

Abstract
Erythropoietin receptor (EpoR) dimerization is an important step in erythrocyte formation. Its transmembrane domain (TMD) and juxtamembrane (JM) region are essential for signal transduction across the membrane. A construct compassing residues S212–P259 and containing the TMD and JM region of the human EpoR was purified and reconstituted in detergent micelles. The solution structure of the construct was determined in dodecylphosphocholine (DPC) micelles by solution NMR spectroscopy. Structural and dynamic studies demonstrated that the TMD and JM region are an α-helix in DPC micelles, whereas residues S212–D224 at the N-terminus of the construct are not structured. The JM region is a helix that contains a hydrophobic patch formed by conserved hydrophobic residues (L253, I257, and W258). Nuclear Overhauser effect analysis, fluorescence spectroscopy, and paramagnetic relaxation enhancement experiments suggested that the JM region is exposed to the solvent. The structures of the TMD and JM region of the mouse EpoR were similar to those of the human EpoR.
Introduction
The erythropoietin (EPO) receptor (EpoR) is a member of the cytokine receptor family, which contains proteins that span the plasma membrane and transfer signals by interacting with cytokine and growth factor proteins (1). Activation of the EpoR by its ligand, EPO, is essential for erythropoiesis (2,3). EPO binding to EpoR activates Janus kinase 2 (JAK2) tyrosine kinase, which autophosphorylates tyrosine residues of JAK2 and phosphorylates residues in the cytoplasmic domain of the EpoR. Phosphorylated tyrosine residues become docking sites for several molecules containing Src homology 2 (SH2) domains that are required for activation of the mitogen-activated protein kinase pathway (4). For example, EpoR can activate signal transducers and activators of transcription (STAT) proteins, which are activated to participate in gene control (5,6).
EpoR is a single-span membrane protein that consists of an extracellular domain, a transmembrane domain (TMD), a juxtamembrane (JM) region, and a cytoplasmic region (Fig. 1 A). The extracellular domain of the EpoR contains two fibronectin type II domains, and crystallographic studies have demonstrated that the extracellular domain exists as a dimer even in the absence of EPO (3,7). The TMD and JM region of the EpoR are important for receptor function (Fig. 1, B and C). One possible mechanism is that two JAK2 proteins associate with a preformed EpoR dimer. To initiate signal transduction, two JAK2 proteins need to be in a close contact, which can be achieved by EPO binding to the extracellular domain to cause a conformational change through the TMD (8,9). Mutagenesis studies have shown that there are two regions in the cytoplasmic region of the EpoR that are required for JAK2 activation (10).
Figure 1.
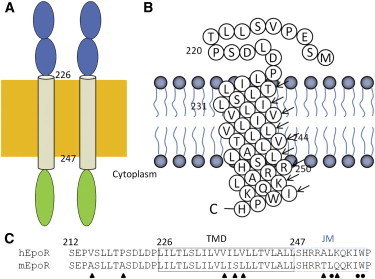
Topology of the EpoR. (A) Diagram of the EpoR. The extracellular region, TMD and JM region, and cytoplasmic region are shown in blue, gray, and green, respectively. The membrane is shown as a brown box. The TMD contains residues L216–L247. (B) Topology of the hEpoR construct used in this study. The hEpoR sequence was obtained from the UniProt Knowledgebase (http://www.uniprot.org; accession number P19235). Sequence numbering does not include the N-terminal 25-residue signal peptide. (C) Sequence alignment of hEpoR and mEpoR. The TMD is highlighted with solid lines and the JM region is highlighted with blue and dashed lines. The different amino acids are indicated with triangles. The conserved hydrophobic residues in the JM region are indicated with black circles. To see this figure in color, go online.
The TMD and JM region of the EpoR are of great interest because of their functions in receptor activation. Constantinescu et al. (11) analyzed ligand-independent association using an immunofluorescence colocalization assay, which showed that the TMD of the EpoR was sufficient to maintain dimerization of the full-length receptor. Using a series of dimeric coiled-coil-containing mutants to constrain the TMD into seven possible relative orientations, Seubert et al. (12) showed that the TMD is important for its orientation and activation of downstream pathways. Using cysteine-scanning mutagenesis, Lu et al. (13) showed that the TMD and JM region of the EpoR are important for dimerization and activation of the EpoR by EPO binding. Using the ToxR interaction screen system, Kubatzky et al. (14) demonstrated that isolated TMDs of both the mouse EpoR (mEpoR) and human EpoR (hEpoR) self-associated. In that study, two leucine residues (L240 and L241) in the TMD were shown to be important for receptor dimerization and signaling. A further thermodynamic study using sedimentation equilibrium analytical ultracentrifugation was carried out for the TMDs of both the hEpoR and mEpoR (9). Both TMDs were demonstrated to be able to dimerize in 3-N,N-dimethylmyristyl-ammonio propanesulfonate (C14SB) micelles, and it was suggested that the mEpoR TMD may have higher dimerization affinity than the hEpoR TMD (9).
Despite the functional and biochemical studies that have been published to date, the atomic structure of the TMD of the EpoR is still not available. Although it is known that the TMD and JM region of the EpoR are important for dimerization, the structure and dynamics of their monomeric form would provide insight into their roles in signal transduction. In this study, we used solution NMR spectroscopy to determine the solution structure of a construct that contained the TMD and JM region of the hEpoR in DPC micelles. The hEpoR was mainly monomeric under our experimental conditions. Information about the structure and dynamics of the TMD and JM region of the EpoR will aid in elucidating their roles in signal transduction.
