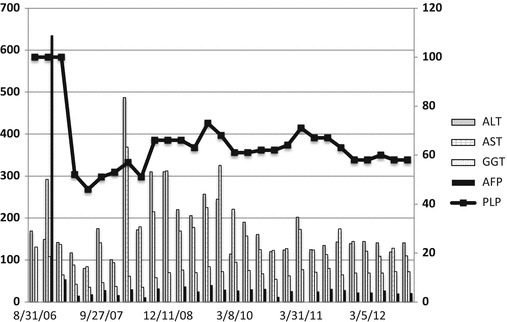
Serial levels of liver enzyme (left axis) and dose of PLP (right axis). AST (U/L) – normal range 10–50 U/L, ALT (U/L) – normal range 0–45 U/L, GGT (U/L) – normal range 0–45 U/L, AFP (U/L) – normal range 0–6 IU/mL, PLP – PLP dose (mg/kg/day)
Managing him at this dose resulted in heightened susceptibility to seizures or encephalopathic episodes. The maintenance dose has since been increased to 60 mg/kg/day. His parents give an extra 400 mg orally for episodes of encephalopathy, and a rectal dose of PLP (400 mg) is used if the episode does not respond to oral PLP (only used on five occasions during his life).
Increases in the dose of PLP beyond 60 m/kg/day have been associated with transient increases in his transaminase levels above his baseline level (Fig. 2). He has persistent mild hepatosplenomegaly, with evidence of hepatic fibrosis and mild portal hypertension.
Currently he has infrequent episodes of encephalopathy usually triggered by missing a dose of PLP or with illness. At the age of 8 years, he is an academically gifted student in a mainstream school.
Discussion
Vitamin B6 consists of three related pyridine derivatives: pyridoxine, pyridoxamine and pyridoxal and their 5′-phosphate esters. PLP is the active form of vitamin B6. After passive intestinal absorption, vitamin B6 is converted to the phosphorylated derivatives in the liver. Pyridoxine-5′-phosphate (PNP) and pyridoxamine-5′-phosphate (PMP) are oxidised to PLP by PNPO. PLP re-enters the circulation bound to albumin. Delivery of active cofactor to the tissues requires hydrolysis of circulating PLP to pyridoxal. Pyridoxal crosses the blood–brain barrier (and enters other tissues) but then needs to be re-phosphorylated to produce the active cofactor (Clayton 2006).
Although most PLP-catalysed reactions regenerate PLP, some enzymes can generate PMP. Thus PNPO plays a role as a metabolite repair enzyme recycling PLP. The turnover of PLP enzymes may also generate PMP. When on treatment with B6, patients with PNPO deficiency have higher levels of pyridoxamine and PMP than patients with antiquitin deficiency (Footitt et al. 2013). The role of PNPO in recycling and trafficking PLP probably explains why PNPO deficiency is much more difficult to treat than antiquitin deficiency.
Compared with other patients with PNPO deficiency reported in the literature, this boy has had a very good neurological outcome. We hypothesise that this results from early introduction of PLP, prompt management of encephalopathy and perhaps the biochemical effects of his mutation. His management entailed the use of very high doses of PLP (to our knowledge the highest dose/weight reported).
There has been only one previous reported case of liver toxicity secondary to high-dose PLP (Yoshida et al. 1985) in a 7-year-old boy with homocystinuria who developed hepatitis with elevated LFTs within 4 days of an increase in his dose of PLP to 1,000 mg/day. Another study reported transient elevation of LFTs in 14/28 patients with infantile spasms treated with PLP (dose range 30–50 mg/kg/day (Takuma and Seki 1996)). The deranged LFTs returned to normal within 2 weeks of reduction or withdrawal of PLP.
In our case, liver dysfunction was identified after the child had been on high doses of PLP for almost 2 years. The improvement in LFTs on reducing the dose of PLP suggests a dose-related response; however, the persistent abnormalities of liver function suggest that there is ongoing hepatic inflammation.
The cause of this toxicity is not known. It is possible that degradation products of PLP in solution contributed to this boy’s liver toxicity. Initially PLP was given as a powder dissolved in milk/yoghurt/water and left in solution before being administered. The solution showed colour change raising concerns about the chemical stability of the preparation. In aqueous solution, PLP is vulnerable to degradation induced by exposure to light and oxygen. In aerobic conditions, the principal breakdown (oxidation) products include 4-pyridoxic acid-5′-phosphate and the dipyridyl α-diketone. In anaerobic conditions, the acyloin is produced (Morrison and Long 1958; Wei et al. 1972; Bazhulina et al. 1974).
