

Abstract
Fibroblasts are activated in heart failure (HF) and produce fibrosis, which plays a role in maintaining atrial fibrillation (AF). The effect of HF on fibroblast ion currents and its potential role in AF are unknown. Here, we used a patch-clamp technique to investigate the effects of HF on atrial fibroblast ion currents, and mathematical computation to assess the potential impact of this remodeling on atrial electrophysiology and arrhythmogenesis. Atrial fibroblasts were isolated from control and tachypacing-induced HF dogs. Tetraethylammonium-sensitive voltage-gated fibroblast current (IKv,fb) was significantly downregulated (by ∼44%), whereas the Ba2+-sensitive inward rectifier current (IKir,fb) was upregulated by 79%, in HF animals versus controls. The fibroblast resting membrane potential was hyperpolarized (−53 ± 2 mV vs. −42 ± 2 mV in controls) and the capacitance was increased (29.7 ± 2.2 pF vs. 17.8 ± 1.4 pF in controls) in HF. These experimental findings were implemented in a mathematical model that included cardiomyocyte-fibroblast electrical coupling. IKir,fb upregulation had a profibrillatory effect through shortening of the action potential duration and hyperpolarization of the cardiomyocyte resting membrane potential. IKv,fb downregulation had the opposite electrophysiological effects and was antifibrillatory. Simulated pharmacological blockade of IKv,fb successfully terminated reentry under otherwise profibrillatory conditions. We conclude that HF induces fibroblast ion-current remodeling with IKv,fb downregulation and IKir,fb upregulation, and that, assuming cardiomyocyte-fibroblast electrical coupling, this remodeling has a potentially important effect on atrial electrophysiology and arrhythmogenesis, with the overall response depending on the balance of pro- and antifibrillatory contributions. These findings suggest that fibroblast K+-current remodeling is a novel component of AF-related remodeling that might contribute to arrhythmia dynamics.
Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia in the general population and is associated with significant cardiovascular morbidity and mortality (1,2). The failure of randomized clinical trials to demonstrate a benefit from sinus-rhythm-maintaining antiarrhythmic therapy, which has often been attributed to the suboptimal efficacy and significant proarrhythmic potential of antiarrhythmic drugs (3), highlights our incomplete understanding of the basic pathophysiology of AF.
At the cellular level, it is well known that AF induces cardiomyocyte electrical remodeling and atrial fibrosis, forming the basis for longer-lasting forms of AF (4–7). Traditionally, fibroblasts were thought to be relevant to arrhythmogenesis primarily because they produce extracellular matrix, which serves as a structural barrier that affects anisotropic conduction and creates conduction block (8–11). However, recent work has modified this paradigm by suggesting that cardiac fibroblasts may couple electrically to atrial cardiomyocytes and modulate their electrophysiological properties to promote various forms of arrhythmogenesis (12). Cardiomyocyte-fibroblast electrical interactions have been studied in cocultured cell systems (13–18) and mathematical models (19,20), and postulated to contribute to the pathogenesis of AF (12,19), although in vivo evidence of such interactions is lacking. Cardiomyocyte ion-channel remodeling is known to play an important role in AF pathophysiology (21). Fibroblasts also possess ion channels, but their function is much less well understood than those of cardiomyocytes (12). The nature and potential significance of fibroblast ion-channel remodeling associated with AF remain to be evaluated. Here, we examined the remodeling of K+ currents in fibroblasts freshly isolated from dogs with an AF substrate associated with heart failure (HF). Freshly isolated fibroblasts were used to preclude the effects of cell culture, which can obscure the fibroblast remodeling phenotype caused by HF (22). We then implemented these experimental findings in a mathematical model based on previous work (23) to investigate the potential effects of fibroblast electrical remodeling on atrial cardiomyocyte electrophysiological properties and arrhythmogenesis, assuming cardiomyocyte-fibroblast electrical coupling.
Materials and Methods
Animal model
Animal care procedures were approved by the Animal Research Ethics Committee of the Montreal Heart Institute and followed National Institutes of Health guidelines. We studied a total of 20 adult mongrel dogs (20–30 kg), divided into control (CTL; n = 10) and 2-week ventricular tachypacing-induced HF (n = 10) groups. HF dogs were anesthetized under diazepam (0.25 mg/kg IV)/ketamine (5.0 mg/kg IV)/halothane (1–2% PI) anesthesia so that two leads could be inserted under fluoroscopy into the right ventricular apex (via the left jugular vein) and connected to a pacemaker (St. Jude Medical, St. Paul, MN) implanted subcutaneously in the neck. After 24 h of postoperative recovery, ventricular pacing was initiated at 240 bpm for 2 weeks. On study days, the dogs were anesthetized with morphine (2 mg/kg SC) and α-chloralose (120 mg/kg IV, followed by 29.25 mg/kg/h) and ventilated mechanically. The atrial effective refractory period (aERP) and mean AF duration were measured at basic cycle lengths (BCLs) of 150, 200 ms, 250 ms, 300 ms, and 350 ms in the right atrium. aERP was determined with 10 basic stimuli (S1) followed by a premature extrastimulus (S2) in 5 ms decrements (the longest S1-S2 interval that failed to capture defined the aERP). The mean of three aERP values at each BCL was used for analysis. AF was induced with atrial burst pacing at 10 Hz and 10 V. The mean AF duration was based on 10 AF inductions in each dog. If the mean duration of the first five episodes of AF was longer than 2 min, AF was only induced five times. After in vivo study, the hearts were removed and immediately immersed in oxygenated Tyrode solution containing (mmol/L) NaCl 136, KCl 5.4, MgCl2 1, CaCl2 2, NaH2PO4 0.33, HEPES 5, and dextrose 10, pH 7.35 (NaOH). Atrial tissue was subjected to enzymatic digestion as described below for cell isolation.
Fibroblast isolation
Atrial fibroblasts were obtained from the left atria of adult mongrel dogs as described previously (24). The heart was removed after intra-atrial injection of heparin (10,000 U) and immersed in Tyrode solution containing 2 mmol/L Ca2+, and the left coronary artery was cannulated. The left atrial (LA) tissue was then perfused with 2 mmol/L Tyrode solution (37°C, 100% O2), then with Ca2+-free Tyrode solution (∼10 min), and finally by ∼60 min perfusion with the same solution containing collagenase II (∼0.48 mg/mL; Worthington, Lakewood, NJ) and 0.1% bovine serum albumin (BSA; Sigma, Sigma-Aldrich Canada Co., Oakville, Ontario, Canada). Cells were dispersed by trituration in Kraftbrühe (KB) solution (for contents, see below). Filtration (500 μm micromesh) was used to remove debris and cells were centrifuged at 800 rpm for 5 min to pellet cardiomyocytes. The supernatant was collected and filtered through 30 μm nanomesh and centrifuged at 1500 rpm for 10 min to pellet fibroblasts. Pelleted, freshly isolated fibroblasts were then separated.