Materials and Methods
Materials
cDNAs encoding the TMD and JM region encompassing residues S212–P259 of the hEpoR and mEpoR were synthesized by GenScript (Piscataway, NJ). pET-29b plasmid was purchased from Merck (San Diego, CA). The SDS-PAGE system, including NuPAGE gels and SDS-PAGE molecular weight standard, were purchased from Invitrogen (Carlsbad, CA). Protein sample loading buffer, SDS-PAGE, and western blot molecular weight standards were purchased from Bio-Rad (Hercules, CA). Bl21 (DE3)-competent cells for protein expression were purchased from Stratagene (Santa Clara, CA). β-D-1-thiogalactopyranoside (IPTG), dithiothreitol (DTT), and detergents (dodecylphosphocholine (DPC), deuterated DPC (D-DPC), lyso-myristoyl phosphatidylcholine (LMPC), lyso-myristoyl phosphatidylglycerol (LMPG), SDS, dihexanoylphosphatidylcholine (DHPC), and dimyristoylphosphatidylcholine (DMPC)) were purchased from Anatrace (Maumee, OH) or Avanti (Birmingham, AL). 15NH4Cl, 13C-glucose, and D2O were purchased from Cambridge Isotope Laboratories (Andover, MA). All other chemicals used in this study were purchased from Sigma (St. Louis, MO).
Expression and purification of the EpoR
The cDNAs of the hEpoR and mEpoR were cloned into the NdeI and XhoI sites of pET29b, respectively. The resulting plasmid pET29-EpoR encodes a protein sequence that contains S212–P259 of hEpoR or mEpoR with six histidine residues (HHHHHH) at its C-terminus for protein purification. The expressed construct contains the TMD and JM region of the EpoR. Plasmid pET29-EpoR was transformed in Escherichia coli-competent cells and plated onto an LB plate containing kanamycin. The EpoR was expressed and purified as described previously (15). Briefly, two to three colonies from the plate were picked up and inoculated in 20 ml of M9 medium and incubated at 37°C overnight with shaking. The overnight culture was then transferred into 1 liter of M9 medium supplied with kanamycin. When the OD600 reached 0.6–0.8, protein was induced overnight at 37°C by adding IPTG to a final concentration of 1 mM. The E. coli cells were harvested and resuspended in a lysis buffer that contained 20 mM Tris-HCl, pH 7.8, 300 mM NaCl, and 2 mM β-mercaptoethanol. The cells were then broken up by sonication in an ice bath. The cell lysate was centrifuged at 20,000 × g for 20 min. The pellet was washed with the lysis buffer and solubilized in a urea buffer that contained 8 M urea, 300 mM NaCl, 10 mM SDS, 20 mM Tris-HCl, pH 7.8. The solution was centrifuged at 48,000 × g for 20 min at room temperature. The supernatant was then loaded in a gravity column that contained nitrilotriacetic acid saturated with nickel (Ni2+-NTA) resin. The resin was washed with 10 column volumes of urea buffer containing 20 mM imidazole. Urea was removed by washing the resin with 10 column volumes of lysis buffer with 10 mM SDS. Protein was eluted using an elution buffer that contained 300 mM imidazole (pH 6.5) and 10 mM SDS (15 mM DPC, 2 mM LMPC, 2 mM LMPG, or 20 mM DHPC) after the resin was washed with 10 column volumes of a washing buffer (lysis buffer containing 2–20 mM detergent). Imidazole in the sample was removed using a PD10 column or by gel filtration chromatography using a buffer that contained 20 mM sodium phosphate (pH 6.5), 1 mM DTT, and 10 mM SDS (15 mM DPC, 2 mM LMPC, 2 mM LMPG, or 2% bicelles containing DHPC and DMPC with a q-value of 0.33). The sample was concentrated to 200–400 μl before it was put into an NMR tube for data acquisition.
Gel filtration experiment
Purified protein from Ni2+-NTA resin was concentrated from 8 ml to 1 ml using a 3 kDa molecular mass cutoff concentrator and loaded on a Superdex 200 10/300 GL column that was pre-equilibrated with a gel filtration buffer containing 20 mM sodium phosphate (pH 6.5), 15 mM DPC, and 1 mM DTT. The flow rate was 0.5 ml/min and purification was conducted at 4°C. The absorbance at 280 and 215 nm was monitored continuously and fractions were collected for SDS-PAGE analysis. Fractions from a single peak were combined and concentrated using a concentrator for further analysis.
Cross-linking experiment
Cross-linking of EpoR samples using glutaraldehyde (GA) was performed as previously described (16) with some modifications. Purified samples in different detergents from E. coli were buffer exchanged to a cross-linking buffer containing 20 mM sodium phosphate (pH 6.5), 0.1 mM DTT, and 15 mM detergent. The protein concentration was diluted to 50 μM to a final volume of 50 μl. GA from a 25% stock was added to the protein solution to a final concentration of 16 mM. The mixture was incubated at room temperature for 10 min. The reaction was stopped by adding SDS-PAGE loading buffer. Samples were analyzed by SDS-PAGE and western blot using an anti-his antibody.
Resonance assignment
A uniformly 13C/15N-labeled sample was prepared using both affinity and gel filtration chromatography and concentrated to 0.5 mM in a buffer that contained 20 mM sodium phosphate (pH 6.5), 1 mM DTT, 200 mM DPC, and 10% D2O. NMR spectra were recorded at 313 K on a Bruker Avance 600 or Avance 700 spectrometers with a cryogenic triple-resonance probe. The pulse programs were obtained from the Topspin 2.1 program library. Data were processed with NMRPipe (17) and analyzed using NMRView (18). Backbone resonances were assigned using 2D-1H-15N-HSQC, 3D HNCACB, HNCA, HN(CO)CA, HN(CO)CACB, HNCO, and HBHACONH experiments. Secondary structure was identified by analysis of 13C secondary chemical shifts (19) and TALOS+ (20). Distance constraints were obtained from a 3D 15N-edited nuclear Overhauser effect spectroscopy (NOESY; mixing time: 100 ms) experiment under the aforementioned conditions. The NOE connection was plotted using CYANA (21).
