

Abstract
Pompe disease is a genetic disorder caused by a deficiency of acid α-glucosidase (GAA). Patients with classic infantile-onset Pompe disease usually present with hypertrophic cardiomyopathy and die before 1 year of age, if not treated with enzyme replacement therapy (ERT). In comparison, patients with late-onset Pompe disease typically do not have hypertrophic cardiomyopathy. However, here we describe five patients who presented with hypertrophic cardiomyopathy but did not fit the criteria of classic infantile-onset Pompe disease. Their ages at diagnosis of cardiomyopathy were 1 month in two patients following detection of an audible cardiac murmur and 2–3 years in the three remaining patients. All patients survived for 5–8 years without ERT. Three patients died before the advent of ERT from causes other than congestive heart failure. One patient had a good response to ERT starting at 5 years of age. The sibling of one patient, who did not receive ERT and died at age seven, was diagnosed prenatally. At 3 months of age, the sibling had hypertrophic cardiomyopathy, and a muscle biopsy at that time revealed glycogen accumulation.
This case series demonstrates that Pompe disease is a continuum of disease, and the development of cardiomyopathy is not limited to classic infantile-onset Pompe disease. These patients do not fit into the discrete phenotypes of infantile- or late-onset Pompe disease, which may suggest reconsidering the nomenclature of Pompe disease.
Introduction
Pompe disease is a lysosomal storage disorder in which a deficiency of acid α-glucosidase (GAA) causes lysosomal glycogen accumulation in multiple tissues and cell types, most notably cardiac, skeletal, and smooth muscle cells (Hirschhorn and Reuser 2001). Pompe disease has been classified into two phenotypes: classic infantile onset (symptoms develop before 1 year of age with hypertrophic cardiomyopathy) and late onset (symptoms develop at or after 1 year of age without hypertrophic cardiomyopathy) (Kishnani et al. 2006b). About 75% of patients with classic infantile-onset Pompe disease die before 12 months of age with a median age at death of 8.7 months (Kishnani et al. 2006a). Patients with the heterogeneous and more slowly progressive late-onset Pompe disease, which includes childhood-, juvenile-, and adult-onset subgroups (Kishnani et al. 2012), typically present with muscle weakness and respiratory failure but no cardiac manifestations (Hirschhorn and Reuser 2001; Kroos et al. 2007). However, Pompe disease is a continuum of disease with variable age of onset, organ involvement, and degree of myopathy, and therefore, not all patients can be categorized according to the aforementioned classification.
Enzyme replacement therapy (ERT) with recombinant human alglucosidase alfa (Myozyme®, Genzyme, Cambridge, MA) is the only approved therapy for Pompe disease. ERT prolonged overall survival and ventilator-free survival and reversed cardiomegaly in infantile-onset Pompe disease (Kishnani et al. 2007). Motor function was maintained when infants with classic infantile-onset Pompe disease were diagnosed through newborn screening and received very early treatment with ERT (Chien et al. 2008; Chien et al. 2009). Primarily based on data from juvenile- and adult-onset patients, ERT for late-onset Pompe disease has been associated with improved motor capability and stabilized pulmonary function (Winkel et al. 2004; Case et al. 2008; van Capelle et al. 2008; Strothotte et al. 2010; van der Ploeg et al. 2010). There is limited data about the effect of ERT in children with Pompe disease (Strothotte et al. 2010; van der Ploeg et al. 2010), who present with symptoms far earlier than juvenile-onset Pompe patients. In these children early symptom presentation is possibly associated with a poor treatment response.
In children, Pompe disease may include at least 3 groups of patients who survive more than one year: (1) nonclassic infantile-onset Pompe disease with cardiomyopathy (Winkel et al. 2005), (2) nonclassic infantile-onset Pompe disease without cardiomyopathy (Spiridigliozzi et al. 2012), and (3) juvenile-onset Pompe disease with delayed motor developmental milestones during childhood. In a review of 225 published cases, survival for nonclassic infantile-onset Pompe disease with cardiomyopathy was usually only 2 to 3 years without ERT (Winkel et al. 2005). However, we observed a group of patients who presented with cardiomyopathy and survived well beyond 1 year without ERT. This finding significantly broadens our understanding about childhood-onset Pompe disease and demonstrates a need to monitor the cardiac status of these children.
Case Presentations
During the first Asia-Pacific Pompe Disease Expert Meeting, held in Taipei in June 2013, cases of infants and young children with an atypical clinical course were noted. Subsequently, we performed a survey to further understand the characteristics of this group of patients. Criteria for enrollment were (1) disease onset during late infancy or early childhood; (2) presence of hypertrophic cardiomyopathy, as defined by a thickened left ventricular wall or interventricular septum; and (3) survival without ERT for a few years. In this case series, we describe five such patients and one sibling who was diagnosed prenatally. The characteristics of these patients are described in Table 1.