Ion current and membrane potential recording
All in vitro recordings were obtained at 37°C. The whole-cell perforated-patch technique was used to record resting membrane potentials (RMPs) in current-clamp mode and the tight-seal patch clamp was used to record K+ current in voltage-clamp mode. Borosilicate glass electrodes (Sutter Instrument) filled with pipette solution were connected to a patch-clamp amplifier (Axopatch 200A; Axon). Electrodes had tip resistances of 6–8 MΩ. For perforated-patch formation, a nystatin-free intracellular solution was in the tip of the pipette by capillary action (∼30 s). The pipettes were then back-filled with a nystatin-containing (600 μg/mL) pipette solution. Currents are expressed as densities (pA/pF). Junction potentials between bath and pipette solutions averaged 10.5 mV and were corrected for RMP measurements only. IKir,fb was quantified as 300 μmol/L Ba2+-sensitive current. IKv,fb was quantified as current sensitive to 10 mmol/L tetraethylammonium (TEA) (24). The bath solution contained (mmol/L) NaCl 136, CaCl2 1.8, KCl 5.4, MgCl2 1, NaH2PO4 0.33, dextrose 10, and HEPES 5, titrated to pH 7.3 with NaOH. The pipette solution contained (mmol/L) GTP 0.1, potassium-aspartate 110, KCl 20, MgCl2 1, MgATP 5, HEPES 10, sodium-phosphocreatine 5, and EGTA 0.005 (pH 7.4, KOH). The KB solution contained (mmol/L) KCl 20, KH2PO4 10, dextrose 10, mannitol 40, L-glutamic acid 70, β-OH-butyric acid 10, taurine 20, EGTA 10, and 0.1% BSA (pH 7.3, KOH).
Atrial cardiomyocyte and fibroblast mathematical models
The Ramirez-Nattel-Courtemanche (RNC) ionic model of canine atrial cardiomyocyte action potentials (APs) (25) was implemented and electrically connected to a variable number (n) of MacCannell model fibroblasts (20) via gap junctions of conductance Ggap. The total ionic current for the RNC model (Iion,RNC) is
where IKir, Ito, IKur,d, IKr, IKs, and IKACh are the inward-rectifier-, transient-outward-, ultrarapid delayed-rectifier-, rapid and slow delayed rectifier-, and acetycholine (ACh)-activated K+ currents respectively; ICaL is the L-type Ca2+ current; IClCa is the Ca2+-activated Cl− current; IpCa is the Ca2+ pump current; INaCa is the Na+/Ca2+ exchange current; INaK is the Na+/K+ pump current; and Ib,Na, Ib,Ca, and Ib,Cl are the background Na+, Ca2+, and Cl− currents, respectively (26). Igap is the gap-junction current and n is the number of fibroblasts coupled to each cardiomyocyte. The total ionic current for the MacCannell fibroblast model (Iion,fb) is calculated as
where IKv,fb and IKir,fb are the fibroblast-delayed-rectifier and inward-rectifier K+ currents, respectively, INaK,fb is the Na+/K+ exchange pump current, Ib,Na,fb is the background Na+ current, and Igap is the gap junction current (20).
Fibroblast electrical remodeling was simulated by scaling the fibroblast IKv,fb and IKir,fb maximal conductance as follows: GKir,fb′ = fKir,fb × GKir,fb and GKv,fb′ = fKv,fb × GKv,fb, where Gx is the baseline conductance, fx is the remodeling factor, and Gx′ is the remodeled conductance. GKir,fb and GKv,fb were set at 0.4822 nS/pF and 0.14 nS/pF, respectively, as described by Ashihara et al. (23). We varied fKir,fb and fKv,fb independently with fKir,fb = {1, 2.5, 5, 10} and fKv,fb = {0.1, 0.25, 0.5, 0.75, 1}.
Single-cell and cable simulations
A single cardiomyocyte with capacitance 100 pF (25) was connected to a fibroblast through a gap junction with a gap-junction conductivity (Ggap) of 3 nS. The cardiomyocyte was paced at a BCL of 250 ms and the cardiomyocyte transmembrane potential, fibroblast transmembrane potential, gap-junction current (Igap), and total fibroblast potassium current (IK,fb) were monitored under CTL and remodeled conditions. We then constructed a 1D monodomain cable of atrial cardiomyocytes connected to fibroblasts and simulated cardiomyocyte and fibroblast transmembrane potential, Igap, IK,fb, and relevant electrophysiological parameters under CTL and remodeled conditions. Numerical integration of the single-cell model was performed using the MATLAB ODE23s ordinary differential equation solver (The MathWorks, Natick, MA).
2D sheet and fibroblast distribution
We performed 2D simulations on a 5 × 10 cm sheet of RNC atrial cells. The tissue contained cardiomyocyte cables (radius 5 μm, resistivity 75 Ω-cm, coupled by resistors of 300 kΩ (length 100 mm) inserted in a brick-wall manner). Fiber resistivity and interfiber resistance values were chosen to match experimental results (26,27). Fig. S1 in the Supporting Material shows the fibroblast distribution used (the results presented here were obtained at intermediate density (0.250), but analyses were conducted with all three fibroblast distribution maps and produced qualitatively similar results). Fibroblast proliferation was generated through a recursive algorithm as described by Ashihara et al. (23). Reentrant spiral waves were initiated using an S1-S2 cross-shock protocol. We investigated the initiation, maintenance, and termination dynamics, and performed a dominant-frequency analysis by computing the frequency domain amplitude of the fast Fourier transform of the pseudo-ECG, as in previous work (28). Both the cable and 2D models were coded in C and calculations were performed with a time step of 25 μs using up to eight processors with an SGI Altix XE high-performance parallel processor (Westgrid).
Results
In vivo measurements
In vivo electrophysiological data and hemodynamic indices are shown in Fig. 1. HF significantly increased aERP at all BCLs (Fig. 1 A). Mean AF duration increased in HF dogs (745 ± 287 vs. 22 ± 10 s in CTL; Fig. 1 B). Systolic and diastolic pressures were reduced in HF dogs, whereas left ventricular (LV) end-diastolic, LA, and right atrial (RA) pressures were increased (Fig. 1, C–E), consistent with the HF phenotype.
Figure 1.
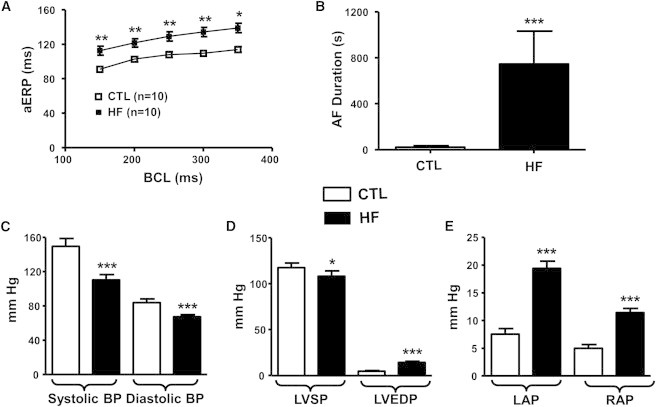
In vivo electrophysiological data and hemodynamic indices. (A) Atrial effective refractory period (aERP) as a function of basic cycle length (BCL). (B) AF duration. (C) Arterial blood pressure. (D) Left ventricular systolic pressure (LVSP) and end-diastolic pressure (LVEDP). (E) Left atrial pressure (LAP) and right atrial pressure (RAP). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus CTL.