H-D exchange experiments
Amide H-D exchange experiments were performed at 40°C using a 15N-labeled sample. Sample aliquots were frozen in liquid nitrogen and lyophilized. Samples were then resuspended in solutions that contained 5%, 10%, 30%, 50%, 70%, and 90% D2O, respectively. 2D-1H-15N-HSQC spectra were obtained after a fixed 30 min incubation time, which was previously shown to be suitable for H-D exchange experiments (22). Data were processed with NMRPipe (17) and analyzed using NMRView (18). Residues with crosspeaks that appeared in 90% D2O were considered to form hydrogen bonds.
Paramagnetic relaxation enhancement and relaxation experiments
A paramagnetic relaxation enhancement (PRE) experiment was conducted using a 15N-labeled sample. The hEpoR was prepared at 0.3 mM concentration in a buffer that contained 20 mM sodium phosphate (pH 6.5), 1 mM DTT, and 120 mM DPC. Diethylenetriaminepentaacetic acid gadolinium (Gd-DTPA) was first dissolved in water to make a 50 mM stock solution. 1H-15N-HSQC spectra of hEpoR in the presence and absence of Gd-DTPA were collected, processed, and compared. R1, R2, and {1H}-15N steady-state NOE values (23) were measured at 313 K on a Bruker Avance II 600 MHz spectrometer. For R1 measurement, data were recorded with relaxation delays of 10, 50, 100, 200, 400, 800, 1200, 1400, 1600, and 1800 ms. For R2 measurement, data were acquired with delays of 16.9, 34, 51, 68, 85, 102, 119, 136, and 153 ms. Steady-state {1H}-15N NOEs were obtained using two data sets that were collected with and without initial proton saturation for a period of 3 s (24).
Structure determination
The hEpoR structure was determined using XPLOR-NIH with the Python interface (25–27). Backbone dihedral angle restraints were generated using TALOS+ (20). The NOE peaks were picked manually from a 3D 15N-edited NOESY and calibrated using NMRview (18). Structure determination was carried out using a randomized template. Simulated annealing and energy minimization were carried out as previously described (28). Simulated annealing was carried out with a starting temperature of 3500 K and 15,000 cooling steps. The structure was energy minimized by Powell energy minimization. Fifty hEpoR structures were obtained and 20 with the lowest energies were selected and deposited in the Protein Data Bank (PDB) under accession number 2MV6.
Fluorescence spectroscopy
Fluorescence measurements was obtained as previously described (29,30). The fluorescence emission spectra were measured in a 96-well plate. Purified hEpoR (100 μM) was prepared in a buffer that contained 20 mM sodium phosphate (pH 6.5) and 40 mM DPC. Samples with a 100 μl volume were subjected to analysis. The excitation wavelength was 280 nm and the emission was scanned from 305 to 400 nm. Experiments were carried out at 25°C.
Results
NMR spectra of the hEpoR in bicelles and micelles
To perform structural studies of the TMD and JM region of the hEpoR, we expressed the hEpoR in E. coli and purified it from inclusion bodies using an on-column refolding method that was used for single-span membrane protein purifications. We showed that the hEpoR construct (Fig. 1 B) was expressed in E. coli and could be reconstituted in both micelles and bicelles (Fig. S1 in the Supporting Material). It was obvious that the yield of the hEpoR in micelles was much higher than in a bicelle system that contained 3:1 (molar ratio) DHPC/DMPC. More than 4 mg of 13C/15N-labed hEpoR could be obtained per liter of culture when four detergent micelles (SDS, LMPC, LMPG, and DPC) were used for protein purification (Fig. S1). A cross-linking experiment was carried out to determine whether hEpoR could form dimers or oligomers when it was purified into micelles or bicelles (Fig. 2). When hEpoR was purified in bicelles, the dimer band of the hEpoR was observed even in the absence of GA (Fig. 2). In the presence of GA, hEpoR dimer was observed in all of the samples. Trimer and oligomers were observed when hEpoR was purified in LMPC and DPC micelles (Fig. 2). We then tested the NMR spectra of the purified hEpoR in these membrane-mimicking systems, which are frequently used in structural studies (31,32). When the hEpoR was reconstituted in LMPC micelles and bicelles, the quality of the 1H-15N-HSQC spectra was poor due to line-broadening (Fig. 3). High-quality spectra were obtained when the hEpoR was purified in LMPG, SDS, and DPC micelles (Figs. 3 and 4 A). Interestingly, the 1H-15N-HSQC spectrum of the hEpoR in SDS micelles exhibited more peaks than expected (Fig. 3), which may arise from conformational exchanges. Taken together with the cross-linking result, these findings suggest that the line-broadening observed for the samples in LMPC and bicelles may be due to sample dimerization or oligomerization (Fig. 3). We then focused on the sample in DPC micelles for further study because hEpoR exhibited the highest-quality spectrum and the capability to form both dimer and oligomers in the cross-linking experiment (Fig. 2). The 1H-15N-HSQC spectrum of hEpoR in DPC micelles exhibited 41 of 45 (excluding prolines and the six C-terminal histidines) expected peaks. In gel filtration chromatography, hEpoR had a retention volume of 15 ml (Fig. S1), which is similar to the monomeric TMD of the insulin receptor (15). In addition, the narrow line widths of the crosspeaks of the purified sample suggested that hEpoR was mainly monomeric in DPC micelles.
Figure 2.
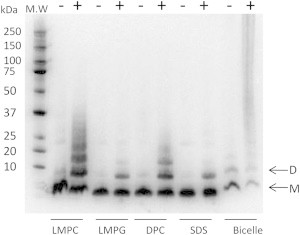
Cross-linking of hEpoR in different membrane systems. Protein was cross-linked using GA. Samples were separated by SDS-PAGE and followed by western blot using an anti-his antibody. The bands above the dimer are trimer or oligomers of the hEpoR. D, dimer; M, monomer.
