

Abstract
Investigations of lipid membranes using NMR spectroscopy generally require isotopic labeling, often precluding structural studies of complex lipid systems. Solid-state 13C magic-angle spinning NMR spectroscopy at natural isotopic abundance gives site-specific structural information that can aid in the characterization of complex biomembranes. Using the separated local-field experiment DROSS, we resolved 13C-1H residual dipolar couplings that were interpreted with a statistical mean-torque model. Liquid-disordered and liquid-ordered phases were characterized according to membrane thickness and average cross-sectional area per lipid. Knowledge of such structural parameters is vital for molecular dynamics simulations, and provides information about the balance of forces in membrane lipid bilayers. Experiments were conducted with both phosphatidylcholine (dimyristoylphosphatidylcholine (DMPC) and palmitoyloleoylphosphatidylcholine (POPC)) and egg-yolk sphingomyelin (EYSM) lipids, and allowed us to extract segmental order parameters from the 13C-1H residual dipolar couplings. Order parameters were used to calculate membrane structural quantities, including the area per lipid and bilayer thickness. Relative to POPC, EYSM is more ordered in the ld phase and experiences less structural perturbation upon adding 50% cholesterol to form the lo phase. The loss of configurational entropy is smaller for EYSM than for POPC, thus favoring its interaction with cholesterol in raftlike lipid systems. Our studies show that solid-state 13C NMR spectroscopy is applicable to investigations of complex lipids and makes it possible to obtain structural parameters for biomembrane systems where isotope labeling may be prohibitive.
Introduction
Lipid-protein interactions (1) and the associated functions of biomembranes (2–6) are known to be significantly influenced by the composition (2,7–11) and structure of the lipid bilayer (6,11–16). Recently, the importance of lipids in cellular membranes and tissues has made lipidomics (17) an emerging field in biomedical research. Essential roles of membrane lipids are brought out by the sphingolipids, which are implicated in human disorders including Tay-Sachs, Niemann-Pick, Gaucher (18), Parkinson’s (19), and Huntington diseases (20). Sphingolipids are attracting much attention to so-called lipid rafts in cellular membranes (4,9,21–27). Moreover, in vitro studies of integral membrane proteins or peptides often require that they be reconstituted with membrane lipids (19,28–30), where bilayer structural dimensions involving hydrophobic matching and area per lipid are significant factors (1,14,31). The constraints imposed by the area per lipid at the membrane aqueous interface (31) also play an important part in validating molecular dynamics (MD) simulations of lipid bilayers (32) and biomembranes (13,33,34). In such cases, it is essential that investigators have a solid understanding of the membrane bilayer structure itself (35).
Despite advances in technology (19,36–45), there is an enduring thirst for new methods on account of the strengths and limitations inherent in existing biophysical techniques. This work is aimed at extending such investigations by using for the first time to our knowledge a combination of separated local-field (SLF) NMR spectroscopy (41,46–48) and statistical mean-torque theory (35). Studies of natural lipids, as well as natural detergents (49), can benefit from extending methods originally developed for 2H NMR spectroscopy (50) to cases where 2H-isotope labeling is impractical. Simple geometrical observations are the basis of a mean-field model used for analysis of both small-angle x-ray scattering (SAXS) and solid-state 2H NMR data (51). The first-order mean-torque model (35) evaluates the average cross-sectional area and volumetric hydrocarbon thickness of the membrane lipids. For multicomponent membranes, the ensemble structures derived from the repeat spacings or electron densities in SAXS do not resolve the contributions from individual lipid species. However, average cross-sectional areas for multicomponent lipid mixtures are obtainable from residual quadrupolar couplings in solid-state 2H NMR spectroscopy, which typically requires isotopically labeled lipids for each component studied (52).
Here, we demonstrate the applicability of mean-field modeling of membrane structure in conjunction with a solid-state 13C NMR technique that does not require isotopic enrichment. Magic-angle spinning (MAS) NMR resolves site-specific details for individual lipid species in membranes (28,47,53–56), thus enabling a range of biologically significant applications. We show how the steric hydrocarbon thickness and average cross-sectional areas are derived from solid-state 13C-1H residual dipolar couplings (RDCs) for sphingolipids and phospholipids in single-component and cholesterol-enriched binary mixtures. Two-dimensional (2D) solid-state 13C NMR spectroscopy (46) can probe the headgroup, backbone, and acyl chain regions for membrane components simultaneously, including cholesterol at natural isotopic abundance (19,46,57). In addition, the membrane phase behavior is further quantified, giving a valuable probe of lipid interactions in bilayers, as well as in natural biomembranes.
Materials and Methods
Preparation of multilamellar lipid dispersions
1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and egg yolk sphingomyelin (EYSM) (predominant species N-(palmitoyl)-sphing-4-enine-1-phosphocholine) were procured from Avanti Polar Lipids (Alabaster, AL), and cholesterol (chol) was from Sigma-Aldrich (St. Louis, MO). The lipids were dissolved in hexane and lyophilized to yield dry powders. Multilamellar lipid dispersions were prepared by hydrating the dry lipid powders with 50 wt % deuterium oxide (Cambridge Isotopes, Cambridge, MA). These dispersions were subjected to three to five freeze-thaw mixing cycles to ensure homogeneity. Samples were tested for ester hydrolysis before and after the experiments using thin-layer chromatography (TLC) by elution with CHCl3/MeOH/H2O (65:30:5), followed by charring with 40% H2SO4 in ethanol.
Implementation of DROSS experiments
Separated local-field 13C MAS NMR experiments were conducted with a magnetic field strength of 11.7 T (500 MHz 1H Larmor frequency) using a Bruker AVANCE-I spectrometer system. The SLF experiment, involving dipolar recoupling on-axis with scaling and shape preservation (DROSS) (46), was implemented on the Bruker Topspin 2.1 software platform (Billerica, MA). A triple-channel MAS NMR probe (DSI-733) from Doty Scientific (Columbia, SC) was used for all experiments. Samples were contained in 4-mm zirconium rotors. Radiofrequency pulses for the 1H and 13C channels were adjusted to exactly the same duration, 3.5 μs for the 90° pulses. Dipolar recoupling at 6–8 kHz MAS frequency was achieved by applying four radiofrequency 180° pulses in one rotor period, with a chemical shift offset of ε = 0 and anisotropy scaling of χP = 0.393 (46,58). The INEPT (insensitive nuclei enhanced by polarization transfer) buildup of anti-phase magnetization and refocusing delays was optimized empirically. The rotor-synchronized sampling of the indirect dimension (t1) was achieved using the States method with a total of 64–128 points. The sampling of the direct time dimension (t2) utilized 8192 points recorded with an interval of 10 μs with 50-kHz 1H SPINAL-32 decoupling (59). Recycle times were 3 s, and between 1000 and 2000 transients were averaged for each t2 record, giving total experiment times ranging from 24–48 h. The fluctuations in rotor spinning speed were controlled to ±2 Hz with a Doty Scientific spin-rate controller. Sample temperature was controlled using a Bruker variable temperature unit and is accurate to ±1°C. The reported 13C NMR chemical shifts were referenced to TMS (external).
Fourier transformation of the t1 and t2 dimensions was carried out and analyzed using the Bruker Topspin software. A 10-Hz Lorentzian broadening was applied in the t2 dimension, and a 50- to 250-Hz Gaussian apodization was applied in the t1 dimension after zero-filling to 128–256 points. Fits to 13C SLF DROSS magnetic dipolar lineshapes were generated using the Topspin solid lineshape analysis (SOLA) software and verified using standard algorithms for the Pake lineshape coded in Matlab (Natick, MA). Segmental order parameters are defined as (50)
| (1) |
where βCH is the instantaneous angle between the 13C-1H bond direction and the bilayer normal. Based on geometrical considerations, the SCH order parameters for a polymethylene chain are negative.
Here, we refer to the absolute order parameters |SCH|, calculated from the relation
| (2) |
In the above formula, χD = (−μ0γHγCℏ/4π2)〈r−3〉 is the dipolar coupling constant (40.783 kHz corresponding to bCH/2π = 20.392 kHz) for an aliphatic 13C-1H bond (46), χP = 0.393 is the pulse-sequence scaling factor (46), and ΔνD is the measured RDC evaluated at the θ = 90° orientation of the lineshape (Pake powder pattern). The estimated random error of the RDCs is ≈±4%, corresponding to a ±4% error of the segmental order parameters. Possible sources of systematic errors include the value of the rigid-lattice (static) dipolar coupling constant as well as the chemical-shift assignments. Note that the above dipolar coupling constant corresponds to an effective equilibrium internuclear 13C-1H distance of 〈r−3〉−1/3 = 1.14 Å due to the correction for dynamic effects, as first shown in our previous work (60). For resonance assignments of the 13C isotropic chemical-shift spectra, ChemBioDraw (PerkinElmer, Waltham, MA)-based simulations based on additivity relations for isotropic liquids (61–65) were initially used. However, because the chemical shifts due to liquid-crystalline phase conformations are not averaged to their isotropic values, we introduced 2H NMR-derived |SCD| order-parameter-based peak assignments (43). In this article, we compare the calculated |SCH| order parameter values with |SCD| order parameters used for the peak assignments. Such methods are helpful in identifying the 13C resonance peaks coming from the hydrocarbon chains of the liquid-crystalline phospholipids.
