

Abstract
Citrulline is among the metabolites measured by expanded newborn screening (NBS). While hypocitrullinemia can be a marker for deficiency of proximal urea cycle enzymes such as ornithine transcarbamylase (OTC), only a handful of state newborn screening programs in the United States officially report a low citrulline value for further work-up due to low positive predictive value. We report a case of a male infant who was found to have hypocitrullinemia on NBS. After excluding proximal urea cycle disorders by DNA sequencing, his NBS result was felt to be a false positive. At 4 months of age, he developed poor feeding, failure to thrive, apnea and infantile spasms with a progression to intractable seizures, as well as persistent hypocitrullinemia. He was diagnosed with Leigh syndrome due to a maternally inherited homoplasmic m.8993T>G mutation in the ATPase 6 gene. His mother, who had previously been diagnosed with cerebral palsy, was concurrently diagnosed with neuropathy, ataxia, and retinitis pigmentosa (NARP) due to heteroplasmy of the same mutation. She had progressive muscle weakness, ataxia, and speech dyspraxia. The m.8993T>G mutation causes mitochondrial ATP synthase deficiency and it is hypothesized to undermine the synthesis of citrulline by CPS1. In addition to proximal urea cycle disorders, the evaluation of an infant with persistent hypocitrullinemia should include testing for the m.8993T>G mutation and other disorders that cause mitochondrial dysfunction.
All states in the United States have implemented expanded NBS using tandem mass spectrometry (MS/MS) which permits a rapid analysis of metabolites based on mass-to-charge ratio. Almost all states screen for the 20 core metabolic conditions recommended by the American College of Medical Genetics and Genomics (ACMG) using MS/MS (ACMG 2006). These include distal urea cycle disorders such as citrullinemia and argininosuccinate aciduria detected by a high citrulline level. A low citrulline level may be incidentally detected by this method and may be used as a marker for deficiencies of a proximal urea cycle enzyme such as N-acetylglutamate synthase (NAGS), carbamoyl phosphate synthetase 1 (CPS1), and ornithine transcarbamylase (OTC). The ACMG recommends that a significantly low citrulline level be reported to rule out OTC or CPS1 deficiency (ACMG 2006), though the two conditions are not included in the recommended universal NBS panel due to a high false-positive rate of hypocitrullinemia (Cavicchi et al. 2009).
We report a case of a male infant who was found to have hypocitrullinemia on NBS and who later was diagnosed with Leigh syndrome due to a maternally inherited homoplasmic m.8993T>G mutation in the MT-ATP6 encoding subunit 6 of mitochondrial ATP synthase. The 24-year-old mother was diagnosed with NARP caused by the same mutation but a reduced mutant load in the peripheral blood of >95%. She also had hypocitrullinemia. Patients with m.8993T>G may benefit from early diagnosis in regard to prognosis and presymptomatic treatment with citrulline, coenzyme Q10, and, perhaps, EPI-743, a promising medication in the pipeline(Enns et al. 2012; Martinelli et al. 2012).
Case Report
A Caucasian male born in Florida at term to a 24-year-old mother after an uncomplicated pregnancy with a birth weight of 3.3 kg was referred to a local metabolic clinic at 5 days of age due to an abnormal NBS result with a low citrulline level (3.37 μmol/L at age 24 h, reference range 4–50 μmol/L), raising concern for a proximal urea cycle disorder. He failed his newborn hearing screen in the right ear. The family denied consanguinity. The mother had been diagnosed with cerebral palsy in childhood attributed to in utero bradycardia/anoxia. An evaluation in Florida included serum amino acid analysis, urine organic acid analysis, and serum ammonia. Serum amino acid analysis revealed persistently low citrulline (3 μmol/L, reference range 9–38), low arginine (24 μmol/L, reference range 29–134), and high alanine (537 μmol/L, reference range 139–474) levels. The patient was on regular formula at the time of the specimen collection. Urine organic acid analysis and serum ammonia levels were normal. The patient was started on protein-restricted formula (2 g/kg/day) and l-citrulline supplement (170 mg/kg/day) for a presumed urea cycle disorder. The patient and the mother relocated to live with the maternal grandmother during the work-up and transferred the care of the child to our service. Sequencing and deletion/duplication analysis of the OTC gene for OTC deficiency, CPS1 gene for CPS I deficiency and the NGS gene for NAGS deficiency were negative for deleterious mutations. The patient was asymptomatic with normal growth and development at the age of 3 months. He was concurrently taken off diet restriction and L-citrulline after the negative genetic test results and a normal citrulline level of 20 μmol/L.