Figure 3.
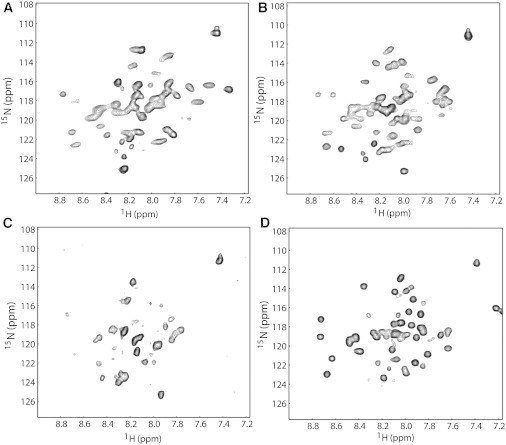
1H-15N-HSQC spectra of the hEpoR in different membrane systems. (A) 1H-15N-HSQC spectrum of 1.2 mM of hEpoR in 30 mM LMPC micelles. (B) 1H-15N-HSQC spectrum of 1.5 mM of hEpoR in 30 mM LMPG micelles. (C) 1H-15N-HSQC spectrum of 0.6 mM hEpoR in 20% DHPC/DMPC bicelles (q = 0.33). (D) 1H-15N-HSQC spectrum of 1.5 mM of hEpoR in 80 mM SDS micelles. All data were acquired at 40°C. The 1H-15N-HSQC spectrum of hEpoR in DPC micelles is shown in Fig. 4A.
Figure 4.
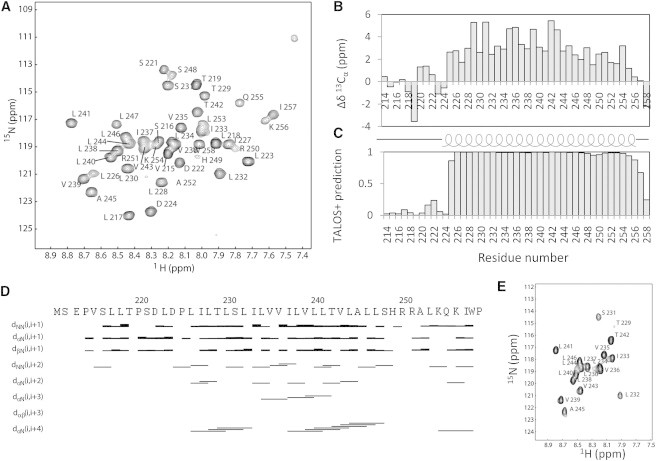
Assignment and secondary structure analysis of hEpoR in DPC micelles. (A) Assigned 1H-15N-HSQC spectrum of hEpoR in DPC micelles. (B) Deviations of the observed Cα chemical-shift values from the corresponding random-coil chemical-shift values. (C) Secondary structure analysis of hEpoR in DPC micelles by TALOS+. The positive value is the possibility (0–1) of the corresponding amino acid being a helix. (D) NOE connections of the hEpoR in DPC micelles. (E) H-D exchange experiment. The 1H-15N-HSQC spectrum of the hEpoR in 90% D2O and assignment of the crosspeaks are shown with the residue name and sequence number.
Secondary structure and dynamics analysis of the hEpoR
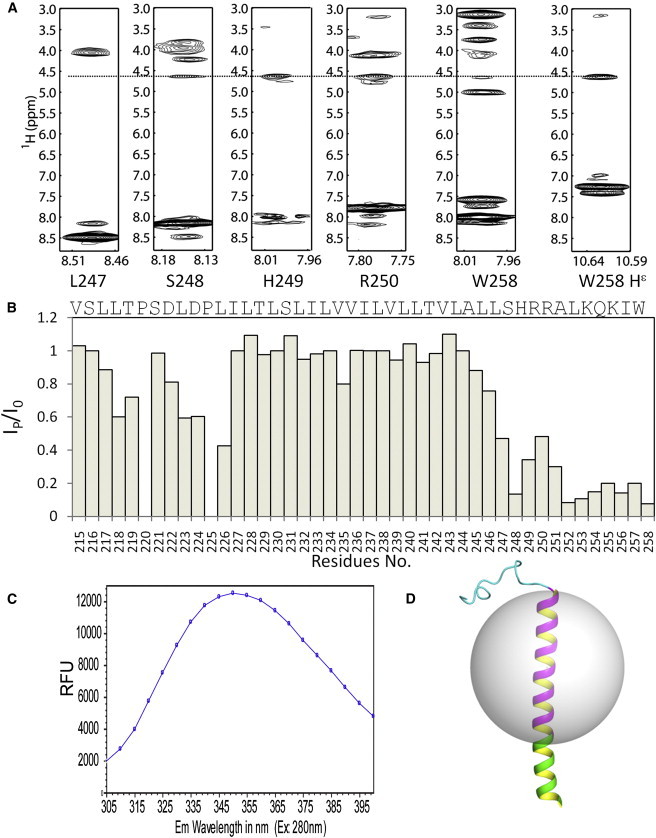
NMR resonances of the hEpoR were assigned using 3D triple-resonance and NOESY experiments (15). Approximately 90% of backbone assignments for the hEpoR monomer in DPC micelles were completed, with the exception of S212 and E213 (Fig. 4 A). The assignment has been deposited in the BioMagResBank under accession number 25079. Secondary structure analysis was conducted using both the Cα chemical-shift difference from random coil values and TALOS+ (Fig. 4, B and C). Both methods gave similar results (Fig. 4). The hEpoR contains only one helix formed by residues 226–257. The JM region also exists as a helix, consistent with a previous prediction (33). Although we could not observe all of the NOEs between Hα of residue i and HN of residue i+4 characterized for a helix, for residues in the TMD, the NOE connection and H-D exchange experiment suggested that these residues are helical (Fig. 4, D and E). Our TALOS analysis showed that the JM region tends to be helical (Fig. 4 C). The lack of a NOE assignment for residues H449–L253 suggested that this region might be flexible or undergo exchanges with the environment. Residues of the JM region, including H249, R250, and R251, are positively charged, which suggests that these residues may have a tendency to interact with the negatively charged membrane surface.