Table 1.
Characteristics of patients with Pompe disease
| No. | Sex | Age at detection of cardiomegaly | Age at onset of weakness | Creatine kinase level (U/L) | Age at start of ERT | Status at start of ERT | Current age (age died) | Current status (at death) | Mutations |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 2 years 6 months | 2 years | 1,235 | 5 years | Walk with support | 13 years | Walk freely | c.875A > G/c.1857C > G |
| 2A | F | 2 years | 5 years | Elevated | – | – | (7 years) | (Walk with support) | c.872T > C/c.1798C > T* |
| 2B | M | 3 months | – | 744 | 3 months | Normal | 2.5 years | Walk freely | c.872T > C/c.1798C > T* |
| 3A | F | 1 month | 1 year 3 months | 741 | 8 years | Walk with support | 15 years | Unable to walk | c.2407_13del/c.1316T > A* |
| 3B | M | 1 month | 5 years | 562 | – | – | (6 years) | (Walk with support) | c.2407_13del/c.1316T > A* |
| 4 | F | 3 years | 3 years | 104 | – | – | (7 years) | (Unable to walk) | c.1082C > T*/c.1432G > A* |
*reported mutation
Patient 1
Patient 1 is a 13-year-old male who displayed poor feeding and decreased activity in the first month after birth (Jeon et al. 2007). He had difficulty with standing at 2 years of age. Upon physical examination for a common cold, at the age of 2 years and 6 months, a cardiac murmur and cardiomegaly were discovered. He was diagnosed with Pompe disease at age 4 based on low GAA activity and the presence of two GAA gene mutations [c.875A>G (p.Tyr292Cys) and c.1857C>G (p.Ser619Arg)]. At the time of diagnosis, he showed Gowers’ sign and waddling gait. His serum creatinine kinase (CK) level was 1,235 U/L (normal < 165 U/L). A chest X-ray revealed cardiomegaly (Fig. 1a), and an echocardiogram revealed hypertrophic cardiomyopathy. The interventricular septal diastolic dimension was 24.8 mm (normal 3.3–6.3 mm). Left ventricular mass index (LVMI) was 484 gm/m2 (normal <65 g/m2). Alglucosidase alfa, 20 mg/kg qow, was started at 5 years of age. After 10 weeks of ERT, he could walk downstairs without using the handrail. He could walk for 1 h after 40 weeks of ERT. Currently, he has been treated for 8 years.
Fig. 1.
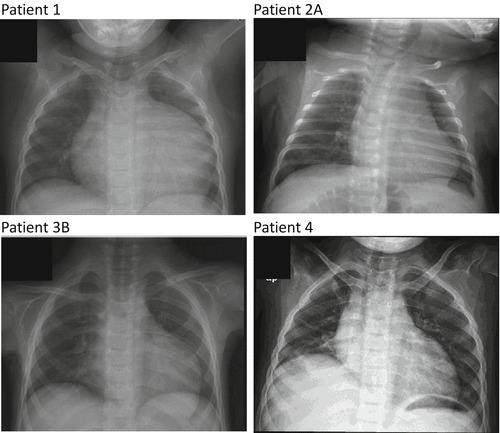
Chest X-ray pictures of four Pompe patients revealed cardiomegaly at ages: (a) 4 years (patient A), (b) 3 months (patient 2A), (c) 1 year and 3 months (patient 3B), and (d) 3 years (patient 4)
Patients 2A and 2B
Patient 2B is a 2.5-year-old male who was diagnosed with Pompe disease through prenatal screening. His elder sister (patient 2A) was noted to have elevated liver enzymes since birth. Elevated CK levels and mild cardiomegaly were documented in patient 2A when she was 2 years old. Patient 2A was prone to falls from the age of 5 years, and she died suddenly in her sleep at age 7. At that time, she had dysarthria (she was hardly understood by her teachers) and walked with support with a myopathic gait. Consequently, the mother received prenatal testing during her pregnancy with patient 2B, which confirmed the presence of two GAA gene mutations [c.872T>C (p.Leu291Pro) and c.1798C>T (p.Arg600Cys)] in the fetus. At birth, a chest X-ray revealed that patient 2B had normal cardiac size. However, at 3 months of age, patient 2B presented with hypotonia, absence of head control, and macroglossia. His CK level was 744 U/L (normal <200 U/L). A chest X-ray revealed cardiomegaly (Fig. 1b), and echocardiography revealed hypertrophic cardiomyopathy. LVMI was 126 g/m2 (normal <65 g/m2). ERT was initiated immediately. After 30 months of ERT, his cardiac size decreased, and improvements in motor function were observed across all motor scales.