In view of the above, we recommend that PLP be consumed as a whole capsule where possible and that any suspension or solution is taken immediately. Until further evidence becomes available, we suggest that the dose of PLP is carefully titrated against symptoms with close monitoring of liver function especially when doses greater than 50 mg/kg/day are used.
Synopsis
Treatment for PNPO deficiency.
Compliance with Ethics Guidelines
Annapurna Sudarsanam, Harry Singh, Bridget Wilcken, Michael Stormon, Susan Arbuckle, Bernhard Schmitt, Peter Clayton, John Earl and Richard Webster declare that they have no conflict of interest.
Peter Clayton is funded by Great Ormond Street Hospital Children’s Charity.
Informed Consent
Informed consent has been obtained from the patient and his family for the publication of this paper.
Details of Contributions of Individual Authors
Annapurna Sudarsanam, Harry Singh and Richard Webster were involved with drafting the article. Richard Webster, Harry Singh, Bridget Wilcken, Michael Stormon, Susan Arbuckle, Bernhard Schmitt, Peter Clayton and John Earl were all involved with the management of the case and critically appraised and revised the article.
Footnotes
Competing interests: None declared
Contributor Information
Richard Webster, Email: richard.webster@health.nsw.gov.au.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Bagci S, Zschocke J, Hoffmann GF, et al. Pyridoxal phosphate-dependent neonatal epileptic encephalopathy. Arch Dis Child Fetal Neonatal Ed. 2008;93(2):F151–F152. doi: 10.1136/adc.2006.115162. [DOI] [PubMed] [Google Scholar]
- Bazhulina NP, Kirpichnikov MP, Morozov YV, Savin FA. Photochemistry of aldehyde forms of pyridoxal, pyridoxal 5′-phosphate, and their derivatives. Mol Photochem. 1974;6(4):367–396. [Google Scholar]
- Clayton PT. B6-responsive disorders: a model of vitamin dependency. J Inherit Metab Dis. 2006;29(2–3):317–326. doi: 10.1007/s10545-005-0243-2. [DOI] [PubMed] [Google Scholar]
- Footitt EJ, Clayton PT, Mills K, et al. Measurement of plasma B6 vitamer profiles in children with inborn errors of vitamin B6 metabolism using an LC-MS/MS method. J Inherit Metab Dis. 2013;36(1):139–145. doi: 10.1007/s10545-012-9493-y. [DOI] [PubMed] [Google Scholar]
- Hoffmann GF, Schmitt B, Windfuhr M, et al. Pyridoxal 5′-phosphate may be curative in early-onset epileptic encephalopathy. J Inherit Metab Dis. 2007;30(1):96–99. doi: 10.1007/s10545-006-0508-4. [DOI] [PubMed] [Google Scholar]
- Kuo MF, Wang HS. Pyridoxal phosphate-responsive epilepsy with resistance to pyridoxine. Pediatr Neurol. 2002;26(2):146–147. doi: 10.1016/S0887-8994(01)00357-5. [DOI] [PubMed] [Google Scholar]
- Mills PB, Surtees RA, Champion MP, et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5′-phosphate oxidase. Hum Mol Genet. 2005;14(8):1077–1086. doi: 10.1093/hmg/ddi120. [DOI] [PubMed] [Google Scholar]
- Morrison AL, Long RF (1958) The photolysis of pyridoxal phosphate. J Chem Soc 211–215
- Schmitt B, Baumgartner M, Mills PB, et al. Seizures and paroxysmal events: symptoms pointing to the diagnosis of pyridoxine-dependent epilepsy and pyridoxine phosphate oxidase deficiency. Dev Med Child Neurol. 2010;52(7):e133–e142. doi: 10.1111/j.1469-8749.2010.03660.x. [DOI] [PubMed] [Google Scholar]
- Takuma Y, Seki T. Combination therapy of infantile spasms with high-dose pyridoxal phosphate and low-dose corticotropin. J Child Neurol. 1996;11(1):35–40. doi: 10.1177/088307389601100109. [DOI] [PubMed] [Google Scholar]
- Wei K-T, Bell RR, Haskell BE. Regarding 4-pyridoxic acid-5′-phosphate in animal tissue. Biochem Biophys Res Commun. 1972;48(6):1671–1674. doi: 10.1016/0006-291X(72)90907-2. [DOI] [PubMed] [Google Scholar]
- Yoshida I, Sakaguchi Y, Nakano M, Yamashita F, Hitoshi T. Pyridoxal phosphate-induced liver injury in a patient with homocystinuria. J Inherit Metab Dis. 1985;8(2):91. doi: 10.1007/BF01801674. [DOI] [PubMed] [Google Scholar]