Fibroblast Kv and Kir current remodeling in AF
Time- and voltage-dependent K+ current (IKv,fb) was elicited by 500 ms voltage-clamp steps to voltages ranging from −110 to +70 mV. IKv,fb activated rapidly and showed little inactivation, and was reversibly suppressed by 30 mmol/L TEA (Fig. 2, A and B). HF strongly downregulated the TEA-sensitive K+ current (Fig. 2 B). Mean current density-voltage relations (Fig. 2 C) showed statistically significant decreases in IKv,fb (by ∼70%) in HF fibroblasts over a wide range of voltages. LA fibroblasts had membrane capacitances of 17.8 ± 1.4 (n = 33) and 29.7 ± 2.2 (n = 33, p < 0.001 versus CTL) pF in CTL and HF, respectively (Fig. 2 D).
Figure 2.
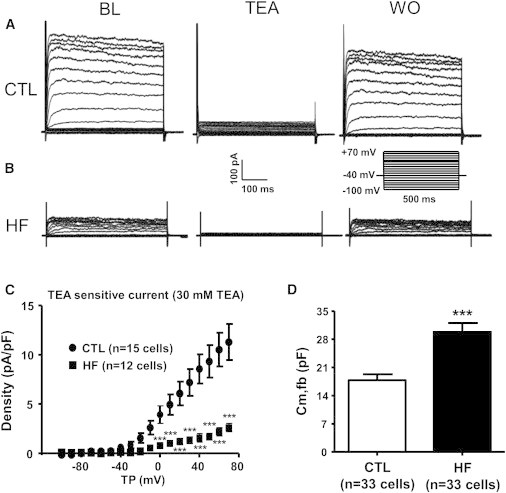
Experimental recordings of the TEA-sensitive current (IKv,fb). (A) Current densities at baseline (BL), after 30 mM TEA, and upon washout (WO) in a CTL atrial fibroblast (voltage protocol in inset). (B) Same as A, but for HF cells. (C) TEA-sensitive current density as a function of test potential (TP). (D) Fibroblast membrane capacitance (Cm,fb). Results are mean ± SEM. ∗∗∗p < 0.001 versus CTL.
Kir currents (IKir,fb) recorded from CTL and HF atrial fibroblasts are shown in Fig. 3 A. Current induced with a ramp depolarization at baseline before Ba2+ and current in the presence of 300 μmol/L Ba2+ are shown for individual cells obtained for each condition. Fig. 3 B shows the mean density of IKir,fb as a function of test voltage. At −120 mV, IKir,fb averaged −1.4 ± 0.6 in CTL (n = 15) and −2.6 ± 0.3 (n = 10) pA/pF in HF fibroblasts, an 86% increase (p < 0.001). Consistent with inward-rectifier current upregulation in HF, RMP averaged −42 ± 2 mV in CTL (n = 16) and −53 ± 2 mV in HF (n = 19, p < 0.01 versus CTL) fibroblasts (Fig. 3 C).
Figure 3.
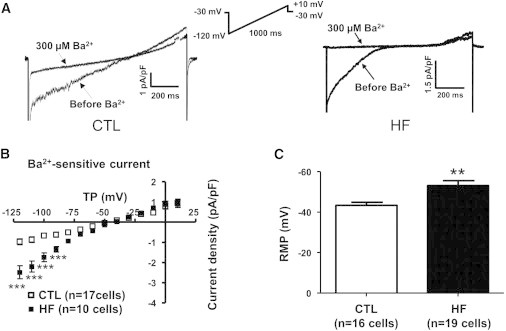
Experimental recordings for Kir current (IKir,fb). (A) Current densities for CTL (left) and HF (right) obtained with a ramp protocol before and after addition of 300 μM Ba2+. (B) Current densities as a function of TP. (C) Resting membrane potential (RMP). Results are mean ± SEM. ∗∗p < 0.01, ∗∗∗p < 0.001 versus CTL.
Effects of fibroblast electrical remodeling on the atrial cardiomyocyte AP
Fig. 4 shows the model-simulated cardiomyocyte transmembrane potential (Vm; Fig. 4 A), fibroblast transmembrane potential (Vm,fb; Fig. 4 B), gap-junction current (Igap; Fig. 4 C) and total fibroblast K+ current (IK,fb; Fig. 4 D), with progressive IKv,fb downregulation for a cardiomyocyte paced at 4 Hz coupled to two fibroblasts by a gap-junction conductivity of 3 nS. Downregulation of fibroblast IKv,fb decreased the total fibroblast K+ current (Fig. 4 D), reducing the fibroblast repolarizing current. This had the effect of increasing the gap-junction current flow from the fibroblast to the cardiomyocyte during phases 2 and 3 of the cardiomyocyte AP (Fig. 4 C), thereby making the fibroblast a source of depolarizing current. This depolarizing current led to progressive cardiomyocyte APD prolongation (Fig. 4 A). IKv,fb downregulation also had a depolarizing effect on the cardiomyocyte RMP.
Figure 4.
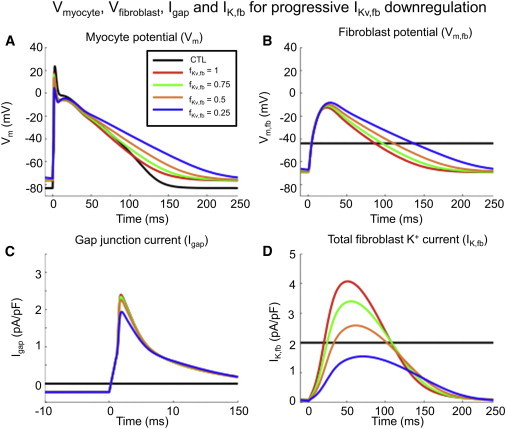
(A–D) Effect of fibroblast IKv,fb downregulation on (A) cardiomyocyte transmembrane potential (Vm), (B) fibroblast transmembrane potential (Vm,fb), (C) gap-junction current (Igap), and (D) total fibroblast K+ current (IK,fb). Progressive IKv,fb downregulation decreased the fibroblast repolarizing K+ current density, making the fibroblast behave as a current source during phases 2 and 3 of the cardiomyocyte AP, leading to APD prolongation. The cardiomyocyte RMP was depolarized by IKv,fb downregulation, leading to partial INa inactivation. Results were obtained with two fibroblasts connected to a cardiomyocyte paced at 4 Hz with a Ggap of 3 nS. CTL = single-cardiomyocyte action potential in the absence of fibroblast coupling. To see this figure in color, go online.
Fig. 5, A–D, show the same variables as in Fig. 4, but for graded IKir,fb upregulation. Upregulation of IKir,fb progressively increased the total fibroblast K+ current (Fig. 5 D), thus increasing the fibroblast repolarizing current. This effect increased the gap-junction current from the cardiomyocyte to the fibroblast, thereby making the fibroblast a current sink for the cardiomyocyte and shortening the cardiomyocyte APD (Fig. 5 A). IKir,fb upregulation also had a hyperpolarizing effect on the RMP, helping to maintain Na+ current availability during phase 0 of the AP.
Figure 5.