The patient had been doing well until 4 months of age, when he was noted to be losing weight by his primary care provider. He was admitted for an evaluation of failure to thrive and hypotonia. A brain MRI and an EEG were obtained for a concern for seizure-like activities. The EEG was initially normal. A brain MRI revealed abnormal signal and restricted diffusion in bilateral lenticular nuclei, the medial thalami and the caudate nuclei, as well as encephalomalacia (Figs. 1 and 2). An MR spectroscopy showed a lactate peak in the left anterior basal ganglia (Fig. 3). These findings were concerning for an underlying mitochondrial disorder, although a hypoxic-ischemic event was also a possibility. A muscle biopsy showed nonspecific changes in SDH, COX, and NADH stains. Electron transport chain enzyme spectrophotometry assay on snap frozen quadriceps muscle was unremarkable except for a reduced enzyme activity of the complex II (31% of mean). Respiratory chain complex V (ATP synthase) activity was not measured due to a technical limitation. A mitochondrial DNA mutation screening panel (leukocytes) was ordered due to the MRI findings and revealed homoplasmic m.8993T>G mutations in the MT-ATP6 gene encoding ATP synthase, leading to the diagnosis of Leigh syndrome. The mutant load was confirmed by real-time allele refractory mutation system (ARMS) quantitative PCR analysis. Mild persistent lactic acidosis with lactate levels in the range of 3–7 mol/L was consistent with the diagnosis. A therapy with coenzyme Q10 was started for the treatment of Leigh syndrome. l-citrulline was restarted at an increased dose of 1.1 g/day (200 mg/kg/day), as serum citrulline level had decreased to 5 μmol/L. An ophthalmologic eye exam revealed bull’s eye maculopathy consistent with the underlying diagnosis (Laird et al. 2006).
Fig. 1.
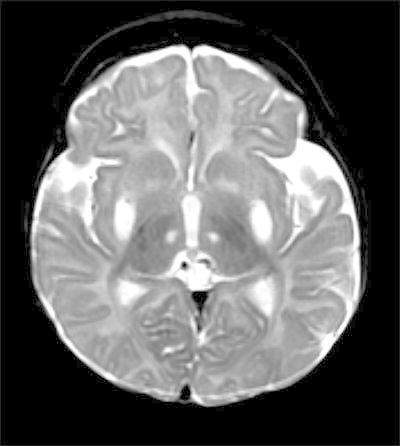
Axial T2. Abnormal increased signal in subganglionic tissues, lenticular nuclei, and medial thalamic nuclei
Fig. 2.
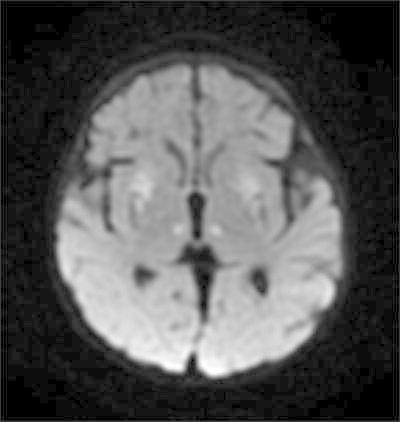
Diffusion restriction in subganglionic tissues, lenticular nuclei, and medial thalamic nuclei
Fig. 3.