Theoretical framework of mean-torque model
For membrane lipids, the dipolar or quadrupolar couplings are reduced from their rigid-lattice (static) values due to segmental and molecular motions as well as collective fluctuations of the entire bilayer (66). The various motions are described by their mean-squared amplitudes and (reduced) spectral densities, and they may include cross correlations due to their statistical (in)dependence (66–68). The segmental order parameters (SCH or SCD) describe the composite motion in terms of the residual couplings (dipolar or quadrupolar) as compared to the static values (43). The order parameters are model-free spectroscopic observables that are statistical averages over all the motions, with correlation times up to the inverse of the rigid-lattice coupling (50). In analogy with liquid crystals, each segment is considered independently, because the spectroscopic observables are site-specific (50,69). Correlations among the motions along the chains and between the chains are included in the order parameters as a function of segmental position (35,70). Interpretation of the segmental order parameters in terms of lipid bilayer structure was first achieved using a diamond-lattice model (71,72). That model assumes discrete orientations of acyl chain segments as the most probable conformations. Although initially it appeared to be in reasonably good agreement with scattering experiments, discrepancies between the area calculations from more accurate 2H NMR and x-ray scattering measurements became evident (73,74). The mean-torque model was proposed as an alternative (35), as it assumes a continuum distribution for the orientations of acyl chain segments. The mean-torque model successfully explains bilayer structural parameters for liquid-crystalline disaturated phospholipid membranes (35).
In this work, we introduce the applicability of the mean-torque model in connection with 13C-NMR-derived segmental order parameters to interpret the membrane structure. Here, the main objective is connecting the lipid structural parameters to hydrocarbon-chain segmental order parameters. Changes due to osmotic stress or to headgroup and acyl chain composition are related to the balance of forces that govern membrane assembly and lipid-protein interactions (1). For disaturated lipids, a plateau is seen in the 2H-NMR-derived segmental order parameter profile close to the headgroup. At a certain depth of the bilayer, the influence of chain terminations becomes important (75,76). Acyl chains on adjacent molecules become more disordered beyond this point to maintain the packing of hydrocarbon density (77). Another observation from 2H NMR order profiles is that the plateau region shows a strong chain-length dependence, whereas nonplateau regions are practically independent of chain length (35). At the molecular scale, each lipid in the membrane occupies a space that on average is related to the volume and length of the hydrocarbon chains according to
| (3) |
where DC is the volumetric thickness of the hydrocarbon layer, and VC is the total volume of an individual acyl chain. In Eq. 3, the volume, VC, is assumed from the densitometry measurements of Nagle and co-workers (78) and is conserved (i.e., constant). Note that the volumetric thickness, DC, and the mean area, 〈A〉, are inversely related by the assumption of constant volume, meaning that the bilayer core has approximately the density of liquid hydrocarbon (77,80). However, DC is not the same as the mean projected acyl length, as discussed by Jansson et al. (79). Due to end effects of the acyl chains, the mean travel away from the aqueous interface is less than the distance to the bilayer midplane, as required for the well-established assumption of constant volume to apply (35). The chain volume at temperature T is found from the methylene volume, , using the expression , where is the isobaric thermal expansion coefficient for methylene groups (35). It is well established that the volume of a methyl group is and that for the methine volume (78,80,81).
When the membrane composition is mixed, neighboring interactions between the lipids can lead to a change in the average cross-sectional area per lipid. To avoid complications from chain upturns (35,70), for estimating the average area per lipid instead of the area of the entire hydrocarbon chain, one can consider relatively ordered acyl segments near the headgroup region. For solid-state 13C NMR (43) the largest RDC corresponds to the plateau region of the 2H NMR order profiles, where it is reasonable to assume that the segmental cross-sectional area and projected length are inversely correlated, as discussed in Jansson et al. (79). The average cross-sectional area per lipid is thus given as (35)
| (4) |
Here is the methylene volume (78) and D is the instantaneous travel of an individual segment along the bilayer normal. This expression can be further rewritten as
| (5) |
where DM = 2.54 Å is the maximum projection of the virtual bonds connecting every second carbon atom in the polymethylene chain to the bilayer normal, and is the lipid cross-sectional area of the extended all-trans conformation (35). The area factor q is defined as 〈1/cos β〉−1, where β is the angle between the virtual bond axis connecting the Ci–1 and Ci+1 carbon atoms and the normal to the lipid bilayer surface.
In Eq. 5, Euclidean geometry is assumed by approximating the shape of a statistical segment by a geometrical prism with constant hydrocarbon volume (50). Consequently, the effective acyl segment length is averaged over the motions, whereas the segmental volume is not. As the motional amplitudes increase, so does the area per lipid, yet the volume per segment spanned in space remains approximately constant. It follows that a Taylor series expansion about the all-trans reference value allows the area factor q to be approximated by q ≈ 3 − 3〈cos βi〉 + 〈cos2 βi〉 up to second order (35). For 13C NMR, the second moment 〈cos2 βi〉 can be obtained directly from the absolute order parameters of a given acyl segment (index i) by
| (6) |
To interpret the |SCH| dipolar order parameters in terms of structural quantities, several models have been developed (35). Because of the inherent complexity of membrane structure, most of these models are confined to simplified statistical treatments of lipid conformations. Calculation of the first moment 〈cos βi〉 with a given value of 〈cos2 βi〉 is further described below.
The mean-torque model assumes that the orientational order for each chain segment relative to the local director is described by an orientational potential, U(β) (potential of mean torque). With the combined effects of thermal fluctuations of the segmental orientations, both segmental and molecular conformations assume a continuous distribution. The probability of finding a statistical segment with a virtual bond orientation β (≡ βCH) at a given instant is given by the Boltzmann distribution,
| (7) |
where the chain index i is also suppressed for clarity. Here, the partition function is
| (8) |
Assuming a first-order mean-torque model (35), the angle-dependent quantities are integrated with the distribution function to give the coupled equations
| (9) |
| (10) |
An analytical solution for 〈cos β〉 can then be obtained by using the approximation coth[U(β)/kBT] ≈ 1, which for an individual segment (index i) yields the relation
| (11) |
It should be noted that for the all-trans conformation of the lipids, 〈cos βi〉 = 〈cos2 βi〉 = 1 and hence q = 1. It follows that Eqs. 3 and 4 give rise to a limiting area of 4VCH2/DM and limiting monolayer thickness of nCDM/2, where nC is the number of carbon atoms in the hydrocarbon chain.
Each molecule contributes to the average thickness for the lipid bilayer ensemble, which includes the headgroup plus backbone thickness in the sense of a Gibbs dividing surface. The volumetric (Luzzati) bilayer thickness is DB = VL/〈A〉 where VL is the lipid volume (78) and DH = 4 Å for DMPC or POPC and 6 Å for EYSM. Alternatively, an effective membrane bilayer thickness can be calculated using the expression,
| (12) |
where DC is the volumetric hydrocarbon thickness of the two acyl chains of the lipid, and DH is the headgroup plus the backbone distance. In the case of phospholipids, DH is 9 Å, and for most sphingolipids it is 7 Å (82–84). Use of the above values of DH and DC corresponds to the steric bilayer thickness DB′ as defined by Nagle et al. (12). Finally, it is important to note that the first-order mean-torque model neglects the effects of collective slow motions (77). The above treatment of a mean-torque model was originally formulated in terms of 2H NMR-derived SCD values. However, we show equivalently that one can use SCH order parameters obtained from natural-abundance 13C NMR spectroscopy to extend the calculation of membrane structural parameters.
Results
The INEPT-based SLF NMR experiment DROSS (46) was implemented for the two glycerophospholipids DMPC and POPC together with the sphingolipid EYSM and their binary mixtures with cholesterol. The DROSS experiment provides measurements for the headgroup, glycerol backbone, and acyl chain order parameters without isotopic enrichment. This is especially useful when comparing responses from these bilayer regions to changes in bilayer composition. However, accurate measurement of small absolute order parameters, as in the case of headgroup segments (85), is challenging in SLF spectroscopy. As an example, Fig. 1 shows a 2D SLF spectrum of a POPC/cholesterol (1:1) binary mixture at 30°C. The representative slices of the 2D DROSS spectrum shown in Fig. 1 a indicate that well-resolved RDCs (Pake doublets) are obtained. The RDCs can be used to obtain dipolar 13C-1H order parameters that are analogous to the C-2H bond order parameters in solid-state 2H NMR spectroscopy (50). An expansion of the 2D DROSS spectrum from the aliphatic fingerprint region is shown in Fig. 1 b. The slices parallel to the F1 frequency axis for each of the chemically shifted (δ) resonances in the F2 dimension give the site-specific RDCs (Pake doublets). Fig. 1 c shows a contour plot of the 2D DROSS spectrum together with the F2 frequency projection corresponding to the 1D chemical-shift spectrum. A number of distinct 13C resonances are observed due to the POPC acyl groups as well as cholesterol. Notably, the (CH2)n envelope includes additional acyl chain resonances that are incompletely resolved due to inhomogeneous line broadening. They can be partially assigned by comparison to the results of solid-state 2H NMR spectroscopy (see below). Further explanation of the SLF experiment is provided in Figs. S1–S4 in the Supporting Material.
Figure 1.
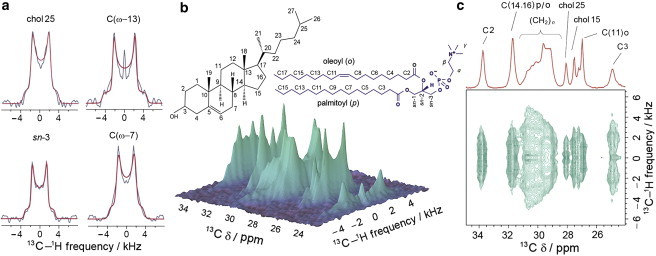
Site-specific 13C–1H RDCs are measured using a 2D DROSS spectrum for the case of the POPC/cholesterol (1:1) binary mixture at 30°C. (a) Selected recoupled powder patterns showing experimental (gray) and simulated (red) lineshapes. (b) Oblique view of the aliphatic fingerprint region of the DROSS spectrum of the binary system POPC/cholesterol. (c) A 2D plane of the spectrum shown in b. The 13C isotropic chemical shift (δ) spectrum is shown along F2 (the horizontal axis) (red). The peak separation of the Pake doublet yields the site-specific 13C-1H dipolar coupling along F1 (the vertical axis). Segmental |SCH| order parameter values are calculated as a function of peak position according to Eq. 2. To see this figure in color, go online.