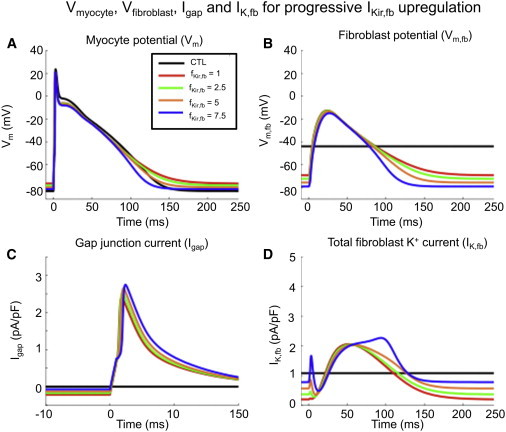
(A–D) Effect of fibroblast IKir,fb upregulation on the (A) cardiomyocyte transmembrane potential (Vm), (B) fibroblast transmembrane potential (Vm,fb), (C) gap-junction current (Igap), and (D) total fibroblast K+ current (IK,fb). IKir,fb upregulation increases fibroblast repolarizing K+ current density, making the fibroblast behave as a current sink during phases 2 and 3 of the myocyte AP, leading to APD shortening. The cardiomyocyte was hyperpolarized by IKir,fb upregulation, preserving Na+ channel availability. Results were obtained with two fibroblasts connected to a cardiomyocyte paced at 4 Hz with a Ggap of 3 nS. CTL = single-cardiomyocyte action potential in the absence of fibroblast coupling. To see this figure in color, go online.
Graded effects of fibroblast electrical remodeling on atrial tissue RMP, APD90, and conduction velocity
We determined the effects of fibroblast ion-current remodeling on cardiomyocyte cellular electrical properties by simulating activation of a cable over a wide range of pacing frequencies to vary the diastolic interval (DI). Fig. S2 shows the RMP (Fig. S2 A), AP duration (APD) at 90% repolarization (APD90; Fig. S2 B), and conduction velocity (CV; Fig. S2 C) as a function of DI for IKv,fb downregulation (red) and IKir,fb upregulation (green) over a 10 cm cable with two fibroblasts per cardiomyocyte and a Ggap of 3 nS. IKv,fb downregulation had a small depolarizing effect on the cardiomyocyte RMP but substantially prolonged APD compared with CTL (e.g., APD90 values at BCL = 500 ms for fKv,fb = 1.0, 0.75, 0.5, and 0.25 were 162 ms, 175 ms, 194 ms, and 225 ms, respectively; RMP values were −76.9 mV, −76.8 mV, −76.6 mV, and −76.4 mV, respectively). Conduction velocities paralleled the conduction velocities for the CTL condition over the spectrum of fKv,fb under investigation.
IKir,fb upregulation had the opposite effect on repolarization and resting potentials, i.e., it significantly hyperpolarized the cardiomyocyte RMP and shortened the APD90 versus CTL (APD90 values at BCL = 500 ms for fKir,fb = 1.0, 2.5, 5.0, and 10 were 162 ms, 152 ms, 137 ms, and 115 ms, respectively, with corresponding RMP values of −76.9 mV, −78.22 mv, −79.7 mV, and −81.2 mV, respectively). IKir,fb upregulation reduced CVs at long DIs compared with CTL, but CVs were increased compared with CTL at DIs of <150 ms, highlighting the biphasic effect of RMPs on the CV and the prolongation of the Na+ channel recovery time constant at more positive RMPs (27) (e.g., at DI ≈ 62 ms, CVs for fKir,fb = 1.0, 2.5, 5.0, and 10 were 72.2 cm/s, 78.4 cm/s, 85.4 cm/s, and 89.5 cm/s, respectively). A 1:1 conduction was sustained at progressively shorter DIs with increasing fKir,fb (Fig. S2 B).
Effect of fibroblast electrical remodeling on 2D spiral-wave dynamics
Fig. 6 shows the APD90 distribution over a 2D sheet of atrial cardiomyocytes with moderate fibrosis density and two fibroblasts per cardiomyocyte, with a Ggap of 3 nS, for progressive IKir,fb upregulation (Fig. 6, top row) and IKv,fb downregulation (Fig. 6, bottom row). In accordance with the single-cell and cable analyses, mean APD90 was progressively shortened by IKir,fb upregulation (mean APD90 values for fKir,fb = 1.0, 2.5, 5.0, and 10 were 121 ms, 116 ms, 107 ms, and 81 ms, respectively; Fig. 6, A–D). The APD90 dispersion, as quantified by the APD90 coefficient of variation, increased with IKir,fb upregulation (e.g., the coefficients of variation for CTL and fKir,fb = 10 were 0.8% and 6.2%, respectively). IKv,fb downregulation (Fig. 6, E–H) prolonged the mean APD90 (mean APD90 values for fKv,fb = 1.0, 0.75, 0.5, and 0.25 were 121 ms, 134 ms, 154 ms, and 159 ms, respectively) and increased the coefficient of variation to an extent similar to that observed for IKir,fb upregulation (coefficients of variation for CTL and fKv,fb = 0.25 were 0.8% and 6.2%, respectively). Fig. S3 illustrates the increasing spatial variability of APD due to heterogeneous distribution of fibroblasts as a function of ion-current remodeling, by showing results as a heatmap of APD deviations around the mean under each condition.
Figure 6.

(A–H) APD90 distribution over a 2D sheet of cardiomyocytes with patchy fibrosis (two fibroblasts with a Ggap of 3 nS per cardiomyocyte) in the 2D model for IKir,fb upregulation (top row, A–D) and IKv,fb downregulation (bottom row, E–H). APD90 was reduced by IKir,fb upregulation and increased by IKv,fb downregulation. Values above each panel are mean ± SD APD90 across the substrate. To see this figure in color, go online.
Fig. 7 A shows snapshots of the cardiomyocyte transmembrane potential as a function of time over a 2D sheet of cardiomyocytes for progressive IKir,fb upregulation with IKv,fb at its CTL value. For the nonremodeled condition (fKir,fb = 1, fKv,fb = 1, first column), the wavefront quickly drifted out of the substrate and became extinguished on the right boundary after completing a single revolution. For intermediate IKir,fb upregulation (fKir,fb = 2.5, second column), the wavefront completed about three revolutions before terminating. At higher levels of IKir,fb upregulation (fKir,fb = 5 and 10), the wavefront was successfully contained within the substrate boundaries, leading to sustained reentry. Fig. 7 B shows the rotor trajectories of the 2D substrate for the corresponding simulations: as fKir,fb was increased, the rotor stabilized into a more compact, quasi-periodic, rosette-like meandering pattern. Fig. S4 shows the power spectra analysis for IKir,fb upregulation: the dominant frequencies of rotors progressively increased with increasing fKir,fb, passing from 5.7 to 6.1 and then 6.7 Hz. The nonremodeled condition (fKir,fb = 1, fKv,fb = 1) terminated after completing a single revolution, precluding a formal calculation of the spiral wave’s dominant frequency (it was estimated to be 4.6 Hz).
Figure 7.