Patients 3A and 3B
Patient 3A is a 15-year-old female who was noted to have a cardiac murmur at 1 month of age (Kim et al. 2009). Echocardiography revealed hypertrophic cardiomyopathy. She started to walk at 15 months of age with her right leg lagging. A follow-up echocardiography revealed asymmetric septal hypertrophy and systolic anterior motion of the mitral valve. LVMI was 94 g/m2 (normal <65 g/m2). Pompe disease was confirmed at 8 years of age based on low GAA activity and the presence of two GAA gene mutations [c.2407_13del and c.1316T>A (p.Met439Lys)]. Her CK level was 741 U/L (normal <165 U/L). At diagnosis, she walked with difficulty due to continued lagging of the right leg, and she could not run. Patient 3A has been receiving ERT since 8 years of age. She is wheelchair bound.
Patient 3B is patient 3A’s younger brother who was found to have Pompe disease at 5 years of age after the diagnosis of his older sister. Although a cardiac murmur was heard at 1 month of age, and CK elevation and hypertrophic cardiomyopathy were noted at 15 months of age (Fig. 1c), LVMI was 142.2 g/m2 (normal <65 g/m2). At the time of diagnosis of Pompe disease, he showed Gowers’ sign, waddling gait, and CK elevation (562 U/L, normal <165 U/L). He died at 6 years of age after an infection.
Patient 4
Patient 4 is a female who started to walk at 15 months of age, but fell easily starting at 3 years of age. At that time, chest X-ray revealed cardiomegaly (Fig. 1d), and echocardiogram and magnetic resonance imaging demonstrated left ventricular hypertrophy with left ventricular outflow tract obstruction. LVMI was 188 g/m2 (normal <95 g/m2). Her CK level was 104 U/L (normal < 170 U/L), and she had two GAA gene mutations [c.1082C>T (p.Pro361Leu) and c.1432G>A (p.Gly478Arg)]. She was able to walk 100 m at 5 years of age but could walk only 30 m at 6 years of age with coarse breath sounds, bilateral wheezing, and decreased oxygen saturation (80%) in room air. She was unable to walk at 7 years of age and died suddenly at age 7 years and 3 months.
Discussion
In this report, we describe a group of children with Pompe disease who presented with cardiomyopathy. Our findings support the concept that, in addition to classic infantile-onset Pompe disease, older children with Pompe disease can also present with cardiomyopathy. These patients do not fit into the discrete phenotypes of infantile- or late-onset Pompe disease, but may be called nonclassic, nontypical, or atypical infantile-onset Pompe disease.
The presence of cardiomyopathy in older children with Pompe disease may not necessarily indicate a poor prognosis similar to classic infantile-onset Pompe disease. In a review of nonclassic Pompe disease, the mean age of death in 32 patients with symptom onset at 0–1 year was 6.1 (0.9–24) years, and 12 of the patients presented with hypertrophic cardiomyopathy (Winkel et al. 2005). Although the cardiomyopathy in our patients occurred as early as 1 month of age, no patients died of congestive heart failure. However, arrhythmias are still a risk and could have contributed to the acute death of patients 2A and 4.
The current nomenclature used to describe individual patients with Pompe disease describes discreet phenotypes like infantile- or late-onset Pompe disease. These phenotypic designations are oversimplified since patients with infantile-onset Pompe disease can either present with cardiomyopathy before 6 months of age and die before 1 year of age (classic infantile-onset Pompe disease) or present with only weakness after 6 months of age and survive well beyond 1 year of age (nonclassic infantile-onset Pompe disease). The nomenclature for nonclassic infantile-onset Pompe disease is also variable. Slonim et al. defined “nontypical” infantile-onset Pompe disease as those with less severe cardiomyopathy and longer survival of 1–2 years (Slonim et al. 2000). Others have used “atypical infantile-onset Pompe disease” to characterize patients who have symptom onset before 1 year of age but with no cardiomyopathy (Bembi et al. 2008; Kishnani et al. 2012). Gungor and Reuser suggested using the term “childhood” Pompe disease to cover the gap between “classic infantile” and “adult” Pompe disease (Gungor and Reuser 2013).
In conclusion, cardiomyopathy cannot be used as a single criterion for the classification of Pompe disease in infants or children. For prospective follow-up of asymptomatic patients with Pompe disease, like those discovered by newborn screening, cardiomegaly needs to be monitored even after 1 year of age.
Take-Home Message
Hypertrophic cardiomyopathy may present in childhood in patients with Pompe disease.
Compliance with Ethics GuidelinesConflict of Interest
Yin-Hsiu Chien has received research grants from Genzyme, a Sanofi company.
Wuh-Liang Hwu has received research grants and traveling funds from Genzyme, a Sanofi company.
Dr Dong-Hwan Lee, Dr Wen-Juan Qiu, and Dr Jeongho Lee have no financial disclosures to make with regard to the development or research of this manuscript and declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained as part of the Pompe Registry study in Soonchunhyang University Hospital and National Taiwan University Hospital. Informed consent was waived for retrospective medical information with no identifying information about patients from Xinhua Hospital included in the article.