Liquid-disordered phase of lipid bilayers
Our findings illustrate that SLF NMR spectroscopy is broadly applicable to a variety of synthetic and natural lipids. Fig. 2 a depicts a 2D plane of a DROSS spectrum for POPC in the liquid-crystalline state at 28°C. The F2 frequency dimension (horizontal) shows the isotropic 13C chemical shift (δ) spectra obtained under MAS. As mentioned above, the DROSS spectra contain Pake doublets corresponding to the 13C-1H dipolar couplings along the F1 frequency axis (vertical). The dipolar slices correspond to each of the isotropic 13C chemical-shift positions. A similar set of spectra representing EYSM at 48°C is shown in Fig. 2 b. Resonances from polar headgroups, glycerol backbone, and acyl chains of the lipids are clearly resolved. The 13C chemical-shift assignments were taken from the literature (86,87) and were verified by simulations based on additivity rules reported for isotropic liquids (61,62,64,65). Even so, in the spectral region between 29.5 ppm and 31.5 ppm there is considerable overlap of the resonances from lipid hydrocarbon chain segments.
Figure 2.
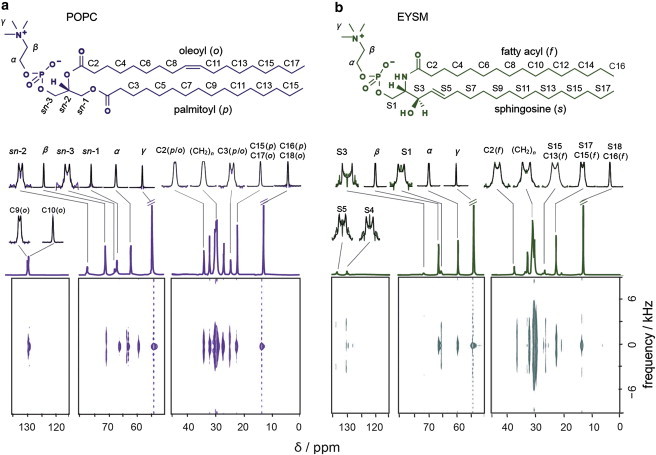
SLF 13C NMR spectroscopy of membrane lipids provides site-specific RDCs in the liquid-crystalline phase. 2D DROSS spectra show isotropic chemical-shift projections in the F2 (horizontal) dimension and 1H-13C dipolar couplings in the F1 (vertical) dimension for POPC at 28°C (a) and EYSM at 48°C (b). The Pake lineshapes corresponding to the dipolar splitting for each carbon chemical-shift position are shown above the isotropic chemical-shift spectra. To see this figure in color, go online.
Because literature chemical shifts are unavailable for individual carbons in this spectral region, initially we assigned those resonance peaks solely depending on 13C chemical shifts of isotropic liquids. For each of the resolved peaks, the spectra obtained for the F1 dimension were fit with Pake lineshapes to extract site-specific RDC values along the hydrocarbon chains, as well as for the lipid backbone and choline headgroup regions. The 1H-13C order parameter values were calculated according to Eq. 2. Results for the calculated |SCH| order parameters were then compared with the corresponding solid-state 2H-NMR-derived |SCD| values. Both the |SCH| and |SCD| order parameters were consistent for those segmental positions having well-defined 13C chemical-shift assignments. However, in the region between 29.5 ppm and 31.5 ppm, we observed a discrepancy between the |SCH| and |SCD| order parameters for the DMPC bilayer at 30°C (Figs. S5 and S6). The discrepancies are more pronounced in the case of POPC lipid bilayers, where overlapping resonances from the palmitoyl and oleoyl hydrocarbon chains further complicate the peak assignments.
Chemical-shift assignments of MAS carbon-13 NMR spectra
With MAS, the 13C chemical shifts of lipid bilayers are not averaged to liquid-phase isotopic chemical shifts. Instead, they correspond to the chemical-shift values averaged over the molecular conformations present in the liquid-crystalline phase. By contrast, the NMR chemical shifts for a particular site (i) in the solution state are averaged over all possible conformations because of the rapid molecular rotations and rotational isomerizations about the chemical bonds. Similar observations in proteins and polypeptides indicate that the 13C chemical shifts strongly depend on the average molecular conformation (88). The observed chemical shifts under MAS can be expressed for a given segment position as . The secondary chemical-shift values Δδ(i) are indicative of the molecular structures. For polymers, proteins, and polypeptides, a statistical analysis of Δδ(i) values provides an intrinsic probe for conformational characterization and secondary structure determination (88–90). An in-depth analysis of conformation-dependent chemical shifts in the case presented here requires theoretical calculations based on molecular orbital theory (91,92), which is beyond the scope of this work. One strategy for assigning the isotropic 13C NMR chemical shifts entails selective 2H isotopic labeling, leading to suppression of cross-polarization from the abundant 1H nuclei (45). An alternative entails comparison of the dipolar order parameters to the quadrupolar order parameters measured in 2H NMR spectroscopy, which we propose as a means of |SCD|-assisted 13C NMR spectral assignments.
In this work, we implemented the |SCD|-assisted 13C resonance assignments (see Figs. S6–S8) by making use of available 2H solid-state NMR data (43). The resulting |SCH| order profiles as a function of carbon position are shown in Fig. 3 for DMPC, POPC, and EYSM bilayers in the liquid-crystalline (also known as the liquid-disordered (ld)) state. The absolute order profiles show a decreasing trend as the peak (carbon) index (i) changes from the headgroup to the acyl chain methyl end. Tethering of the acyl chains to the aqueous interface results in order parameters that are higher near the headgroups than near the methyl ends of the acyl chain (93). In the case of 13C NMR for DMPC and EYSM (Fig. 3, a and d), the order parameters for the individual acyl chains were not separately identified. However, 13C NMR for POPC (Fig. 3, b and c) shows that the unsaturated carbons (C9 and C10) and allylic carbons (C8 and C11), as well as additional carbon positions from the sn-2 acyl chain, were distinct from the sn-1 chain 13C resonances. It follows that separate order profiles were determined for the palmitoyl and oleoyl chains (Fig. 3, b and c). Comparison of the dipolar |SCH| order parameters to the quadrupolar |SCD| order parameters is given in Figs. S7 and S8 for POPC in the ld phase. For EYSM, assignments of the chemical shifts to the sphingosine backbone carbon positions were limited to the C1, C3, C4, and C5 sites. The C2 position was not observed, due either to line broadening caused by quadrupolar coupling to the 14N nucleus or to its close chemical-shift proximity to the intense γ choline methyl position.
Figure 3.
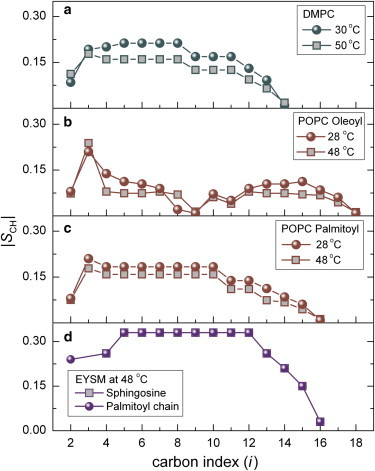
Segmental |SCH| order parameters are derived from SLF 13C NMR spectroscopy for lipid bilayers in the ld phase. Absolute order profiles are plotted with magnitude decreasing as a function of peak (carbon) index (i). Results are shown for DMPC (a), the sn-2 oleoyl chain of POPC (b), the sn-1 palmitoyl chain of POPC (c), and the sphingosine and palmitoyl chains of EYSM (d). Order parameters for the individual chains are not separately presented for DMPC and EYSM. See individual profiles for symbol codes. Relatively high order parameters for the EYSM sphingosine and palmitoyl chains indicate that it is less flexible than the other lipids in the liquid-crystalline (ld) state. The |SCH| order parameters represent the 13C-1H bond orientational distributions with respect to the membrane normal and are used to calculate the area per lipid within the bilayer. To see this figure in color, go online.
The above findings show that solid-state 13C NMR at natural abundance is complementary to 2H NMR spectroscopy of membrane lipid bilayers (50). Bilayer structural properties including the volumetric hydrocarbon thickness and area per lipid can be obtained without the need for 2H-labeling, which can be time-consuming, expensive, and otherwise prohibitive (see below). The 13C chemical-shift assignments, together with the dipolar couplings and segmental order parameters, are summarized in Tables S1–S5. Fig. 4 shows a comparison between the DROSS-derived |SCH| and |SCD| values for DMPC. For the DMPC bilayer at 50°C, in the liquid-crystalline (ld) state, the nearly unit slope for the plot of |SCD| versus |SCH| (Fig. 4, a and b) indicates the consistence of both the 2H solid-state NMR and 13C SLF NMR results in calculating the lipid segmental order parameters. The |SCD|-assisted assignments of the solid-state 13C NMR chemical shifts lead to dipolar |SCH| order parameters that are generally in good agreement with the results of corresponding 2H NMR experiments (35). However, the dynamic timescales that average out these interactions are dissimilar, owing to the difference between the rigid-lattice coupling constants (40.7 kHz for 13C nuclei and 167 kHz for 2H nuclei) (60,94). For DMPC at 30°C and for POPC, there are some additional deviations that could be due either to misassignment of some of the peaks or to different motional scalings of the lineshapes that may require future investigation (Figs. S5, S7, and S8).
Figure 4.