To assess hEpoR flexibility in the picosecond-to-nanosecond timescale, we obtained R1, R2, and 15N-{1H} heteronuclear NOE (hetNOE) values (Fig. 5). The N-terminus of the EpoR construct in this study was flexible, as characterized by the high R1 and low R2 and hetNOE values (Fig. 5). It is not surprising that the TMD (L226–L247) exhibited low R1 and high R2 and hetNOE values (Fig. 5). R2/R1 analysis demonstrated that there were three regions with different values for the hEpoR construct. The residues in the N-terminal region exhibited the lowest R2/R1 values due to its high flexibility. The residues from the TMD exhibited the highest R2/R1 values because it was embedded in DPC micelles. The residues from the JM region exhibited values between those of the N-terminal domain and the TMD, suggesting that the JMD was not buried in the micelles. A few residues, such as H245, R252, and A253, exhibited slightly lower hetNOE values than residues in the JM region, which may be due to the interactions with micelles (Fig. 5). These positively charged residues may have a tendency to interact with the membrane surface. As shown in Fig. 4 A, H249 and R250 had lower peak intensities than other residues, which may be due to the interaction with the phosphate groups of DPC micelles.
Figure 5.

Relaxation measurements (600 MHz) of the hEpoR in DPC micelles. 15N R1, R2, and hetNOE values of the hEpoR were obtained at 313 K. R1, R2, R2/R1, and hetNOE values are shown as a function of residue number.
Structure of the hEpoR in DPC micelles
We determined the solution structure of the hEpoR in DPC micelles using restraints that contained dihedral angles derived from TALOS+, hydrogen-bond restraints from H-D exchange experiments (Fig. S2), and short-range NOEs (Fig. 6). The structure has been deposited in the PDB under accession number 2MV6. Table 1 shows the statistics of the determined hEpoR structure. The hEpoR contains one helix formed by residues 226–257 (Fig. 6, A–C). The N-terminal fragment of the hEpoR is not structured (Fig. 6 C). The pairwise root mean-square deviation (RMSD) of the TMD and JM region formed by residues 226–257 of the hEpoR was 0.657 Å for the backbone atoms and 1.119 Å for the heavy atoms (Fig. 6 A). Surface charge representation of the hEpoR showed that the TMD was mainly hydrophobic, which is a common feature of TM regions because they contain mainly hydrophobic residues (Fig. 6 D). The length of the TM helix was ∼32.1 Å. Although the JM region exhibits like an amphipathic helix containing a hydrophobic surface formed by hydrophobic residues such as L253 and I257 and a hydrophilic surface formed by hydrophilic residues, the hydrophobic surface was small (Fig. 6 D). A helix-wheel representation of the JM region (Fig. 6 E) and structural analysis suggested that these three hydrophobic residues formed a hydrophobic patch, which might be important for receptor function (Fig. 6 G).
Figure 6.
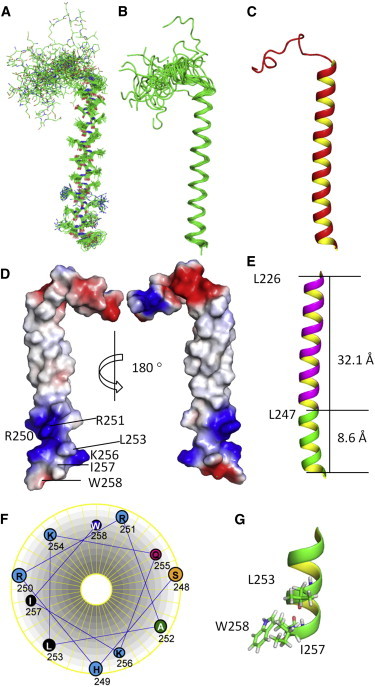
Structure of the hEpoR in DPC micelles. (A) Twenty structures of the hEpoR are superimposed. The side chains of the amino acids are shown in sticks. Carbon, nitrogen, and oxygen atoms are shown in green, blue, and red, respectively. The structures have been deposited in the PDB under accession number 2MV6. (B) The 20 superimposed structures are shown with backbone atoms. (C) Ribbon representation of the hEpoR for the lowest-energy conformer. The N-terminal residues are flexible. The TMD and JM region form one helix. (D) Color-coded electrostatic surface potential for the hEpoR. Positive and negative potentials are shown in blue and red, respectively. Positively charged residues and the three conserved hydrophobic residues in the JM region are labeled with the residue name and sequence number. (E) Ribbon representation of the TMD and JM region of the hEpoR. The TMD and JM region are shown in purple and green, respectively. The heights of the TMD and JM region are shown. The calculation and all of the figures were made using PyMOL (http://www.pymol.org). (F) Helix-wheel representation of the JM region. (G) Ribbon presentation of the JM region. The three hydrophobic residues are shown in sticks.
Table 1.