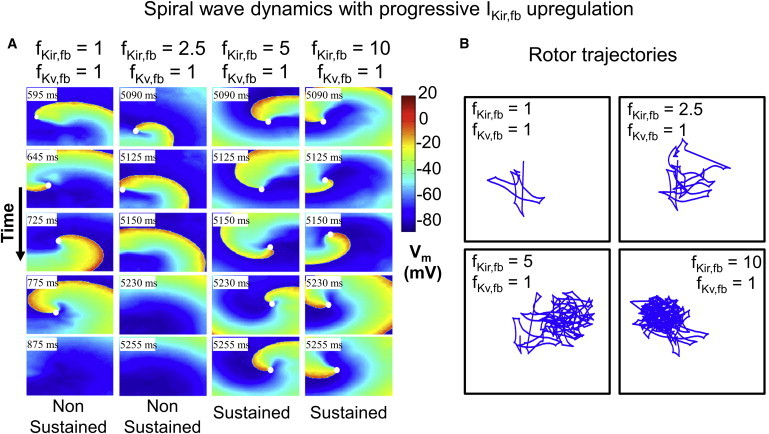
(A) Spiral-wave dynamics over time for progressive IKir,fb upregulation and fixed IKv,fb. White dots identify the rotor-tip phase singularities. In the CTL condition (fKir,fb = fKv,fb = 1), the phase singularity drifts rapidly and is extinguished on a boundary. With fKir,fb = 2.5, the spiral wave completes one complete revolution before it is extinguished. With fKir,fb = 5 and 10, reentry is sustained. (B) Rotor trajectory for the corresponding simulations in A. To see this figure in color, go online.
Fig. S5 A follows the same format as Fig. 7, but for IKv,fb downregulation while keeping IKir,fb at the CTL value. As described above, for the CTL condition (fKir,fb = 1, fKv,fb = 1, leftmost column), the wavefront rapidly became extinguished on a substrate boundary. Similar dynamics are observed with fKv,fb = 0.75 (fKir,fb = 1, fKv,fb = 0.75, second column). With 2- and 4-fold IKv,fb downregulation, the wavefront failed to depolarize neighboring cardiomyocytes, also leading to propagation failure and reentry termination with qualitatively similar dynamics (fKir,fb = 1, fKv,fb = 0.5 and 0.25, third and fourth columns). Fig. S5B shows the rotor trajectories for the corresponding simulations; all three trajectories had the same morphology. Hence, IKv,fb downregulation did not facilitate reentry, and reentry was not sustained long enough to allow for determination of the dominant frequency.
We then investigated the effect of combined IKir,fb upregulation and IKv,fb downregulation on reentry dynamics. Fig. 8, A and B, show representative examples of the dynamics observed under CTL conditions (fKir,fb = 1, fKv,fb = 1), with IKv,fb downregulation (fKir,fb = 1, fKv,fb = 0.25), IKir,fb upregulation (fKir,fb = 5, fKv,fb = 1), and IKir,fb upregulation with the addition of IKv,fb suppression (fKir,fb = 5, fKv,fb = 0), along with corresponding rotor trajectories. IKir,fb upregulation stabilized the primary rotor, leading to sustained reentry (Fig. 8 A, third column versus first column). Keeping IKir,fb at proarrhythmic conditions but simulating the addition of an IKv,fb blocker by IKv,fb downregulation (Fig. 8 A, fourth column) successfully terminated reentry, and the rotor dynamics (Fig. 8 B) became qualitatively similar to the CTL condition. In summary, the arrhythmogenic effect of fibroblast remodeling is strongly dependent on the relative degree of IKv,fb downregulation and IKir,fb upregulation, and IKv,fb suppression is able to terminate AF under proarrhythmic conditions.
Figure 8.
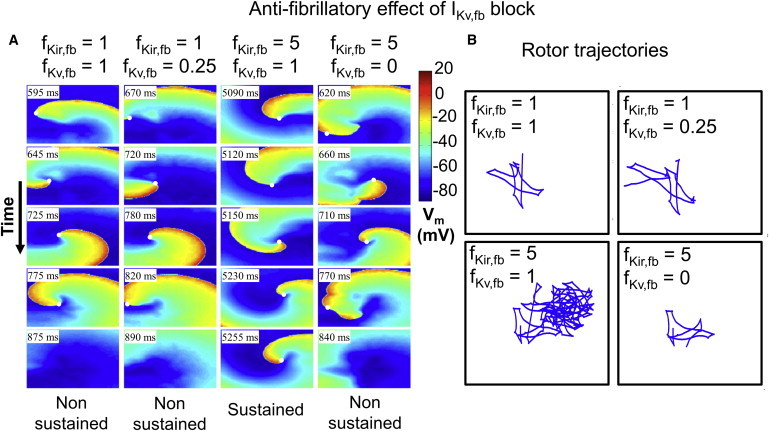
(A) Spiral-wave dynamics for representative cases of mixed IKir,fb upregulation and IKv,fb downregulation. Downregulation of IKv,fb terminates reentry after fewer revolutions under CTL conditions. Conversely, IKir,fb upregulation leads to sustained reentry. Simulating the addition of an IKv,fb blocker under proarrhythmic conditions (last column) demonstrates the potential antiarrhythmic effect of IKv,fb blockade, with reentry termination occurring promptly where reentry had previously been sustained. (B) Trajectories of spiral-wave rotor-core tip corresponding to cases in A. To see this figure in color, go online.
Fig. S6 shows the model-predicted effects of the average experimentally observed change in IKir,fb and IKv,fb remodeling on AF vulnerability. With two fibroblasts per cardiomyocyte, the spiral-wave dynamics were similar to those in the nonremodeled condition, with reentry terminating shortly after initiation. However, with four fibroblasts per cardiomyocyte, the HF-induced fibroblast ionic remodeling effect was greater, leading to more complex reentry dynamics and greater spiral-wave persistence compared with CTL. HF also increased fibroblast capacitance (Cm,fb), at least in part by increasing fibroblast size (Fig. S7). This increase would be expected to enhance the electrotonic influence of fibroblasts on coupled cardiomyocytes. The result of reproducing experimental HF-induced Cm,fb increases is also shown in Fig. S6. The HF-induced Cm,fb remodeling prolonged the time to termination and qualitatively increased the rotor trajectory complexity for two fibroblasts per cardiomyocyte, whereas it had the opposite effect with four fibroblasts per cardiomyocyte. These discrepant changes result from the fact that Cm,fb increases enhance the effects of both profibrillatory IKir,fb increases and antifibrillatory IKv,fb decreases, with the net change determined by the balance between the two.
Discussion
The main findings from this study are that 1), HF induces fibroblast ion-current remodeling by downregulating IKv,fb and upregulating IKir,fb; 2), in the presence of appreciable cardiomyocyte-fibroblast electrical coupling, fibroblast ionic remodeling affects the electrophysiology of coupled cardiomyocytes; and 3), atrial arrhythmogenesis is altered by fibroblast ion-current remodeling, with IKv,fb downregulation having an antifibrillatory effect and IKir,fb upregulation having a profibrillatory effect. To our knowledge, this is the first report to describe fibroblast ionic current remodeling in the setting of HF and investigate the potential effects of this remodeling on atrial arrhythmogenesis.
Relation to previous studies of fibroblast ion currents
The properties of IKir,fb and IKv,fb described in this work are grossly similar to those reported in prior studies of rat (29), canine (22,24), and human (30) fibroblasts. The downregulation of IKv,fb reported here is similar to our observations in previous work, both in fibroblasts from HF animals and occurring spontaneously under cell-culture conditions (22,24). We are not aware of any previous reports of IKir,fb remodeling in HF. The fibroblast RMP was hyperpolarized in HF compared with CTL (−53 ± 2.1 vs. −42 ± 1.9 mV) because of upregulation of IKir,fb, as the inward-rectifier current is the primary determinant of the fibroblast RMP (31). Our simulated nonremodeled fibroblast RMP closely matched the experimental measurements (experimental and simulated RMPs were −42 ± 1.9 vs. −43.2 mV, respectively). In our experiments, the baseline Cm,fb was somewhat larger than that previously reported for rat ventricular fibroblasts (17.8 ± 1.4 vs. 6.3 pF) (29) and greatly increased in HF animals (29.7 ± 2.2 pF), closer to the value of 53 pF reported for rat ventricular myofibroblasts (29) and passages 2–6 human fibroblasts (30). The differences among these studies may be due to the species, tissues, and study conditions used.