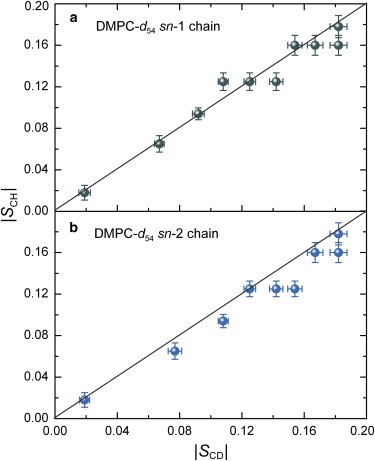
SLF 13C NMR spectroscopy gives segmental |SCH| order parameters that agree with 2H solid-state NMR-derived |SCD| order parameters. Data are shown for the sn-1 (a) and sn-2 (b) chains of DMPC-d54 at 50°C in the liquid-crystalline (ld) phase. The solid line represents a unit slope. Data for the C2 segment position are not included because of the different order parameters for the initial segment of the acyl chain. The error bars correspond to the standard deviations in the measured RDCs in multiple DROSS experiments. To see this figure in color, go online.
Cholesterol-containing lipid bilayers in the lo phase
One striking feature we consider in this work is the drastic increase of absolute order parameters for acyl chain segments of liquid-ordered (lo) bilayers containing cholesterol versus ld bilayers (95). Here, it is possible that the cholesterol may exert its effects through either the chains, the headgroup, or possibly both regions of the lipids. To better understand the structural perturbations indicated by the RDCs, we performed DROSS experiments on POPC and EYSM bilayers in the lo phase containing 50 mol % cholesterol. Using the INEPT polarization transfer for liquid-crystalline samples, chemically shifted resonances are prominent for the distal alkyl chain positions because of the effective magnetization transfer. For relatively immobile molecular regions with large static dipolar couplings, several sterol ring positions are detected by Hartmann-Hahn cross-polarization techniques (96,97) that helped in assigning many positions of the sterol ring and alkyl chain chemical shifts of cholesterol (54). In general, the RDC lineshape quality is limited by the signal/noise ratio afforded by the chemically shifted resonances.
Representative order-parameter profiles showing the influence of cholesterol for the lo phases of POPC and EYSM are provided in Fig. 5. The higher magnitudes of |SCH| values for both lipid bilayers at various carbon positions are an indication of the lo phase (8,98–101) due to interaction with cholesterol (95,102,103). The nonequivalence of the segments of the sn-1 and sn-2 chains observed in 2H solid-state NMR experiments (104) is clearly reflected in the case of POPC (Fig. 5, a and b). There is a decreasing trend in |SCH| from the upper acyl positions toward the distal end of the chain. At the uppermost acyl sites (C2 segment of oleoyl and palmitoyl chains), an initial downturn effect corresponds to RDCs that are reduced compared with nearest-neighbor carbon positions. In addition, monounsaturation of the oleoyl chain at the C9 and C10 sites renders the two vinyl 13C-1H positions orientationally nonequivalent both to each other and to the other saturated chain segments (104). The terminal methyl groups of the acyl chains exhibit a very small RDC because of the reorientation and threefold symmetry of the methyl 13C-1H bonds. For EYSM, most notable are the |SCH| values associated with the sphingosine backbone sites (Fig. 5, c and d). The largest couplings of these sites are observed at the upper C3 position, which may participate in interfacial exchange-type C3-OH hydrogen bonding and/or in C3-OH acceptor and NH donor hydrogen bonding. The large value is suggestive of a static backbone orientation assisted by hydrogen-bonding in the ld phase and stabilized through lipid packing.
Figure 5.
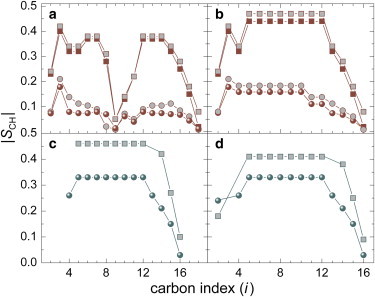
Segmental order parameter |SCH| profiles from SLF 13C NMR spectroscopy indicate a lipid-specific loss of conformational disorder due to cholesterol. Order profiles are plotted in terms of decreasing absolute magnitude for the sn-2 oleoyl chain of POPC (a), the sn-1 palmitoyl chain of POPC (b), the sn-2 sphingosine chain of EYSM (c), and the sn-1 palmitoyl chain of EYSM (d). Circles represent pure lipids and squares represent lipid mixtures with cholesterol (1:1). For POPC, data are shown at two temperatures, 28°C (gray-filled symbols) and 48°C (solid symbols), and for EYSM at 48°C. Note that upon adding cholesterol the |SCH| order parameters increase more in POPC than in EYSM. To see this figure in color, go online.
Discussion
Separated local-field spectroscopy (45–47) at natural 13C abundance expands the range of applications of solid-state NMR in membrane biophysics in significant new ways. The development and use of biophysical methods for biomembranes can benefit by extending the 2H NMR approach (50) to cases where 2H-isotope labeling is impractical. Sphingolipids and other natural lipids can be investigated, together with their interactions both with cholesterol in raftlike lipid mixtures and with membrane proteins. It has been proposed that in complexes with cholesterol and/or receptor proteins, sphingolipids form functional microdomains in putative lipid rafts (9,19,105,106). Such membrane lipids in the brain and other organs are associated with second-messenger events implicated in intracellular signaling and cholesterol shuttling (23,107,108). Applications of solid-state 13C NMR methods to polyunsaturated lipid bilayers (72,109) have also been described. In these examples and others, we are interested in the relationship between molecular properties of membrane lipids and their biological functions within the broader context of structural biophysics (1,5,6,11).
13C-1H dipolar couplings allow calculations of lipid bilayer structure
The DROSS experimental method (46) allows measurement of the direct 13C-1H dipolar couplings in liquid-crystalline systems, such as lipid bilayers at natural isotopic abundance. Through-space direct 13C-1H dipolar interactions report on the orientation of the individual 13C-1H bonds with respect to the bilayer normal, and they are mathematically isomorphous to the C-2H bond order parameters measured by 2H solid-state NMR spectroscopy. Moreover, unlike 2H solid-state NMR, this technique makes it possible to obtain the average orientation of C-H bond segments without isotopic labeling. Analogous studies have also been performed using switched-angle spinning and off-MAS experiments (110). However, DROSS has multiple advantages over the other experiments mentioned here. For instance, implementation of DROSS does not require specialized hardware to control the orientation of the rotor during the experiment. Another advantage is the ability of DROSS to define the sign of the dipolar coupling with a single set of scaling factors (46). The experiment takes advantage of the larger chemical-shift dispersion of the 13C nucleus that enables assignment to be made to specific carbon atoms. Despite these advantages, however, the DROSS experiment for lipid membranes suffers from significant 13C signal superposition, thus limiting the precision of peak assignments and measurement of assigned order parameters. On the other hand, ω–3 polyunsaturated lipid acyl groups show better resolution of 13C chemical shifts (111), so that DROSS can be conveniently applied as shown by Gawrisch and co-workers (47).
Dipolar order parameters reflect structural and dynamic features of phospholipids and sphingolipids
These studies aim at determining the membrane structural parameters using the largest absolute order parameter as typical of the lipid segments near the headgroup. Because DROSS can determine these order parameters unambiguously, it is a suitable technique for such structural studies. The DROSS experiment enables the order parameters to be assigned to specific carbon atoms in the ld phase at natural isotopic abundance (see Fig. 1). In the case of the disaturated lipid DMPC, there is significant overlap of 13C resonances from the middle of the hydrocarbon chain, yet we could resolve four to five peaks for eight of the carbons (C4–C11). Detailed assignments are not available for this crowded spectral region. First, we assigned these peaks by calculating the chemical shifts using the additivity rules proposed for isotropic liquids (61–65). With such assignments, the measured |SCH| order parameters for DMPC lipids agreed well with solid-state 2H-NMR-determined |SCD| values (43,112). The overlapped methylene carbon resonances around 30.5 ppm are also incompletely resolved in 2H NMR, and lead to a plateau region in the order profile. A similar assignment strategy did not work for POPC, as the signals from saturated and unsaturated acyl chains significantly overlap. However, by comparing the absolute |SCH| order parameters with 2H-NMR-derived |SCD| order parameters, we could identify the carbon chemical-shift positions. For POPC, the carbon resonances of the unsaturated region (C9 and C10) and the immediately adjacent allylic carbons (C8 and C11) were clearly identified. The order parameter values for the unambiguously identified carbon positions of the palmitoyl and oleoyl chains were consistent with the corresponding |SCD| values. The lower order parameter values for the unsaturated carbon positions compare favorably with those from 2H NMR and MD simulations (93). Notably, EYSM showed higher order parameters than the other lipid bilayers studied, indicating its lower flexibility in the single-component bilayer. Complete peak assignments were not essential for the structural parameter calculations. Thus, the highest order parameter shown by the acyl segments, which corresponds to the plateau value of the 2H NMR order profile, was chosen for structural calculations.
The mean-torque model explains ordering of sphingolipids versus phospholipids
Next, we used dipolar 13C-1H order parameters in combination with the mean-torque model (35) to determine the average cross-sectional area per lipid, 〈A〉, and volumetric hydrocarbon chain thickness, DC, of the DMPC bilayer in the ld phase, and for POPC and EYSM in both the ld and lo phases. This structural model has previously been used successfully with 2H NMR order parameters (35,74,113). To avoid complications from chain upturns, the largest dipolar order parameters are used for solid-state 13C NMR; these values correspond to the plateau value used in treating the 2H NMR data. Comparison of the bilayer structural parameters from 13C NMR shows good agreement with the corresponding 2H NMR data (Fig. 4). Referring to Fig. 6, for DMPC in excess water at 30°C, we find an average cross-sectional area per lipid of 〈A〉 = 59.5 Å2. Note that this value compares favorably to those found using x-ray scattering and 2H solid-state NMR spectroscopy (83). Such agreement for the canonical DMPC bilayer allows us to extend this technique to additional lipid bilayer systems. The main structural results for the different samples are summarized in Table 1. For POPC, we obtain an area per lipid, 〈A〉, of 70.5 Å2 at 48°C. By including contributions from the lipid polar headgroups of ≈9 Å/monolayer leaflet (83,84), according to Eq. 4 we arrive at an effective POPC bilayer thickness of DB′ ≈ 46.6 Å at 48°C. Note that the choice of 9 Å corresponds to the steric bilayer thickness as defined by Nagle et al. (12). In the study described here, we found that EYSM exhibits a strikingly smaller area per lipid of 〈A〉 = 51.5 Å2 at 48°C. This value is significantly larger than the reported x-ray values but close to the complementary 2H NMR result of 54.8 Å2 (8). The total bilayer thickness calculated was DB′ = 49.0 Å, which compares favorably with both small-angle x-ray measures (114) and 2H NMR values (8). The discrepancy between the 13C-1H and x-ray cross-sectional areas likely reflects the assumptions implicit in the determination of the areas from electron densities (12). Fig. 6, a and b, compares the structural measurements 〈A〉 and DB′ in the ld phases of DMPC, POPC, and EYSM lipid bilayers determined using the different techniques. A nearly unit slope for the plots of solid-state 13C versus 2H NMR-derived values is a clear indication of the consistence of these two techniques in determining lipid structural parameters.