Summary of the 20 structures of the hEpoR in DPC micelles
| Number of unambiguous NOEs | |
|---|---|
| Short-range (|i−j|≤ 1) | 218 |
| Medium-range (2 ≤|i−j|< 5) | 42 |
| Long-range (|i−j| > 5) | 0 |
| Number of dihedral angle constraints | 80 |
| Number of hydrogen-bond restraints | 17 |
| Number of restraint violationsa | |
| Total number of restraint violations > 0.5 Å | 0 |
| Total number of dihedral angle constraints > 5° | 0 |
| Ramachandran plot statisticsb (%) | |
| Residues in most favored regions | 84.1 |
| Residues in additionally allowed regions | 13.6 |
| Residues in generously allowed regions | 2.3 |
| Residues in disallowed regions | 0 |
| Average RMSD to mean (Å) | |
| Backbone (residues 953–981) | 0.66 ± 0.16 Å |
| Heavy atoms (residues 953–981) | 1.12 ± 0.22 Å |
There are no distance violations greater than 0.5 Å or dihedral angle violations greater than 5°.
The Ramachandran plot was obtained using PROCHECK-NMR (34) based on the conformer with the lowest energy.
Solvent exposure of the JM region
Although the TMD of the hEpoR could be easily identified by sequence analysis, we performed an additional analysis to identify residues that are exposed to the solvent or buried in the micelles. The N-terminal and C-terminal residues of the hEpoR exhibited NOEs between those of water protons and amide protons, suggesting that these residues are exposed to the solvent (Fig. 7 A). To further identify residues that are exposed to solvent, we carried out a PRE experiment using Gd-DTPA. If a residue is buried in micelles, the peak intensity will not be affected when Gd-DTPA is added (35). The most affected residues were those at the C-terminus (Fig. 7 B). This demonstrated that the JM region is exposed to the solvent. Although the N-terminal residues of the construct are not structured, some residues are protected from exposure to Gd-DTPA, suggesting that this region may interact with micelles. The JM region of the EpoR contains three conserved hydrophobic residues: L253, I257, and W258. Tryptophan residue in an integral membrane protein is important for interaction with the membrane due to its amphipathic nature (36). The fluorescence emission spectra of tryptophan residues are very sensitive to the environment and are used to probe residue and membrane interactions (29,36). When the tryptophan residue is exposed to an aqueous solution, the emission maximum is close to 350 nm. On the other hand, the emission maximum will be shifted to a lower wavelength if it is buried in a hydrophobic environment such as micelles (29,36). The hEpoR construct contains only one tryptophan residue (W258) in the JM region, which makes it straightforward to elucidate its interaction with micelles. The florescence emission spectrum of the hEpoR was collected in DPC micelles and the emission maximum was ∼350 nm, indicating that W258 is exposed to the solvent (Fig. 7 C). The indole amide proton of W258 exhibited NOEs with water protons, which further confirmed that this residue is not buried in the micelles. Based on the above results, we propose a model of the hEpoR in a micelle (Fig. 7 D). In this model, a micelle ∼32 Å in diameter covers the TMD. The JM region is exposed to the solvent. Although the JM contains a tryptophan, this residue does not interact with DPC micelles (Fig. 7 D).
Figure 7.
Model of the hEpoR in a micelle. (A) NOEs between water protons and amide protons of residues at the C-terminus of the hEpoR. Slices from a 3D 1H-15N-HSQC-NOESY experiment are shown. The signal from water protons is indicated with a dotted line. (B) PRE experiment for the hEpoR. The ratio of peak intensities of a residue in the presence (Ip) and absence (I0) of 2 mM Gd-DTPA is plotted against the residue number. (C) Fluorescence spectroscopy of the hEpoR in DPC micelles. (D) Model of the hEpoR in a micelle. The micelle is drawn as a sphere with a diameter of 32 Å. The N-terminus of the construct is shown in cyan. The TMD is shown as a purple helix and the JM region is shown as a green helix. This figure was made using PyMOL (http://www.pymol.org). To see this figure in color, go online.
Structural investigation of the mEpoR
The TMDs and JM regions of the hEpoR and mEpoR share high sequence homology (Fig. 1 C). The mEpoR was expressed and purified into DPC micelles. Although there were seven amino acids that differed between these two constructs (Fig. 1 C), the dispersion of crosspeaks in the 1H-15N-HSQC spectra was different (Fig. 8 A). We then obtained the backbone assignment of mEpoR in DPC micelles (Fig. 8 B; Table S1). We obtained almost complete amide proton and amide nitrogen assignments, except for residues L226 and R251. Interestingly, multiple crosspeaks in the 1H-15N-HSQC spectrum were observed for the N-terminal residues encompassing S212–L218 (Fig. 8 B). This may arise from multiple conformations. For the residues in the TMD region, residue S238 showed two crosspeaks in the spectrum, which may reflect the fact that the construct contained a dimer population and S238 might be in the dimer interface. The GA cross-linking experiment showed that mEpoR formed more dimers than hEpoR (Fig. S4). Secondary structure analysis based on Cα chemical shifts demonstrated that the TMD and JM region of mEpoR are helical and the N-terminus is flexible, just as observed for hEpoR (Fig. 8 C). A Cα chemical-shift comparison of the identical residues between the hEpoR and mEpoR constructs used in this study showed that residues at the N-terminus underwent significant chemical-shift changes, whereas the changes of the residues within the TMD and JM region were minor (Fig. 8 D).
Figure 8.
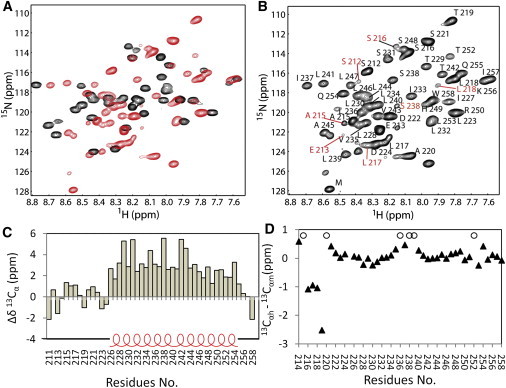
NMR study of the mEpoR. (A) Superimposed 1H-15N-HSQC spectra of hEpoR (black) and mEpoR (red). (B) Assigned 1H-15N-HSQC spectrum of mEpoR in DPC micelles. (C) Deviations of the observed mEpoR Cα chemical-shift values from the corresponding random-coil chemical-shift values. (D) 13Cα chemical-shift difference between the mEpoR (Cαm) and hEpoR (Cαh). Residues that differ between the hEpoR and mEpoR are shown as open circles. To see this figure in color, go online.