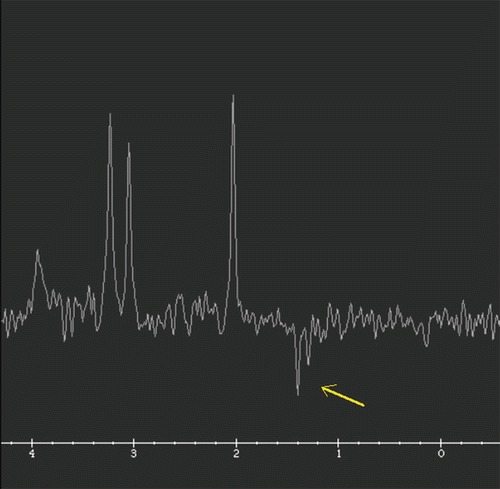
MR spectroscopy TE 144 confirms inverted lactate duplet
The patient required a G-tube for feeding for hypotonia and a failed swallow study, Nissen fundoplication for severe reflux and intermittent urinary catheterization for a neurogenic bladder. He remained on therapy with l-citrulline and coenzyme Q10 (100 mg/kg/day) supplement. He continued to have seizure-like activities. Long-term monitoring EEG at 5 months of age revealed frequent focal seizures arising from each hemisphere. He developed intractable seizures requiring multiple admissions with mechanical ventilation despite being on three antiseizure medications. Due to frequent apneic episodes, he was on home oxygen therapy. At 6.5 months of age, he developed infantile spasms. The EEG showed amplitude suppression in the bilateral anterior head regions, a disorganized and slow background, posteriorly dominant multifocal spikes, and infantile spasms associated with a broad high amplitude slow wave and electrodecrement. His infantile spasms resolved within one day of starting vigabatrin. Although his background abnormalities and multifocal spikes persisted, an overnight EEG performed three weeks after starting vigabatrin confirmed the clinical remission of infantile spasms. However, the frequency of his focal seizures increased, he became increasingly lethargic, and the family agreed to hospice care. He died at 11 months of age at home related to ongoing seizures and apnea. He was enrolled for a safety and efficacy clinical trial of EPI-743 designed for children with Leigh syndrome (NCT01721733) when he was 10 months of age, but he died before the therapy was started.
Testing of the mother with real-time ARMS quantitative PCR revealed that she had the m.8993T>G mutation with a mutation load of >95% in the blood; she was diagnosed with NARP based on the mutation and clinical presentation. The maternal grandmother was also tested for the familial mutation, but no mutation was detected in a blood specimen by real-time ARMS quantitative PCR. Neither the patient nor the mother has any siblings. The mother was a product of an uncomplicated pregnancy and perinatal hypoxia. She was diagnosed with cerebral palsy at the age of 14 months. She did not walk until 15–16 months and required special education throughout school. At the time of diagnosis with NARP, the mother was found to have low levels of multiple amino acids in the serum including citrulline (3 μmol/L), and l-citrulline supplement was started. She did not tolerate coenzyme Q10 due to headaches. An ophthalmologic eye exam did not reveal any evidence of pigmentary retinopathy. A brain MRI revealed increased signals in the bilateral basal ganglia in FLAIR and diffusion sequences. Her lactate, alanine, and proline levels were within the normal range. Since giving birth to the proband, she has demonstrated progressively worsening neurologic symptoms including deterioration in speech, swallowing function, and ambulation.
Discussion
Although citrulline level is measured in NBS to detect distal urea cycle disorders leading to hypercitrullinemia (ACMG 2006), an NBS protocol for low hypocitrullinemia is not established because of low sensitivity of hypocitrullinemia for proximal urea cycle disorders (Cavicchi et al. 2009). Persistent hypocitrullinemia initially identified by NBS warrants a work-up beyond proximal urea cycle disorders.
Persistent hypocitrullinemia has been seen in patients with mitochondrial disorders (Atkuri et al. 2009) including patients with Pearson syndrome (OMIM 557000) (Ribes et al. 1993), m.8993T>G-associated Leigh syndrome/NARP (OMIM 516060) (Rabier et al. 1998; Parfait et al. 1999; Enns et al. 2006; Debray et al. 2010; Henriques et al. 2012), and mitochondrial encephalomyopathy, lactic acidosis with stroke-like episodes syndrome (MELAS, OMIM 540000) (Perry et al. 1989; Koga et al. 2005; Naini et al. 2005). Hypocitrullinemia is also associated with secondary mitochondrial respiratory chain dysfunction caused by organic acidemias (Atkuri et al. 2009), deficiency of mitochondrial pyrroline-5-carboxylate synthase (P5C) (Rabier and Kamoun 1995; Baumgartner et al. 2000), or intestinal malrotation in newborns (Cavicchi et al. 2009). Citrulline is synthesized in the mitochondrial matrix of enterocytes in the small intestine catalyzed by pyrroline-5-carboxylate synthase (P5C), CPS1, and OTC (Naini et al. 2005). Hepatocytes also synthesize citrulline, but they cannot export the product. Hypocitrullinemia is thought to be a nonspecific marker of impaired oxidative phosphorylation in the enterocyte. Reduced mitochondrial ATP or enterocyte dysfunction can lead to reduced activity of P5C or CPS1 in enterocytes, leading to reduced synthesis of circulating citrulline (Parfait et al. 1999; Munnich and Rustin 2001). Thus, persistent citrullinemia can be a marker for dysfunction of enterocytes, mitochondrial ATP production, or mitochondrial P5C and urea cycle enzymes. The prevalence of hypocitrullinemia among infants with mtDNA disorders including m.8993T>G is unknown and the predictive power of hypocitrullinemia for these disorders cannot be determined. In a review of six children with Leigh syndrome due to the m.8993T>G mutation, only one child was found with hypocitrullinemia (Morava et al. 2006).