Figure 6.
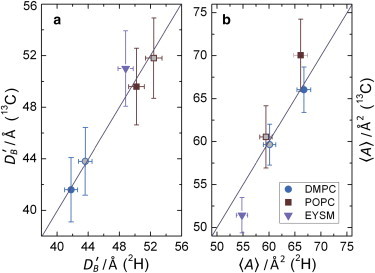
Bilayer structural parameters obtained from SLF 13C NMR spectroscopy agree with results from solid-state 2H NMR spectroscopy of lipids that have perdeuterated acyl groups. Parameter values obtained using 2H solid-state NMR are compared with those obtained using the SLF 13C NMR experiment DROSS for bilayer thickness, DB′ (a), and area per lipid, 〈A〉 (b). For DMPC and POPC bilayers, structural data are shown at temperatures of 30°C (solid circles) and 50°C (gray-filled circles), and at 28°C (squares) and 48°C (gray-filled squares), respectively. Cross-sectional lipid areas are calculated using the plateau |SCD| value in the case of 2H NMR and the highest |SCH| value of the acyl segments in the case of 13C NMR spectroscopy. The unit slope indicates excellent agreement of the two methods. Note that isotopic labeling is not required in the case of solid-state 13C NMR spectroscopy. The estimated error bars correspond to the standard deviations in the measured RDCs in multiple DROSS experiments. To see this figure in color, go online.
Table 1.
Structural parameters for lipid bilayers obtained from solid-state 13C and 2H NMR spectroscopy
| Lipid | T (°C) |
DC (Å)a |
DB′ (Å)a |
〈A〉 (Å2)a |
|||
|---|---|---|---|---|---|---|---|
| 13C | 2H | 13C | 2H | 13C | 2H | ||
| DMPC | 30 | 12.9 ± 0.5 | 12.8 ± 0.3 | 43.8 ± 2.6 | 43.6 ± 0.9 | 59.5 ± 3.6 | 60.0 ± 1.2 |
| 50 | 11.4 ± 0.4 | 11.9 ± 0.2 | 41.6 ± 2.5 | 41.8 ± 0.8 | 68.2 ± 4.0 | 66.1 ± 1.3 | |
| POPC | 28 | 14.8 ± 0.6b | 13.6 ± 0.3b | 51.8 ± 3.1 | 52.4 ± 1.1 | 60.4 ± 3.6 | 59.3 ± 1.2 |
| 16.7 ± 0.7c | 15.3 ± 0.4c | ||||||
| 48 | 12.8 ± 0.5b | 14.2 ± 0.3b | 46.6 ± 2.8 | 50.2 ± 1.0 | 70.5 ± 4.2 | 66.2 ± 1.3 | |
| 14.4 ± 0.6c | 16.0 ± 0.3c | ||||||
| EYSM | 48 | 17.5 ± 0.7b | 16.5 ± 0.4 | 51.0 ± 3.1 | 48.8 ± 1.0 | 51.5 ± 2.1 | 54.9 ± 1.1 |
| 17.0 ± 0.7c | — | ||||||
| POPC/Cholesterol (1:1) | 48 | 17.8 ± 0.7 | — | 57.8 ± 3.5 | — | 45.1 ± 0.9 | — |
| 19.4 ± 0.8 | — | ||||||
| EYSM/Cholesterol (1:1) | 48 | 17.5 ± 0.7 | — | 53.5 ± 3.2 | — | 45.5 ± 2.7 | — |
| 18.0 ± 0.7 | — | ||||||
Error bars correspond to the standard deviations propagated from multiple experimental measurements of RDCs.
sn-1 hydrocarbon chain.
sn-2 hydrocarbon chain.
Configurational entropy governs mixing of sphingolipids and phospholipids with cholesterol
Solid-state 2H NMR spectroscopy (85,115) has previously established an umbrellalike model for cholesterol-phospholipid interactions, where the cholesterol C3-OH group is situated beneath the phospholipid headgroups, so that it acts as a spacer molecule as originally proposed by Brown and Seelig (85). Thermodynamically, the cholesterol interactions with DMPC, POPC, and/or EYSM membranes are driven by the hydrophobic effect plus van der Waals interactions between the acyl chains and sterol ring system, giving an lo phase beyond a threshold cholesterol concentration (116–118). Cholesterol is found to significantly increase the hydrocarbon thickness of the POPC bilayer. Such large volumetric thicknesses are characteristic of both the POPC and EYSM lo phases. The average bilayer thicknesses for cholesterol-enriched lo phases are DB′ ≈ 56.8 Å and 50.0 Å for POPC and EYSM, respectively. The average cross-sectional areas in the lo phase are reduced owing to the condensing effect of cholesterol, as discussed by McConnell and co-workers (119). For POPC bilayers containing cholesterol (1:1) we find that 〈A〉 = 45.1 Å2, and for EYSM, 〈A〉 = 45.5 Å2 at 48°C (Table 1). The cholesterol ordering effect on lipid bilayer stiffening is limited by the maximum acyl length. Furthermore, membranes comprising EYSM/cholesterol may be affected by sphingosine backbone hydrogen bonding and packing interfacial hydrogen bonding involving the NH donor and cholesterol C3-OH acceptor. Such interactions are observed in MD simulations (120,121), although supporting experimental evidence remains elusive (122–127). Another observation is that the sphingosine backbone possesses both an OH hydrogen-bond acceptor and an NH hydrogen-bond donor, which may lead to interlipid hydrogen-bonding and possibly to super-lattice formation in the ld phase (128).
Readers should note that the cholesterol-mediated structural perturbations are clearly less pronounced for the sphingolipid EYSM than for POPC in the lo phase. Upon addition of 50 wt % cholesterol, the increase in absolute segmental order parameters is ≈0.25 for POPC and ≈0.12 for EYSM, as indicated in Fig. 5, b and c (comparisons are for the maximum |SCH| values due to the plateau region of the order profiles). Correspondingly, the increase in the hydrocarbon thickness and condensation of area per lipid is less for EYSM than for POPC bilayers. Such a remarkable difference indicates that EYSM is in a relatively ordered state in the single-component membrane. The higher acyl segmental order parameters in single-component bilayers at a given temperature for EYSM relative to POPC indicates the high propensity of self-association for the hydrophobic moieties of sphingomyelin lipids (9,27). Notably, these observations suggest that the entropic loss upon adding cholesterol is less pronounced for EYSM than for POPC. A greater loss of conformational entropy for POPC versus EYSM may explain selective enrichment of sphingomyelin in putative raftlike microdomains. Mixing of cholesterol is more favorable for sphingolipids compared to phosphatidylcholines, potentially driving the formation of lipid rafts in multicomponent biomembranes (129–131). That is to say, like dissolves like—as we learn in our introductory chemistry courses.
Conclusions
Natural-abundance 13C separated local-field NMR together with a mean-torque model gives lipid structural parameters that are complementary to those from solid-state 2H NMR spectroscopy. The 13C-1H residual dipolar couplings of membrane lipids were successfully used to calculate the area per lipid and bilayer thickness. Differences in molecular interactions are resolved by site-specific 13C chemical shifts and |SCH| dipolar order parameters, giving insight into interfacial molecular packing and hydrophobic interactions. The behavior of glycerophospholipids and sphingolipids in the ld and lo phases reflects the balance of compositional heterogeneity and chemical structure in biological systems. Despite a common thermodynamic lo phase for both lipids, their molecular interactions with cholesterol vary significantly. The higher order of EYSM lipids versus POPC lipids implies that the entropy loss due to interactions with cholesterol is less, favoring the association of EYSM over POPC in raftlike lipid mixtures. Moreover, SLF NMR experiments can address key membrane lipid roles of polyunsaturated lipid bilayers and lipid mixtures containing biologically active peptides or proteins. An important question for future research is how the average material properties emerge from the atomic-level interactions in lipid bilayers as investigated by solid-state NMR spectroscopy and related biophysical methods.
Acknowledgments
This article is dedicated to Professor Harden McConnell.
Support of this research by the National Institutes of Health and by the Arizona Biomedical Research Commission is gratefully acknowledged.
Footnotes
Avigdor Leftin and Trivikram R. Molugu contributed equally to this work.
Avigdor Leftin’s present address is Department of Chemical Physics, Weizmann Institute of Science, Rehovot 76100, Israel.
Supporting Material
Supporting Citations
Reference (132) appears in the Supporting Material.