Discussion
Studies have shown that ligand-induced JAK2 activation by EpoR depends on communication between the extracellular and cytoplasmic domains of the receptor (37). The efficiency of signaling transduction across the membrane to activate JAK2 relies on the cytoplasmic region of the receptor because different ligands have different effects on kinase activation (37,38). The ligand-binding sites of the EpoR are far away from the TMD. Residues S212–D224 are localized between the cytoplasmic region and the TMD of the EpoR. This region was demonstrated to be unstructured in DPC micelles (Figs. 5 and 6). This region might be important for transfering the ligand-induced conformational changes of the cytoplasmic domains to the TMD due to its physical location, which transfers signal across the cell membrane. A previous x-ray structure showed that residues V215–T219 form a β-strand (38). It is not surprising that this region was unstructured, because the extracellular domain was not included in the hEpoR construct (Fig. 1 B). There are two prolines (P220 and P225) in the hEpoR, which might prevent it from forming a helix (Fig. 1 C). This region may exist as an extended strand in the entire receptor and act as a linker between the cytoplasmic region and the TMD (Fig. 9), and it may be flexible even in the entire receptor (Fig. 9). Ligand-induced conformational changes of the extracellular region may affect the orientation of residues P220–P225, which would induce TMD dimerization or change its orientation. The mEpoR TMD was shown to thermodynamically dimerize with a higher affinity than human TMD in the C14SB micelles (9). Our GA cross-linking experiment also showed that mEpoR had a tendency to form more dimers than hEpoR (Fig. S3), which is consistent with a previous study (9). The NMR spectrum of the mEpoR demonstrated that the residues at the N-terminus of the mEpoR exhibited multiple crosspeaks in the 1H-15N-HSQC spectrum (Fig. 8), suggesting that these residues may have a propensity to change their conformations or have exchanges during TMD dimerization.
Figure 9.
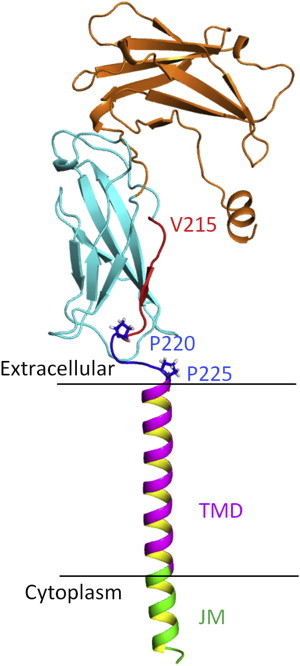
Model of the EpoR extracellular domain, TMD, and JM region. Only a single molecule is shown. The model was generated by manually linking two structures (PDB ID 1EBP and the structure determined in this study) in PyMOL. The two proline residues are shown in sticks. The extracellular domain, TMD, and JM region are shown in different colors. The membrane is indicated as two lines. To see this figure in color, go online.
To activate JAK2, the cytoplasmic region of the EpoR must be brought into close contact upon ligand binding (39). For the JM region, an Ala insertion study showed that receptor function requires a specific structure and orientation of the TMD (33). The three positively charged residues (H249–R251) may undergo exchanges with the environment, as indicated by the line-broadening observed in the HSQC spectrum (Fig. 3). These charged residues may only be important for membrane anchoring, and not for JAK2 activation (33). The three conserved hydrophobic residues in the JM region (L253, I257, and W258) are indispensable for JAK2 activation (33). The orientation of the three hydrophobic residues was shown to be important for signal transduction (33). Our results show that the JM region is helical, supporting a previous prediction that the JM region forms a helical structure (Fig. 6). The three hydrophobic residues could form a hydrophobic surface, and this hydrophobic patch might be important for positioning JAK2 correctly for downstream signaling (33). The tryptophan residue has a tendency to interact with the cell membrane due to its structure. Based on the florescence emission spectrum, W258 was shown to be exposed to the solvent (Fig. 7 C). Further NOE analysis also demonstrated that this residue has NOEs with water protons (Fig. 7 B). Our results suggest that W258 in the hEpoR may not be functional as a membrane-anchoring residue, but may be important for interaction with JAK2, as proposed in a previous study (33).