Potential role of fibroblast electrical remodeling in atrial arrhythmogenesis
There is extensive evidence from in vitro and simulation models that fibroblast coupling to cardiomyocytes can alter cardiac electrical activity and promote arrhythmogenesis (12), although clear in vivo evidence is lacking. Fibroblasts can couple cardiomyocytes over extended distances, producing arrhythmogenic coupling delays (15). Cardiac injury promotes fibroblast-cardiomyocyte interactions, enhancing their ability to contribute to arrhythmia formation (32). Coculture of myofibroblasts with cardiomyocytes is associated with electrotonic coupling that is highly arrhythmogenic (18). Mathematical modeling work suggests that cardiomyocyte-fibroblast interactions contribute to conduction abnormalities in arrhythmogenic LA posterior walls from HF animals (11) and to complex fractionated electrogram patterns in patients (23).
Mathematical modeling has been applied extensively to assess the potential electrophysiological consequences of fibroblast-cardiomyocyte coupling. Key determinants include the size of individual fibroblasts relative to cardiomyocytes, the number of fibroblasts coupled to cardiomyocytes, and fibroblast density (18–20,34–36). Here, we introduced a determinant that to our knowledge has not been examined before: changes in fibroblast K+-channel properties resulting from phenotypic alterations induced by cardiac pathology. We investigated the functional effect of fibroblast electrical remodeling on atrial arrhythmogenesis by independently upregulating IKir,fb and downregulating IKv,fb. IKir,fb upregulation proved to be profibrillatory, whereas IKv,fb downregulation was antifibrillatory. As summarized in Fig. S8, the profibrillatory effect of fibroblast IKir,fb upregulation was mediated through an increased fibroblast repolarizing current. This made the fibroblast act as a current sink for the cardiomyocyte, leading to 1), hyperpolarization of the atrial cardiomyocyte RMP and increased cardiomyocyte INa availability; and 2), shortening of the atrial cardiomyocyte APD, thereby preserving cardiomyocyte excitability and facilitating conduction and reentry at higher frequencies (such as those of AF). In contrast, the antifibrillatory effect of IKv,fb downregulation was mediated through a decrease in fibroblast repolarizing current. This made the fibroblast act as a current source for the cardiomyocyte, leading to 1), depolarization of the atrial cardiomyocyte RMP and decreased INa availability; and 2), prolongation of the APD, thereby decreasing cardiomyocyte excitability and impeding reentry.
Our findings are conceptually consistent with previous work on AF-induced cardiomyocyte remodeling by Pandit et al. (37), in which they described the rotor-stabilizing effects of atrial cardiomyocyte IKir,fb-equivalent (IK1) upregulation. They found that cardiomyocyte IK1 upregulation hyperpolarized the RMP, thereby increasing INa availability and shortening the APD. This is similar to what we observed with IKir,fb upregulation facilitating reentry and maintenance of the rotor underlying AF. In our study, the cardiomyocyte was hyperpolarized through a novel (to our knowledge) indirect mechanism (i.e., fibroblast electrical remodeling and cardiomyocyte-fibroblast electrical interaction) in the absence of intrinsic cardiomyocyte IK1 upregulation.
To clarify their individual effects, we performed most of our analyses of IKir,fb and IKv,fb remodeling by independently up- or downscaling individual current conductances. However, the experimental data show that IKv,fb and IKir,fb remodeling occurred simultaneously. Therefore, we investigated the effect of simultaneous IKv,fb downregulation and IKir,fb upregulation (Fig. S6), and found that the balance between these opposing effects determines the consequences of fibroblast ion-current remodeling on AF properties. Using experimentally obtained values for IKir,fb upregulation and IKv,fb downregulation, we found that HF-induced fibroblast ionic remodeling has the capacity to promote AF (Fig. S6) while prolonging the effective refractory period, as observed experimentally (Tables S1 and S2). However, for a given upregulated fKir,fb, one could observe sustained reentry, nonsustained reentry, or no reentry at all depending on the degree of IKv,fb downregulation. With our first-order model, it is difficult to directly correlate the magnitude of the experimentally observed remodeling with mathematical-model predictions regarding atrial arrhythmogenesis. Nevertheless, IKv,fb had to be downregulated to extreme values before it could counterbalance modest IKir,fb upregulation. Atrial cardiomyocyte ionic-current remodeling also occurs in HF (38). Implementing both cardiomyocyte and fibroblast ionic modeling (Tables S1 and S2), we found that the cardiomyocyte ionic remodeling previously reported to prolong the APD and ERP (38) makes reentry maintenance more difficult, but does not otherwise alter the qualitative properties of AF associated with fibroblast ionic remodeling as described here (Fig. S9).
Novelty and potential importance
The notion that atrial ion-current remodeling plays an important role in AF promotion has been established for almost 20 years (39). Atrial ion-current remodeling has always been associated with cardiomyocyte ion currents, but here we show for the first time (to our knowledge) that the fibroblast ion-current remodeling occurring with AF-promoting pathologies might affect AF susceptibility. The remodeling of ion currents in fibroblasts coupled to cardiomyocytes was able to contribute to the AF-maintaining substrate via electrotonic interactions that modified cardiomyocyte electrical function in profibrillatory ways. We also found that reducing IKv,fb could counter the profibrillatory effect of IKir,fb upregulation and lead to reentry termination (Fig. 8), providing a proof-of-principle for the plausibility of targeting the ion currents of cardiomyocyte-coupled fibroblasts for antiarrhythmic purposes. Conversely, one could target IKir,fb, as upregulation of this current was clearly found to facilitate reentry. Traditional cardiomyocyte K+-channel blockers are moderately effective antiarrhythmic agents; however, their use is severely limited by ventricular proarrhythmic risk (e.g., torsade de pointes), resulting from APD and QTc prolongation (40). A selective blocker of fibroblast Kv or Kir current with little action on corresponding cardiomyocyte currents might convey antiarrhythmic activity, with the benefit of interacting with atrial tissue predominantly in areas of fibrosis, thereby theoretically avoiding the risk of K+ channel blocker-induced ventricular proarrhythmia. If significant atrial fibroblast-cardiomyocyte coupling is confirmed in vivo, it might be of interest to search for molecular and/or functional differences between cardiomyocyte and fibroblast K+ channels as a basis for developing fibroblast-selective pharmacological agents.
Targeting fibroblast ionic currents for antiarrhythmic drug therapy might have applications beyond rhythm control of AF. Ventricular tachyarrhythmias in patients with ischemic cardiomyopathies are a major source of cardiovascular morbidity and a leading cause of sudden cardiac death (41). These arrhythmias often originate in postinfarction scars with complex networks of viable and hibernating myocardium imbedded within the area of infarction, making such highly proarrhythmic lesions difficult to treat via catheter ablation (42). Targeting ventricular fibroblast ion currents to produce antiarrhythmic electrophysiological changes in and around the infarct site may provide a new paradigm for the management of complex ventricular arrhythmias (43). Evidence suggests that modulating the fibroblast ATP-regulated K+ current may be exploited to alter border-zone electrophysiology in infarcted hearts (44).