References
- 1.Brown M.F. Curvature forces in membrane lipid-protein interactions. Biochemistry. 2012;51:9782–9795. doi: 10.1021/bi301332v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gibson N.J., Brown M.F. Lipid headgroup and acyl chain composition modulate the MI-MII equilibrium of rhodopsin in recombinant membranes. Biochemistry. 1993;32:2438–2454. doi: 10.1021/bi00060a040. [DOI] [PubMed] [Google Scholar]
- 3.Brown M.F. Influence of non-lamellar forming lipids on rhodopsin. In: Epand R., editor. Lipid Polymorphism and Membrane Properties. Academic Press; New York: 1997. pp. 285–356. [Google Scholar]
- 4.Simons K., Toomre D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 5.Lee A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Phillips R., Ursell T., Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature. 2009;459:379–385. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Botelho A.V., Gibson N.J., Brown M.F. Conformational energetics of rhodopsin modulated by nonlamellar-forming lipids. Biochemistry. 2002;41:6354–6368. doi: 10.1021/bi011995g. [DOI] [PubMed] [Google Scholar]
- 8.Bartels T., Lankalapalli R.S., Brown M.F. Raftlike mixtures of sphingomyelin and cholesterol investigated by solid-state 2H NMR spectroscopy. J. Am. Chem. Soc. 2008;130:14521–14532. doi: 10.1021/ja801789t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lingwood D., Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 10.Sanders C.R., Mittendorf K.F. Tolerance to changes in membrane lipid composition as a selected trait of membrane proteins. Biochemistry. 2011;50:7858–7867. doi: 10.1021/bi2011527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soubias O., Gawrisch K. The role of the lipid matrix for structure and function of the GPCR rhodopsin. Biochim. Biophys. Acta. 2012;1818:234–240. doi: 10.1016/j.bbamem.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagle J.F., Tristram-Nagle S. Structure of lipid bilayers. Biochim. Biophys. Acta. 2000;1469:159–195. doi: 10.1016/s0304-4157(00)00016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gumbart J., Wang Y., Schulten K. Molecular dynamics simulations of proteins in lipid bilayers. Curr. Opin. Struct. Biol. 2005;15:423–431. doi: 10.1016/j.sbi.2005.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Botelho A.V., Huber T., Brown M.F. Curvature and hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys. J. 2006;91:4464–4477. doi: 10.1529/biophysj.106.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Künze G., Barré P., Huster D. Binding of the three-repeat domain of tau to phospholipid membranes induces an aggregated-like state of the protein. Biochim. Biophys. Acta. 2012;1818:2302–2313. doi: 10.1016/j.bbamem.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teague W.E., Jr., Soubias O., Gawrisch K. Elastic properties of polyunsaturated phosphatidylethanolamines influence rhodopsin function. Faraday Discuss. 2013;161:383–395. doi: 10.1039/c2fd20095c. discussion 419–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shevchenko A., Simons K. Lipidomics: coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 2010;11:593–598. doi: 10.1038/nrm2934. [DOI] [PubMed] [Google Scholar]
- 18.Wolf C., Quinn P.J. Lipidomics in diagnosis of lipidoses. Subcell. Biochem. 2008;49:567–588. doi: 10.1007/978-1-4020-8831-5_22. [DOI] [PubMed] [Google Scholar]
- 19.Leftin A., Job C., Brown M.F. Solid-state 13C NMR reveals annealing of raft-like membranes containing cholesterol by the intrinsically disordered protein α-synuclein. J. Mol. Biol. 2013;425:2973–2987. doi: 10.1016/j.jmb.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Block R.C., Dorsey E.R., Shoulson I. Altered cholesterol and fatty acid metabolism in Huntington disease. J. Clin. Lipidol. 2010;4:17–23. doi: 10.1016/j.jacl.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scarlata S. Regulation of the lateral association of phospholipase Cβ2 and G protein subunits by lipid rafts. Biochemistry. 2002;41:7092–7099. doi: 10.1021/bi025625j. [DOI] [PubMed] [Google Scholar]
- 22.Hong H., Tamm L.K. Elastic coupling of integral membrane protein stability to lipid bilayer forces. Proc. Natl. Acad. Sci. USA. 2004;101:4065–4070. doi: 10.1073/pnas.0400358101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korade Z., Kenworthy A.K. Lipid rafts, cholesterol, and the brain. Neuropharmacology. 2008;55:1265–1273. doi: 10.1016/j.neuropharm.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Day C.A., Kenworthy A.K. Tracking microdomain dynamics in cell membranes. Biochim. Biophys. Acta. 2009;1788:245–253. doi: 10.1016/j.bbamem.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiessling V., Wan C., Tamm L.K. Domain coupling in asymmetric lipid bilayers. Biochim. Biophys. Acta. 2009;1788:64–71. doi: 10.1016/j.bbamem.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golebiewska U., Scarlata S. The effect of membrane domains on the G protein-phospholipase Cβ signaling pathway. Crit. Rev. Biochem. Mol. Biol. 2010;45:97–105. doi: 10.3109/10409231003598812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simons K., Gerl M.J. Revitalizing membrane rafts: new tools and insights. Nat. Rev. Mol. Cell Biol. 2010;11:688–699. doi: 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- 28.Hong M., Zhang Y., Hu F. Membrane protein structure and dynamics from NMR spectroscopy. Annu. Rev. Phys. Chem. 2012;63:1–24. doi: 10.1146/annurev-physchem-032511-143731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das N., Murray D.T., Cross T.A. Lipid bilayer preparations of membrane proteins for oriented and magic-angle spinning solid-state NMR samples. Nat. Protoc. 2013;8:2256–2270. doi: 10.1038/nprot.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H.X., Cross T.A. Influences of membrane mimetic environments on membrane protein structures. Annu. Rev. Biophys. 2013;42:361–392. doi: 10.1146/annurev-biophys-083012-130326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kinnun J.J., Mallikarjunaiah K.J., Brown M.F. Elastic deformation and area per lipid of membranes: atomistic view from solid-state deuterium NMR spectroscopy. Biochim. Biophys. Acta. 2014 doi: 10.1016/j.bbamem.2014.06.004. Published online June 16, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pastor R.W., Venable R.M., Feller S.E. Lipid bilayers, NMR relaxation, and computer simulations. Acc. Chem. Res. 2002;35:438–446. doi: 10.1021/ar0100529. [DOI] [PubMed] [Google Scholar]
- 33.Krepkiy D., Mihailescu M., Swartz K.J. Structure and hydration of membranes embedded with voltage-sensing domains. Nature. 2009;462:473–479. doi: 10.1038/nature08542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leioatts N., Mertz B., Brown M.F. Retinal ligand mobility explains internal hydration and reconciles active rhodopsin structures. Biochemistry. 2014;53:376–385. doi: 10.1021/bi4013947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petrache H.I., Dodd S.W., Brown M.F. Area per lipid and acyl length distributions in fluid phosphatidylcholines determined by 2H NMR spectroscopy. Biophys. J. 2000;79:3172–3192. doi: 10.1016/S0006-3495(00)76551-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feller S.E. Molecular dynamics simulations of lipid bilayers. Curr. Opin. Colloid Interface Sci. 2000;5:217–223. [Google Scholar]
- 37.Dietrich C., Bagatolli L.A., Gratton E. Lipid rafts reconstituted in model membranes. Biophys. J. 2001;80:1417–1428. doi: 10.1016/S0006-3495(01)76114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tristram-Nagle S., Liu Y., Nagle J.F. Structure of gel phase DMPC determined by x-ray diffraction. Biophys. J. 2002;83:3324–3335. doi: 10.1016/S0006-3495(02)75333-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tristram-Nagle S., Nagle J.F. Lipid bilayers: thermodynamics, structure, fluctuations, and interactions. Chem. Phys. Lipids. 2004;127:3–14. doi: 10.1016/j.chemphyslip.2003.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cegelski L., Rice C.V., Schaefer J. Mapping the locations of estradiol and potent neuroprotective analogues in phospholipid bilayers by REDOR. Drug Dev. Res. 2006;66:93–102. [Google Scholar]
- 41.Dvinskikh S.V., Castro V., Sandström D. Efficient solid-state NMR methods for measuring heteronuclear dipolar couplings in unoriented lipid membrane systems. Phys. Chem. Chem. Phys. 2005;7:607–613. doi: 10.1039/b418131j. [DOI] [PubMed] [Google Scholar]
- 42.Kučerka N., Nagle J.F., Katsaras J. Lipid bilayer structure determined by the simultaneous analysis of neutron and x-ray scattering data. Biophys. J. 2008;95:2356–2367. doi: 10.1529/biophysj.108.132662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leftin A., Brown M.F. An NMR database for simulations of membrane dynamics. Biochim. Biophys. Acta. 2011;1808:818–839. doi: 10.1016/j.bbamem.2010.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mallikarjunaiah K.J., Leftin A., Brown M.F. Solid-state ²H NMR shows equivalence of dehydration and osmotic pressures in lipid membrane deformation. Biophys. J. 2011;100:98–107. doi: 10.1016/j.bpj.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferreira T.M., Coreta-Gomes F., Topgaard D. Cholesterol and POPC segmental order parameters in lipid membranes: solid state 1H-13C NMR and MD simulation studies. Phys. Chem. Chem. Phys. 2013;15:1976–1989. doi: 10.1039/c2cp42738a. [DOI] [PubMed] [Google Scholar]