Although there is no current cure for mitochondrial disorders or Leigh syndrome, coenzyme Q10 may have beneficial effects related to its antioxidant properties. A coenzyme Q10 derivative EPI-743 is believed to be more potent than coenzyme Q10, readily crosses the blood–brain barrier (Enns et al. 2012), and has shown promising results with no side effects in four patients (Enns et al. 2012). A randomized clinical trial of EPI-743 for children with Leigh syndrome is underway (NCT01721733).
When hypocitrullinemia is detected in NBS and persists in a confirmatory test, differential diagnoses should not be limited to proximal urea cycle disorders but also include m.8993T>G as well as other disorders that can impair P5C or CPS1 function including MELAS and Pearson syndrome. Serum lactate level may be high in these disorders but is not a sensitive marker, especially when the patient is asymptomatic.
Conclusions
We described a case of m.8993T>G-associated Leigh syndrome who initially presented with hypocitrullinemia on NBS. Persistent hypocitrullinemia detected on NBS with no defect in NAGS, CPS1, or OTC may result from other disorders causing respiratory chain dysfunction. Although additional study is needed, a presymptomatic diagnosis of mitochondrial disorders may be of therapeutic benefit. Further studies are needed to establish a protocol for hypocitrullinemia including a cut-off value.
Synopsis
When hypocitrullinemia is detected in NBS and persists in a confirmatory test, differential diagnoses should not be limited to proximal urea cycle disorders but also include m.8993T>G as well as other disorders that can impair P5C or CPS1 function including MELAS and Pearson syndrome.
Compliance with Ethics Guidelines
Mari Mori, John R. Mytinger, Lisa C. Martin, Dennis Bartholomew, and Scott Hickey declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
Contribution of Authors
Mari Mori: Dr. Mori cared for the patient during hospital admittance and in the genetics clinic. She enrolled the patient in the EPI-743 clinical study. Dr. Mori drafted the initial draft and approved the final manuscript as submitted.
Scott E. Hickey: Dr. Hickey diagnosed the patient with Leigh syndrome and his mother with NARP, cared for the patient during hospital admittance and in the genetics clinic, aided with enrollment into the EPI-743 clinical study, aided with draft revision, and approved the final manuscript as submitted.
Dennis Bartholomew: Dr. Bartholomew reviewed the newborn screening results, performed the initial work-up, aided in draft revision, and approved the final manuscript as submitted.
Dr. John R. Mytinger: Dr. Mytinger cared for the patient during hospital admittance and in neurology clinic, aided in draft revision and approved the final manuscript as submitted.
Dr. Lisa C. Martin: Dr. Martin interpreted the brain MRI in radiology, aided in preparing the figure, and approved the final manuscript as submitted.