Potential limitations
A significant limitation of this work is the lack of in vivo evidence of cardiomyocyte-fibroblast electrical coupling. Several studies have demonstrated the existence of cardiomyocyte-fibroblast electrical coupling in experimental cocultured media. Nevertheless, with the exception of work on the sinoatrial node (16), up to now, it has not been technically possible to determine whether such coupling occurs in vivo. Despite extensive experimentation with a variety of in vitro systems, this fundamental question remains to be resolved to establish the in vivo relevance of cardiomyocyte-fibroblast electrical interactions in the heart. Establishing the significance of fibroblast electrical remodeling in the pathogenesis of AF remains critically dependent on experimental validation of electrical coupling between cardiomyocytes and fibroblasts in the atrium in situ.
In this work, we only considered cardiomyocyte-fibroblast coupling (single-sided connections in Kohl and Camelliti’s (46) terminology), and did not consider that fibroblasts may couple to more than one cardiomyocyte at a time, creating electrical connections between previously disconnected cardiomyocytes (double-sided connections). We used a single specific value for Ggap, knowing that the magnitude of this parameter can have a significant impact on the dynamics of cardiomyocyte-fibroblast coupling (19). We selected a Ggap that is well within experimentally reported values, which range from 0.31 to 8 nS (13,14). In addition, we examined limited combinations of conditions of fibroblast coupling and numbers of fibroblasts coupled to cardiomyocytes (Nfb). Fig. S10 shows a further examination of the parameter space, varying Nfb, Ggap, and Cm,fb. Some of the parameter manipulations had the expected effects (e.g., decreasing Nfb or Cm,fb attenuated the effects of ionic remodeling, reducing Cm,fb, and reducing Ggap virtually eliminated the effect of IKv,fb downregulation). Increasing these variables had no clear effect, perhaps because the default values were already sufficiently large that further increases produced little additional change. We also did not include the HF-induced cardiomyocyte ionic and capacitance remodeling. A limited set of simulations with remodeled cardiomyocytes did not qualitatively change the nature of our results (Fig. S9). Furthermore, we used a 2D sheet to study the effect of fibroblast electrical remodeling on reentry dynamics. This choice led to the introduction of geometrical constraints that can potentially bias arrhythmia sustainability in situations of increased meandering, leading to rotor extinction on substrate boundaries, with substrate size being an important determinant of reentry maintenance.
Finally, we used a fibroblast ionic model that was developed based on observations in ventricular fibroblasts (20). Burstein et al. (47) showed that atrial and ventricular fibroblasts behave and proliferate differently. However, they did not identify any ion-channel subunit expression differences by genome-wide analysis, and recent work suggests that atrial and ventricular IKv,fb differ significantly only at physiologically nonrelevant voltages positive to +60 mV (24). In addition, there were no differences in IKv,fb for atrial versus ventricular fibroblasts from HF dogs (24).
Conclusions
HF induces fibroblast ion-current remodeling: fibroblast Kv current is downregulated, whereas fibroblast Kir current is upregulated. Mathematical modeling indicates antifibrillatory effects of IKv,fb downregulation and profibrillatory effects of IKir,fb upregulation, and reveals the underlying electrophysiological mechanisms. Therefore, the outcome of fibroblast electrical remodeling in atria with a disease-induced AF substrate will critically depend on the balance between pro- and antifibrillatory changes. If efficient electrical coupling between atrial fibroblasts and cardiomyocytes can be confirmed in vivo, pharmacological modulation of fibroblast K+ currents might be useful as a potential AF-selective target for antiarrhythmic drug therapy. These findings provide new insights (to our knowledge) into the potential role of fibroblasts in the pathogenesis and treatment of AF.
Acknowledgments
The authors thank Nathalie L’Heureux and Chantal St-Cyr for technical help, and France Thériault for secretarial assistance with the manuscript.
This work was supported by the Canadian Institutes of Health Research (grant 43565) and the Heart and Stroke Foundation of Canada.
Footnotes
Philippe Comtois and Stanley Nattel contributed equally to this work.
Supporting Material
References
- 1.Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415:219–226. doi: 10.1038/415219a. [DOI] [PubMed] [Google Scholar]
- 2.Ehrlich J.R., Nattel S. Novel approaches for pharmacological management of atrial fibrillation. Drugs. 2009;69:757–774. doi: 10.2165/00003495-200969070-00001. [DOI] [PubMed] [Google Scholar]
- 3.Roy D., Talajic M., Waldo A.L., Atrial Fibrillation and Congestive Heart Failure Investigators Rhythm control versus rate control for atrial fibrillation and heart failure. N. Engl. J. Med. 2008;358:2667–2677. doi: 10.1056/NEJMoa0708789. [DOI] [PubMed] [Google Scholar]
- 4.Wijffels M.C., Kirchhof C.J., Allessie M.A. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–1968. doi: 10.1161/01.cir.92.7.1954. [DOI] [PubMed] [Google Scholar]
- 5.Wijffels M.C., Kirchhof C.J., Allessie M.A. Electrical remodeling due to atrial fibrillation in chronically instrumented conscious goats: roles of neurohumoral changes, ischemia, atrial stretch, and high rate of electrical activation. Circulation. 1997;96:3710–3720. doi: 10.1161/01.cir.96.10.3710. [DOI] [PubMed] [Google Scholar]
- 6.Li D., Fareh S., Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation. 1999;100:87–95. doi: 10.1161/01.cir.100.1.87. [DOI] [PubMed] [Google Scholar]
- 7.Sun H., Chartier D., Nattel S. Intracellular calcium changes and tachycardia-induced contractile dysfunction in canine atrial myocytes. Cardiovasc. Res. 2001;49:751–761. doi: 10.1016/s0008-6363(00)00294-7. [DOI] [PubMed] [Google Scholar]
- 8.Carver W., Nagpal M.L., Terracio L. Collagen expression in mechanically stimulated cardiac fibroblasts. Circ. Res. 1991;69:116–122. doi: 10.1161/01.res.69.1.116. [DOI] [PubMed] [Google Scholar]
- 9.Kawara T., Derksen R., de Bakker J.M. Activation delay after premature stimulation in chronically diseased human myocardium relates to the architecture of interstitial fibrosis. Circulation. 2001;104:3069–3075. doi: 10.1161/hc5001.100833. [DOI] [PubMed] [Google Scholar]
- 10.Spach M.S., Heidlage J.F., Barr R.C. Mechanism of origin of conduction disturbances in aging human atrial bundles: experimental and model study. Heart Rhythm. 2007;4:175–185. doi: 10.1016/j.hrthm.2006.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka K., Zlochiver S., Kalifa J. Spatial distribution of fibrosis governs fibrillation wave dynamics in the posterior left atrium during heart failure. Circ. Res. 2007;101:839–847. doi: 10.1161/CIRCRESAHA.107.153858. [DOI] [PubMed] [Google Scholar]
- 12.Yue L., Xie J., Nattel S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc. Res. 2011;89:744–753. doi: 10.1093/cvr/cvq329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rook M.B., Jongsma H.J., de Jonge B. Single channel currents of homo- and heterologous gap junctions between cardiac fibroblasts and myocytes. Pflugers Arch. 1989;414:95–98. doi: 10.1007/BF00585633. [DOI] [PubMed] [Google Scholar]