- 46.Gross J.D., Warschawski D.E., Griffin R.G. Dipolar recoupling in MAS NMR: a probe for segmental order in lipid bilayers. J. Am. Chem. Soc. 1997;119:796–802. [Google Scholar]
- 47.Gawrisch K., Eldho N.V., Polozov I.V. Novel NMR tools to study structure and dynamics of biomembranes. Chem. Phys. Lipids. 2002;116:135–151. doi: 10.1016/s0009-3084(02)00024-5. [DOI] [PubMed] [Google Scholar]
- 48.Castro V., Stevensson B., Maliniak A. NMR investigations of interactions between anesthetics and lipid bilayers. Biochim. Biophys. Acta. 2008;1778:2604–2611. doi: 10.1016/j.bbamem.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 49.Soberón-Chávez G., Maier R.M. Biosurfactants: a general overview. In: Soberón-Chávez G., editor. Biosurfactants. Springer; Heidelberg: 2011. pp. 1–11. [Google Scholar]
- 50.Brown M.F. Membrane structure and dynamics studied with NMR spectroscopy. In: Merz K.M. Jr., Roux B., editors. Biological Membranes: A Molecular Perspective from Computation and Experiment. Birkhäuser; Basel: 1996. pp. 175–252. [Google Scholar]
- 51.Petrache H.I., Brown M.F. X-ray scattering and solid-state deuterium nuclear magnetic resonance probes of structural fluctuations in lipid membranes. In: Dopico A.M., editor. Methods in Molecular Biology. Humana; Totowa, NJ: 2007. pp. 341–353. [DOI] [PubMed] [Google Scholar]
- 52.Otten D., Brown M.F., Beyer K. Softening of membrane bilayers by detergents elucidated by deuterium NMR spectroscopy. J. Phys. Chem. B. 2000;104:12119–12129. [Google Scholar]
- 53.Sefcik M.D., Schaefer J., Brown M.F. Lipid bilayer dynamics and rhodopsin-lipid interactions: new approach using high-resolution solid-state 13C NMR. Biochem. Biophys. Res. Commun. 1983;114:1048–1055. doi: 10.1016/0006-291x(83)90668-x. [DOI] [PubMed] [Google Scholar]
- 54.Brown M.F., Chan S.I. Bilayer membranes: deuterium & carbon-13 NMR. In: Harris R.K., Grant D.M., editors. Encyclopedia of Magnetic Resonance. John Wiley & Sons; Chichester: 2007. Published online: 15 March 2007. [Google Scholar]
- 55.McDermott A. Structure and dynamics of membrane proteins by magic angle spinning solid-state NMR. Annu. Rev. Biophys. 2009;38:385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 56.Leftin A., Xu X., Brown M.F. Phospholipid bilayer membranes: deuterium and carbon-13 NMR spectroscopy. eMagRes. 2014 In press. [Google Scholar]
- 57.Warschawski D.E., Devaux P.F. Order parameters of unsaturated phospholipids in membranes and the effect of cholesterol: a 1H-13C solid-state NMR study at natural abundance. Eur. Biophys. J. 2005;34:987–996. doi: 10.1007/s00249-005-0482-z. [DOI] [PubMed] [Google Scholar]
- 58.Tycko R., Dabbagh G., Mirau P.A. Determination of chemical shift anisotropy lineshapes in a two dimensional magic angle spinning NMR experiment. J. Magn. Reson. 1989;85:265–274. [Google Scholar]
- 59.Fung B.M., Khitrin A.K., Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 60.Brown M.F. Unified picture for spin-lattice relaxation of lipid bilayers and biomembranes. J. Chem. Phys. 1984;80:2832–2836. [Google Scholar]
- 61.Fürst A., Pretsch E. A computer program for the prediction of 13C-NMR chemical shifts of organic compounds. Anal. Chim. Acta. 1990;229:17–25. [Google Scholar]
- 62.Pretsch E., Fürst A., Munk M.E. C13shift: a computer program for the prediction of 13C NMR spectra based on an open set of additivity rules. J. Chem. Inf. Comput. Sci. 1992;32:291–295. [Google Scholar]
- 63.Schaller R.B., Pretsch E. A computer program for the automatic estimation of 1H NMR chemical shifts. Anal. Chim. Acta. 1994;290:295–302. [Google Scholar]
- 64.Schaller R.B., Arnold C., Pretsch E. New parameters for predicting 1H NMR chemical shifts of protons attached to carbon-atoms. Anal. Chim. Acta. 1995;312:95–105. [Google Scholar]
- 65.Schaller R.B., Munk M.E., Pretsch E. Spectra estimation for computer-aided structure determination. J. Chem. Inf. Comput. Sci. 1996;36:239–243. [Google Scholar]
- 66.Brown M.F. Theory of spin-lattice relaxation in lipid bilayers and biological membranes. 2H and 14N quadrupolar relaxation. J. Chem. Phys. 1982;77:1576–1599. [Google Scholar]
- 67.Nevzorov A.A., Brown M.F. Dynamics of lipid bilayers from comparative analysis of 2H and 13C nuclear magnetic resonance relaxation data as a function of frequency and temperature. J. Chem. Phys. 1997;107:10288–10310. [Google Scholar]
- 68.Xu X., Struts A.V., Brown M.F. Generalized model-free analysis of nuclear spin relaxation experiments. eMagRes. 2014 In press. [Google Scholar]
- 69.Brown M.F., Seelig J., Häberlen U. Structural dynamics in phospholipid bilayers from deuterium spin-lattice relaxation time measurements. J. Chem. Phys. 1979;70:5045–5053. [Google Scholar]
- 70.Nagle J.F. Area/lipid of bilayers from NMR. Biophys. J. 1993;64:1476–1481. doi: 10.1016/S0006-3495(93)81514-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seelig A., Seelig J. The dynamic structure of fatty acyl chains in a phospholipid bilayer measured by deuterium magnetic resonance. Biochemistry. 1974;13:4839–4845. doi: 10.1021/bi00720a024. [DOI] [PubMed] [Google Scholar]
- 72.Salmon A., Dodd S.W., Brown M.F. Configurational statistics of acyl chains in polyunsaturated lipid bilayers from 2H NMR. J. Am. Chem. Soc. 1987;109:2600–2609. [Google Scholar]
- 73.Koenig B.W., Strey H.H., Gawrisch K. Membrane lateral compressibility determined by NMR and x-ray diffraction: effect of acyl chain polyunsaturation. Biophys. J. 1997;73:1954–1966. doi: 10.1016/S0006-3495(97)78226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petrache H.I., Tu K., Nagle J.F. Analysis of simulated NMR order parameters for lipid bilayer structure determination. Biophys. J. 1999;76:2479–2487. doi: 10.1016/S0006-3495(99)77403-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dill K.A., Flory P.J. Interphases of chain molecules: monolayers and lipid bilayer membranes. Proc. Natl. Acad. Sci. USA. 1980;77:3115–3119. doi: 10.1073/pnas.77.6.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dill K.A., Flory P.J. Molecular organization in micelles and vesicles. Proc. Natl. Acad. Sci. USA. 1981;78:676–680. doi: 10.1073/pnas.78.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brown M.F., Ribeiro A.A., Williams G.D. New view of lipid bilayer dynamics from 2H and 13C NMR relaxation time measurements. Proc. Natl. Acad. Sci. USA. 1983;80:4325–4329. doi: 10.1073/pnas.80.14.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nagle J.F., Wilkinson D.A. Lecithin bilayers. Density measurement and molecular interactions. Biophys. J. 1978;23:159–175. doi: 10.1016/S0006-3495(78)85441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jansson M., Thurmond R.L., Brown M.F. Deuterium NMR study of intermolecular interactions in lamellar phases containing palmitoyllysophosphatidylcholine. J. Phys. Chem. 1992;96:9532–9544. [Google Scholar]
- 80.Petrache H.I., Feller S.E., Nagle J.F. Determination of component volumes of lipid bilayers from simulations. Biophys. J. 1997;72:2237–2242. doi: 10.1016/S0006-3495(97)78867-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Armen R.S., Uitto O.D., Feller S.E. Phospholipid component volumes: determination and application to bilayer structure calculations. Biophys. J. 1998;75:734–744. doi: 10.1016/S0006-3495(98)77563-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maulik P.R., Atkinson D., Shipley G.G. X-ray scattering of vesicles of N-acyl sphingomyelins. Determination of bilayer thickness. Biophys. J. 1986;50:1071–1077. doi: 10.1016/S0006-3495(86)83551-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Petrache H.I., Tristram-Nagle S., Nagle J.F. Fluid phase structure of EPC and DMPC bilayers. Chem. Phys. Lipids. 1998;95:83–94. doi: 10.1016/s0009-3084(98)00068-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kučerka N., Tristram-Nagle S., Nagle J.F. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 2005;208:193–202. doi: 10.1007/s00232-005-7006-8. [DOI] [PubMed] [Google Scholar]
- 85.Brown M.F., Seelig J. Influence of cholesterol on the polar region of phosphatidylcholine and phosphatidylethanolamine bilayers. Biochemistry. 1978;17:381–384. doi: 10.1021/bi00595a029. [DOI] [PubMed] [Google Scholar]
- 86.Sears B. 13C nuclear magnetic resonance studies of egg phosphatidylcholine. J. Membr. Biol. 1975;20:59–73. doi: 10.1007/BF01870628. [DOI] [PubMed] [Google Scholar]
- 87.Volke F., Waschipky R., Welzel P. Characterisation of antibiotic moenomycin A interaction with phospholipid model membranes. Chem. Phys. Lipids. 1997;85:115–123. doi: 10.1016/s0009-3084(96)02649-7. [DOI] [PubMed] [Google Scholar]
- 88.Baldus M. Correlation experiments for assignment and structure elucidation of immobilized polypeptides under magic angle spinning. Prog. Nucl. Magn. Reson. Spectrosc. 2002;41:1–47. [Google Scholar]
- 89.Saitô H., Ando I., Ramamoorthy A. Chemical shift tensor - the heart of NMR: insights into biological aspects of proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2010;57:181–228. doi: 10.1016/j.pnmrs.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ando I. Some aspects of the NMR chemical shift/structure correlation in the structural characterization of polymers and biopolymers. Polym. J. 2012;44:734–747. [Google Scholar]
- 91.Kondo M., Ando I., Nishioka A. Theoretical calculation of carbon-13 NMR shielding constants and their tensors in hydrocarbons by finite perturbation method. J. Magn. Reson. 1976;24:315–326. [Google Scholar]