Author Contribution to the Manuscript
Dong-Hwan Lee and Wuh-Liang Hwu planned and conducted this research, and Wen-Juan Qiu, Jeongho Lee, and Yin-Hsiu Chien contributed to the cases.
Footnotes
Competing interests: None declared
Contributor Information
Wuh-Liang Hwu, Email: hwuwlntu@ntu.edu.tw.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Bembi B, Cerini E, et al. Diagnosis of glycogenosis type II. Neurology. 2008;71(23 Suppl 2):S4–11. doi: 10.1212/WNL.0b013e31818da91e. [DOI] [PubMed] [Google Scholar]
- Case LE, Koeberl DD, et al. Improvement with ongoing Enzyme Replacement Therapy in advanced late-onset Pompe disease: a case study. Mol Genet Metab. 2008;95(4):233–235. doi: 10.1016/j.ymgme.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Chien YH, Chiang SC, et al. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics. 2008;122(1):e39–45. doi: 10.1542/peds.2007-2222. [DOI] [PubMed] [Google Scholar]
- Chien YH, Lee NC, et al. Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics. 2009;124(6):e1116–1125. doi: 10.1542/peds.2008-3667. [DOI] [PubMed] [Google Scholar]
- Gungor D, Reuser AJ. How to describe the clinical spectrum in Pompe disease? Am J Med Genet Part A. 2013;161A(2):399–400. doi: 10.1002/ajmg.a.35662. [DOI] [PubMed] [Google Scholar]
- Hirschhorn R, Reuser A (2001) Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Scriver C, Beaudet A, Sly W, Valle D (eds) The metabolic and molecular bases of inherited disease, vol 3. McGraw-Hill, New York pp 3389–3420
- Jeon YH, Eun BL, et al. Clinical improvement in a case of atypical infantile onset Pompe disease with enzyme replacement therapy. Korean J Pediatr. 2007;50(2):213–217. doi: 10.3345/kjp.2007.50.2.213. [DOI] [Google Scholar]
- Kim EH, Ko JM, et al. Two patients with atypical infantile Pompe disease presenting with hypertrophic cardiomyopathy. J Genet Med. 2009;6(2):161–165. [Google Scholar]
- Kishnani P, Hwu W, et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148(5):671–676. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Steiner RD, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8(5):267–288. doi: 10.1097/01.gim.0000218152.87434.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani P, Corzo D, et al. Recombinant human acid {alpha}-glucosidase. Major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68(2):99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Beckemeyer AA, et al. The new era of Pompe disease: advances in the detection, understanding of the phenotypic spectrum, pathophysiology, and management. Am J Med Genet Part C Seminars Med Genet. 2012;160(1):1–7. doi: 10.1002/ajmg.c.31324. [DOI] [PubMed] [Google Scholar]
- Kroos MA, Pomponio RJ, et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology. 2007;68(2):110–115. doi: 10.1212/01.wnl.0000252798.25690.76. [DOI] [PubMed] [Google Scholar]
- Slonim AE, Bulone L, et al. Identification of two subtypes of infantile acid maltase deficiency. J Pediatr. 2000;137(2):283–285. doi: 10.1067/mpd.2000.107112. [DOI] [PubMed] [Google Scholar]
- Spiridigliozzi GA, Heller JH, et al. Cognitive and adaptive functioning of children with infantile Pompe disease treated with enzyme replacement therapy: long-term follow-up. Am J Med Genet Part C Seminars Med Genet. 2012;160(1):22–29. doi: 10.1002/ajmg.c.31323. [DOI] [PubMed] [Google Scholar]
- Strothotte S, Strigl-Pill N, et al. Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J Neurol. 2010;257(1):91–97. doi: 10.1007/s00415-009-5275-3. [DOI] [PubMed] [Google Scholar]
- van Capelle CI, Winkel LP, et al. Eight years experience with enzyme replacement therapy in two children and one adult with Pompe disease. Neuromuscul Disord. 2008;18(6):447–452. doi: 10.1016/j.nmd.2008.04.009. [DOI] [PubMed] [Google Scholar]
- van der Ploeg AT, Clemens PR, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362(15):1396–1406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- Winkel LP, Van den Hout JM, et al. Enzyme replacement therapy in late-onset Pompe's disease: a three-year follow-up. Ann Neurol. 2004;55(4):495–502. doi: 10.1002/ana.20019. [DOI] [PubMed] [Google Scholar]
- Winkel LP, Hagemans ML, et al. The natural course of non-classic Pompe’s disease; a review of 225 published cases. J Neurol. 2005;252(8):875–884. doi: 10.1007/s00415-005-0922-9. [DOI] [PubMed] [Google Scholar]