- 14.Rook M.B., van Ginneken A.C., Jongsma H.J. Differences in gap junction channels between cardiac myocytes, fibroblasts, and heterologous pairs. Am. J. Physiol. 1992;263:C959–C977. doi: 10.1152/ajpcell.1992.263.5.C959. [DOI] [PubMed] [Google Scholar]
- 15.Gaudesius G., Miragoli M., Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ. Res. 2003;93:421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 16.Camelliti P., Green C.R., Kohl P. Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ. Res. 2004;94:828–835. doi: 10.1161/01.RES.0000122382.19400.14. [DOI] [PubMed] [Google Scholar]
- 17.Miragoli M., Gaudesius G., Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ. Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 18.Zlochiver S., Muñoz V., Jalife J. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys. J. 2008;95:4469–4480. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacquemet V., Henriquez C.S. Loading effect of fibroblast-myocyte coupling on resting potential, impulse propagation, and repolarization: insights from a microstructure model. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H2040–H2052. doi: 10.1152/ajpheart.01298.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacCannell K.A., Bazzazi H., Giles W.R. A mathematical model of electrotonic interactions between ventricular myocytes and fibroblasts. Biophys. J. 2007;92:4121–4132. doi: 10.1529/biophysj.106.101410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wakili R., Voigt N., Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Invest. 2011;121:2955–2968. doi: 10.1172/JCI46315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dawson K., Wu C.T., Nattel S. Congestive heart failure effects on atrial fibroblast phenotype: differences between freshly-isolated and cultured cells. PLoS ONE. 2012;7:e52032. doi: 10.1371/journal.pone.0052032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashihara T., Haraguchi R., Trayanova N.A. The role of fibroblasts in complex fractionated electrograms during persistent/permanent atrial fibrillation: implications for electrogram-based catheter ablation. Circ. Res. 2012;110:275–284. doi: 10.1161/CIRCRESAHA.111.255026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu C.T., Qi X.Y., Nattel S. Disease and region-related cardiac fibroblast potassium current variations and potential functional significance. Cardiovasc. Res. 2014;102:487–496. doi: 10.1093/cvr/cvu055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramirez R.J., Nattel S., Courtemanche M. Mathematical analysis of canine atrial action potentials: rate, regional factors, and electrical remodeling. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H1767–H1785. doi: 10.1152/ajpheart.2000.279.4.H1767. [DOI] [PubMed] [Google Scholar]
- 26.Kneller J., Zou R., Nattel S. Cholinergic atrial fibrillation in a computer model of a two-dimensional sheet of canine atrial cells with realistic ionic properties. Circ. Res. 2002;90:E73–E87. doi: 10.1161/01.res.0000019783.88094.ba. [DOI] [PubMed] [Google Scholar]
- 27.Xie Y., Garfinkel A., Qu Z. Effects of fibroblast-myocyte coupling on cardiac conduction and vulnerability to reentry: a computational study. Heart Rhythm. 2009;6:1641–1649. doi: 10.1016/j.hrthm.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Comtois P., Sakabe M., Nattel S. Mechanisms of atrial fibrillation termination by rapidly unbinding Na+ channel blockers: insights from mathematical models and experimental correlates. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H1489–H1504. doi: 10.1152/ajpheart.01054.2007. [DOI] [PubMed] [Google Scholar]
- 29.Chilton L., Ohya S., Giles W.R. K+ currents regulate the resting membrane potential, proliferation, and contractile responses in ventricular fibroblasts and myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H2931–H2939. doi: 10.1152/ajpheart.01220.2004. [DOI] [PubMed] [Google Scholar]
- 30.Li G.R., Sun H.Y., Lau C.P. Characterization of multiple ion channels in cultured human cardiac fibroblasts. PLoS ONE. 2009;4:e7307. doi: 10.1371/journal.pone.0007307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shibukawa Y., Chilton E.L., Giles W.R. K+ currents activated by depolarization in cardiac fibroblasts. Biophys. J. 2005;88:3924–3935. doi: 10.1529/biophysj.104.054429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vasquez C., Mohandas P., Morley G.E. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ. Res. 2010;107:1011–1020. doi: 10.1161/CIRCRESAHA.110.227421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reference deleted in proof.
- 34.Sachse F.B., Moreno A.P., Abildskov J.A. A model of electrical conduction in cardiac tissue including fibroblasts. Ann. Biomed. Eng. 2009;37:874–889. doi: 10.1007/s10439-009-9667-4. [DOI] [PubMed] [Google Scholar]
- 35.Xie Y., Garfinkel A., Qu Z. Cardiac alternans induced by fibroblast-myocyte coupling: mechanistic insights from computational models. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H775–H784. doi: 10.1152/ajpheart.00341.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDowell K.S., Arevalo H.J., Trayanova N.A. Susceptibility to arrhythmia in the infarcted heart depends on myofibroblast density. Biophys. J. 2011;101:1307–1315. doi: 10.1016/j.bpj.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pandit S.V., Berenfeld O., Jalife J. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys. J. 2005;88:3806–3821. doi: 10.1529/biophysj.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li D., Melnyk P., Nattel S. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation. 2000;101:2631–2638. doi: 10.1161/01.cir.101.22.2631. [DOI] [PubMed] [Google Scholar]
- 39.Yue L., Feng J., Nattel S. Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ. Res. 1997;81:512–525. doi: 10.1161/01.res.81.4.512. [DOI] [PubMed] [Google Scholar]
- 40.Tamargo J., Caballero R., Delpón E. Pharmacology of cardiac potassium channels. Cardiovasc. Res. 2004;62:9–33. doi: 10.1016/j.cardiores.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 41.Zheng Z.J., Croft J.B., Mensah G.A. Sudden cardiac death in the United States, 1989 to 1998. Circulation. 2001;104:2158–2163. doi: 10.1161/hc4301.098254. [DOI] [PubMed] [Google Scholar]
- 42.Josephson M.R. 4th ed. Lippincott Williams & Wilkins; Philadelphia: 2008. Clinical Cardiac Electrophysiology: Techniques and Interpretations. [Google Scholar]
- 43.Feld Y., Melamed-Frank M., Gepstein L. Electrophysiological modulation of cardiomyocytic tissue by transfected fibroblasts expressing potassium channels: a novel strategy to manipulate excitability. Circulation. 2002;105:522–529. doi: 10.1161/hc0402.102661. [DOI] [PubMed] [Google Scholar]
- 44.Benamer N., Vasquez C., Morley G.E. Fibroblast KATP currents modulate myocyte electrophysiology in infarcted hearts. Am. J. Physiol. Heart Circ. Physiol. 2013;304:H1231–H1239. doi: 10.1152/ajpheart.00878.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reference deleted in proof.
- 46.Kohl P., Camelliti P. Cardiac myocyte-nonmyocyte electrotonic coupling: implications for ventricular arrhythmogenesis. Heart Rhythm. 2007;4:233–235. doi: 10.1016/j.hrthm.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 47.Burstein B., Libby E., Nattel S. Differential behaviors of atrial versus ventricular fibroblasts: a potential role for platelet-derived growth factor in atrial-ventricular remodeling differences. Circulation. 2008;117:1630–1641. doi: 10.1161/CIRCULATIONAHA.107.748053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.