- 92.Ando I., Saitô H., Ozaki T. Conformation-dependent 13C NMR chemical shifts of poly(L-alanine) in the solid state: FPT INDO calculation of N-acetyl-N′-methyl-L-alanine amide as a model compound of poly(L-alanine) Macromolecules. 1984;17:457–461. [Google Scholar]
- 93.Huber T., Rajamoorthi K., Brown M.F. Structure of docosahexaenoic acid-containing phospholipid bilayers as studied by 2H NMR and molecular dynamics simulations. J. Am. Chem. Soc. 2002;124:298–309. doi: 10.1021/ja011383j. [DOI] [PubMed] [Google Scholar]
- 94.Brown M.F. Theory of spin-lattice relaxation in lipid bilayers and biological membranes. Dipolar relaxation. J. Chem. Phys. 1984;80:2808–2831. [Google Scholar]
- 95.Ipsen J.H., Mouritsen O.G., Bloom M. Relationships between lipid membrane area, hydrophobic thickness, and acyl-chain orientational order. The effects of cholesterol. Biophys. J. 1990;57:405–412. doi: 10.1016/S0006-3495(90)82557-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Forbes J., Husted C., Oldfield E. High-field, high-resolution proton “magic-angle” sample-spinning nuclear magnetic resonance spectroscopic studies of gel and liquid crystalline lipid bilayers and the effects of cholesterol. J. Am. Chem. Soc. 1988;110:1059–1065. [Google Scholar]
- 97.Holland G.P., Alam T.M. Multi-dimensional 1H-13C HETCOR and FSLG-HETCOR NMR study of sphingomyelin bilayers containing cholesterol in the gel and liquid crystalline states. J. Magn. Reson. 2006;181:316–326. doi: 10.1016/j.jmr.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 98.Martinez G.V., Dykstra E.M., Brown M.F. NMR elastometry of fluid membranes in the mesoscopic regime. Phys. Rev. E. 2002;66:0509021-1–0509021-4. doi: 10.1103/PhysRevE.66.050902. [DOI] [PubMed] [Google Scholar]
- 99.Martinez G.V., Dykstra E.M., Brown M.F. Lanosterol and cholesterol-induced variations in bilayer elasticity probed by 2H NMR relaxation. Langmuir. 2004;20:1043–1046. doi: 10.1021/la036063n. [DOI] [PubMed] [Google Scholar]
- 100.Warschawski D.E., Devaux P.F. 1H-13C polarization transfer in membranes: a tool for probing lipid dynamics and the effect of cholesterol. J. Magn. Reson. 2005;177:166–171. doi: 10.1016/j.jmr.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 101.Lai A.L., Freed J.H. HIV gp41 fusion peptide increases membrane ordering in a cholesterol-dependent fashion. Biophys. J. 2014;106:172–181. doi: 10.1016/j.bpj.2013.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen Z., Rand R.P. The influence of cholesterol on phospholipid membrane curvature and bending elasticity. Biophys. J. 1997;73:267–276. doi: 10.1016/S0006-3495(97)78067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Filippov A., Orädd G., Lindblom G. The effect of cholesterol on the lateral diffusion of phospholipids in oriented bilayers. Biophys. J. 2003;84:3079–3086. doi: 10.1016/S0006-3495(03)70033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Seelig J., Seelig A. Lipid conformation in model membranes and biological membranes. Q. Rev. Biophys. 1980;13:19–61. doi: 10.1017/s0033583500000305. [DOI] [PubMed] [Google Scholar]
- 105.Kaiser H.-J., Lingwood D., Simons K. Order of lipid phases in model and plasma membranes. Proc. Natl. Acad. Sci. USA. 2009;106:16645–16650. doi: 10.1073/pnas.0908987106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Quinn P.J. Structure of sphingomyelin bilayers and complexes with cholesterol forming membrane rafts. Langmuir. 2013;29:9447–9456. doi: 10.1021/la4018129. [DOI] [PubMed] [Google Scholar]
- 107.Barenholz Y., Thompson T.E. Sphingomyelin: biophysical aspects. Chem. Phys. Lipids. 1999;102:29–34. doi: 10.1016/s0009-3084(99)00072-9. [DOI] [PubMed] [Google Scholar]
- 108.Epand R.M., Epand R.F. Non-raft forming sphingomyelin-cholesterol mixtures. Chem. Phys. Lipids. 2004;132:37–46. doi: 10.1016/j.chemphyslip.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 109.Wassall S.R., Stillwell W. Polyunsaturated fatty acid-cholesterol interactions: domain formation in membranes. Biochim. Biophys. Acta. 2009;1788:24–32. doi: 10.1016/j.bbamem.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 110.Hong M., Schmidt-Rohr K., Pines A. NMR measurement of signs and magnitudes of C-H dipolar couplings in lecithin. J. Am. Chem. Soc. 1995;117:3310–3311. [Google Scholar]
- 111.Brown M.F., Deese A.J., Dratz E.A. Proton, carbon-13, and phosphorus-31 NMR methods for the investigation of rhodopsin-lipid interactions in retinal rod outer segment membranes. Methods Enzymol. 1982;81:709–728. doi: 10.1016/s0076-6879(82)81098-7. [DOI] [PubMed] [Google Scholar]
- 112.Oldfield E., Meadows M., Jacobs R. Spectroscopic studies of specifically deuterium labeled membrane systems. Nuclear magnetic resonance investigation of the effects of cholesterol in model systems. Biochemistry. 1978;17:2727–2740. doi: 10.1021/bi00607a006. [DOI] [PubMed] [Google Scholar]
- 113.Petrache H.I., Salmon A., Brown M.F. Structural properties of docosahexaenoyl phospholipid bilayers investigated by solid-state 2H NMR spectroscopy. J. Am. Chem. Soc. 2001;123:12611–12622. doi: 10.1021/ja011745n. [DOI] [PubMed] [Google Scholar]
- 114.Maulik P.R., Shipley G.G. N-palmitoyl sphingomyelin bilayers: structure and interactions with cholesterol and dipalmitoylphosphatidylcholine. Biochemistry. 1996;35:8025–8034. doi: 10.1021/bi9528356. [DOI] [PubMed] [Google Scholar]
- 115.Brown M.F. Anisotropic nuclear spin relaxation of cholesterol in phospholipid bilayers. Mol. Phys. 1990;71:903–908. [Google Scholar]
- 116.Ipsen J.H., Karlström G., Zuckermann M.J. Phase equilibria in the phosphatidylcholine-cholesterol system. Biochim. Biophys. Acta. 1987;905:162–172. doi: 10.1016/0005-2736(87)90020-4. [DOI] [PubMed] [Google Scholar]
- 117.Tsamaloukas A., Szadkowska H., Heerklotz H. Thermodynamic comparison of the interactions of cholesterol with unsaturated phospholipid and sphingomyelins. Biophys. J. 2006;90:4479–4487. doi: 10.1529/biophysj.105.080127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van Meer G., Voelker D.R., Feigenson G.W. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McConnell H.M., Radhakrishnan A. Condensed complexes of cholesterol and phospholipids. Biochim. Biophys. Acta. 2003;1610:159–173. doi: 10.1016/s0005-2736(03)00015-4. [DOI] [PubMed] [Google Scholar]
- 120.Hofsäβ C., Lindahl E., Edholm O. Molecular dynamics simulations of phospholipid bilayers with cholesterol. Biophys. J. 2003;84:2192–2206. doi: 10.1016/S0006-3495(03)75025-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pandit S.A., Vasudevan S., Scott H.L. Sphingomyelin-cholesterol domains in phospholipid membranes: atomistic simulation. Biophys. J. 2004;87:1092–1100. doi: 10.1529/biophysj.104.041939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kan C.-C., Ruan Z.-s., Bittman R. Interaction of cholesterol with sphingomyelin in bilayer membranes: evidence that the hydroxy group of sphingomyelin does not modulate the rate of cholesterol exchange between vesicles. Biochemistry. 1991;30:7759–7766. doi: 10.1021/bi00245a013. [DOI] [PubMed] [Google Scholar]
- 123.Bittman R., Kasireddy C.R., Slotte J.P. Interaction of cholesterol with sphingomyelin in monolayers and vesicles. Biochemistry. 1994;33:11776–11781. doi: 10.1021/bi00205a013. [DOI] [PubMed] [Google Scholar]
- 124.Slotte J.P. Sphingomyelin-cholesterol interactions in biological and model membranes. Chem. Phys. Lipids. 1999;102:13–27. doi: 10.1016/s0009-3084(99)00071-7. [DOI] [PubMed] [Google Scholar]
- 125.Guo W., Kurze V., Hamilton J.A. A solid-state NMR study of phospholipid-cholesterol interactions: sphingomyelin-cholesterol binary systems. Biophys. J. 2002;83:1465–1478. doi: 10.1016/S0006-3495(02)73917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Niemelä P., Hyvönen M.T., Vattulainen I. Structure and dynamics of sphingomyelin bilayer: insight gained through systematic comparison to phosphatidylcholine. Biophys. J. 2004;87:2976–2989. doi: 10.1529/biophysj.104.048702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Holopainen J.M., Metso A.J., Kinnunen P.K.J. Evidence for the lack of a specific interaction between cholesterol and sphingomyelin. Biophys. J. 2004;86:1510–1520. doi: 10.1016/S0006-3495(04)74219-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Somerharju P., Virtanen J.A., Cheng K.H. Lateral organisation of membrane lipids. The superlattice view. Biochim. Biophys. Acta. 1999;1440:32–48. doi: 10.1016/s1388-1981(99)00106-7. [DOI] [PubMed] [Google Scholar]
- 129.Ohvo-Rekilä H., Ramstedt B., Slotte J.P. Cholesterol interactions with phospholipids in membranes. Prog. Lipid Res. 2002;41:66–97. doi: 10.1016/s0163-7827(01)00020-0. [DOI] [PubMed] [Google Scholar]
- 130.Edidin M. The state of lipid rafts: from model membranes to cells. Annu. Rev. Biophys. Biomol. Struct. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- 131.Ramstedt B., Slotte J.P. Sphingolipids and the formation of sterol-enriched ordered membrane domains. Biochim. Biophys. Acta. 2006;1758:1945–1956. doi: 10.1016/j.bbamem.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 132.Shipley G.G., Avecilla L.S., Small D.M. Phase behavior and structure of aqueous dispersions of sphingomyelin. J. Lipid Res. 1974;15:124–131. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.