Footnotes
Competing interests: None declared
Contributor Information
Mari Mori, Email: Mari.Mori@nationwidechildrens.org.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- ACMG Newborn screening: toward a uniform screening panel and system. Genet Med. 2006;8(Suppl 1):1S–252S. doi: 10.1097/01.gim.0000223891.82390.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkuri KR, Cowan TM, Kwan T, Ng A, Herzenberg LA, Enns GM. Inherited disorders affecting mitochondrial function are associated with glutathione deficiency and hypocitrullinemia. Proc Natl Acad Sci U S A. 2009;106(10):3941–3945. doi: 10.1073/pnas.0813409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner MR, Hu CA, Almashanu S, et al. Hyperammonemia with reduced ornithine, citrulline, arginine and proline: a new inborn error caused by a mutation in the gene encoding delta(1)-pyrroline-5-carboxylate synthase. Hum Mol Genet. 2000;9(19):2853–2858. doi: 10.1093/hmg/9.19.2853. [DOI] [PubMed] [Google Scholar]
- Cavicchi C, Malvagia S, la Marca G, et al. Hypocitrullinemia in expanded newborn screening by LC-MS/MS is not a reliable marker for ornithine transcarbamylase deficiency. J Pharm Biomed Anal. 2009;49(5):1292–1295. doi: 10.1016/j.jpba.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Debray FG, Lambert M, Allard P, Mitchell GA. Low citrulline in Leigh disease: still a biomarker of maternally inherited Leigh syndrome. J Child Neurol. 2010;25(8):1000–1002. doi: 10.1177/0883073809351983. [DOI] [PubMed] [Google Scholar]
- Enns GM, Bai RK, Beck AE, Wong LJ. Molecular-clinical correlations in a family with variable tissue mitochondrial DNA T8993G mutant load. Mol Genet Metab. 2006;88(4):364–371. doi: 10.1016/j.ymgme.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Enns GM, Kinsman SL, Perlman SL, et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab. 2012;105(1):91–102. doi: 10.1016/j.ymgme.2011.10.009. [DOI] [PubMed] [Google Scholar]
- Henriques M, Diogo L, Garcia P, Pratas J, Simoes M, Grazina M. Mitochondrial DNA 8993T>G mutation in a child with ornithine transcarbamylase deficiency and leigh syndrome: an unexpected association. J Child Neurol. 2012;27(8):1059–1061. doi: 10.1177/0883073811431015. [DOI] [PubMed] [Google Scholar]
- Koga Y, Akita Y, Nishioka J, et al. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64(4):710–712. doi: 10.1212/01.WNL.0000151976.60624.01. [DOI] [PubMed] [Google Scholar]
- Laird PW, Mohney BG, Renaud DL. Bull's-eye maculopathy in an infant with Leigh disease. Am J Ophthalmol. 2006;142(1):186–187. doi: 10.1016/j.ajo.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Martinelli D, Catteruccia M, Piemonte F, et al. EPI-743 reverses the progression of the pediatric mitochondrial disease–genetically defined Leigh Syndrome. Mol Genet Metab. 2012;107(3):383–388. doi: 10.1016/j.ymgme.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Morava E, Rodenburg RJ, Hol F, et al. Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations. Am J Med Genet A. 2006;140(8):863–868. doi: 10.1002/ajmg.a.31194. [DOI] [PubMed] [Google Scholar]
- Munnich A, Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am J Med Genet. 2001;106(1):4–17. doi: 10.1002/ajmg.1391. [DOI] [PubMed] [Google Scholar]
- Naini A, Kaufmann P, Shanske S, Engelstad K, De Vivo DC, Schon EA. Hypocitrullinemia in patients with MELAS: an insight into the “MELAS paradox”. J Neurol Sci. 2005;229–230:187–193. doi: 10.1016/j.jns.2004.11.026. [DOI] [PubMed] [Google Scholar]
- Parfait B, de Lonlay P, von Kleist-Retzow JC, et al. The neurogenic weakness, ataxia and retinitis pigmentosa (NARP) syndrome mtDNA mutation (T8993G) triggers muscle ATPase deficiency and hypocitrullinaemia. Eur J Pediatr. 1999;158(1):55–58. doi: 10.1007/s004310051009. [DOI] [PubMed] [Google Scholar]
- Perry TL, Hansen S, Booth FA, Penn AM, Jones K, Dilling LA. An unusual aminoacidopathy associated with mitochondrial encephalomyopathy. J Inherit Metab Dis. 1989;12(1):23–32. doi: 10.1007/BF01805527. [DOI] [PubMed] [Google Scholar]
- Rabier D, Kamoun P. Metabolism of citrulline in man. Amino Acids. 1995;9(4):299–316. doi: 10.1007/BF00807268. [DOI] [PubMed] [Google Scholar]
- Rabier D, Diry C, Rotig A, et al. Persistent hypocitrullinaemia as a marker for mtDNA NARP T 8993G mutation? J Inherit Metab Dis. 1998;21(3):216–219. doi: 10.1023/A:1005391300203. [DOI] [PubMed] [Google Scholar]
- Ribes A, Riudor E, Valcarel R, et al. Pearson syndrome: altered tricarboxylic acid and urea-cycle metabolites, adrenal insufficiency and corneal opacities. J Inherit Metab Dis. 1993;16(3):537–540. doi: 10.1007/BF00711675. [DOI] [PubMed] [Google Scholar]