The entire EpoR forms dimers in the absence of ligand (11), and the TMD of the receptor was shown to be sufficient to maintain dimerization of the entire receptor (11). Although there are only three amino acids that differ within the TMDs of the hEpoR and mEpoRs (Fig. 1), the TMD of the mEpoR may have a stronger association affinity than that of the hEpoR (9). Unlike other membrane proteins, the TM-TM dimer interface of the mEpoR may contain residues S231, S238, T242, and S248 (12,40). In this study, we found that S238 of the mEpoR exhibited two crosspeaks in the 1H-15N-HSQC spectrum, suggesting that this residue may be important for dimerization (Fig. 8 A). An asparagine-scanning experiment showed that the TM-TM interface of the EpoR was based on a leucine zipper-like heptad repeat pattern (14,41). Residue L241 was the most import residue for TMD dimerization (42). Cysteine-scanning mutagenesis studies showed that residues such as L223 and L226 are important for EpoR dimerization (5,13). Fusion of the leucine zipper coiled coil from the Put3 transcription factor with EpoR in combination with molecular simulation studies identified active and inactive orientations of the TMD of the EpoR dimer (12). Residues from the JM region, such as T242 (mEpoR) and S248, were shown to be important for dimerization (12). A previous study also showed that the detergent/protein ratio affected TMD dimerization (9). We compared the 1H-15N-HSQC spectra of hEpoR samples with different protein/DPC ratios (Fig. S4). Interestingly, we observed line-broadening of crosspeaks for residues L223 and D224 of the N-terminal region of the hEpoR; residues L226, L228, L241, and L247 of the TMD; and residues S248, L253, Q255, and K256 of the JM region (Fig. S4). This result suggested that these residues might be important for dimerization if the hEpoR forms dimers when the detergent/protein ratio is decreased, but it has to be noted that the line-broadening of crosspeaks may also be due to nonspecific dimerization or aggregation (43). Although several studies have been conducted to elucidate the TM-TM interface of the TMD of the EpoR, the TM dimer structure is not available. For the TMD of the Thrombopoietin receptor (TpoR, a homology of EpoR), cysteine cross-linking, alanine-scanning, and computational simulations confirmed that there are three different stable, rotationally related conformations, suggesting that signaling induced by TpoR occurs through TMD rotation (44). It is also possible that rotation of the TMD during ligand binding is one of the mechanisms underlying EpoR signaling, which means that the EpoR may have different dimerization interfaces. Our cross-linking study showed that the hEpoR could form trimers or oligomers in LMPC and DPC micelles, suggesting that the hEpoR may have multiple dimerization interfaces (Fig. 2). The amino acid sequence in the TMD of the hEpoR suggests that its dimer interface is different from that of other single-span transmembrane proteins, such as glycophorin A (45) and the Eph receptors (46). Further structural studies of the TMD dimer of the EpoR would be very useful. We show that the TMD and JM region of the hEpoR and mEpoR form a helix in DPC micelles (Fig. 6). Due to the short sequence of the constructs used in this study, the structures of the TMD and JM region in DPC micelles are close to the physical structures. To study the dimeric structure, other membrane systems, such as bicelles, should be considered because the hEpoR has a tendency to form dimers even in the absence of the GA cross-linker (Fig. 2). The bicelle system has been shown to be ideal for studying the structure and dynamics of membrane proteins using NMR (47,48). Further assignment strategies or condition optimization may be needed, however, because signals from the TMD of the hEpoR in bicelles were broadened (Fig. 3). Nevertheless, this study presents the monomeric structure of the hEpoR. Information about the structure and dynamics of the TMD and JM region of the EpoRs will provide insight into their functional mechanism. The results obtained in this study will be useful for further structural and functional investigations of the receptors.
In summary, we have conducted a dynamic study and determined the solution structure of the TMD and JM region of the hEpoR in DPC micelles. The TMD and JM region form one helix. The JM region is exposed to the solvent. Both the TMD and the JM region of the EpoR may be important for receptor dimerization. The structure of the TMD and JM region of the mEpoR is similar to that of the hEpoR, but has a different 1H-15N-HSQC spectrum.
Acknowledgments
We thank Prof. Ho Sup Yoon and Dr. Hong Ye from Nanyang Technological University for their help with the NMR experiments.
This work was supported by the Agency for Science, Technology and Research (A∗STAR) and partially supported by a grant from A∗STAR JCO (1331A028).
Footnotes
Qingxin Li and Ying Lei Wong contributed equally to this work.
Supporting Material
References
- 1.Atanasova M., Whitty A. Understanding cytokine and growth factor receptor activation mechanisms. Crit. Rev. Biochem. Mol. Biol. 2012;47:502–530. doi: 10.3109/10409238.2012.729561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Andrea A.D., Lodish H.F., Wong G.G. Expression cloning of the murine erythropoietin receptor. Cell. 1989;57:277–285. doi: 10.1016/0092-8674(89)90965-3. [DOI] [PubMed] [Google Scholar]
- 3.Richmond T.D., Chohan M., Barber D.L. Turning cells red: signal transduction mediated by erythropoietin. Trends Cell Biol. 2005;15:146–155. doi: 10.1016/j.tcb.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Elliott S., Pham E., Macdougall I.C. Erythropoietins: a common mechanism of action. Exp. Hematol. 2008;36:1573–1584. doi: 10.1016/j.exphem.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Kubatzky K.F., Liu W., Constantinescu S.N. Structural requirements of the extracellular to transmembrane domain junction for erythropoietin receptor function. J. Biol. Chem. 2005;280:14844–14854. doi: 10.1074/jbc.M411251200. [DOI] [PubMed] [Google Scholar]
- 6.Darnell J.E., Jr. STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 7.Livnah O., Stura E.A., Wilson I.A. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science. 1999;283:987–990. doi: 10.1126/science.283.5404.987. [DOI] [PubMed] [Google Scholar]
- 8.Rawlings J.S., Rosler K.M., Harrison D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 9.Ebie A.Z., Fleming K.G. Dimerization of the erythropoietin receptor transmembrane domain in micelles. J. Mol. Biol. 2007;366:517–524. doi: 10.1016/j.jmb.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 10.Pelletier S., Gingras S., Ihle J.N. Two domains of the erythropoietin receptor are sufficient for Jak2 binding/activation and function. Mol. Cell. Biol. 2006;26:8527–8538. doi: 10.1128/MCB.01035-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Constantinescu S.N., Keren T., Lodish H.F. Ligand-independent oligomerization of cell-surface erythropoietin receptor is mediated by the transmembrane domain. Proc. Natl. Acad. Sci. USA. 2001;98:4379–4384. doi: 10.1073/pnas.081069198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seubert N., Royer Y., Constantinescu S.N. Active and inactive orientations of the transmembrane and cytosolic domains of the erythropoietin receptor dimer. Mol. Cell. 2003;12:1239–1250. doi: 10.1016/s1097-2765(03)00389-7. [DOI] [PubMed] [Google Scholar]
- 13.Lu X., Gross A.W., Lodish H.F. Active conformation of the erythropoietin receptor: random and cysteine-scanning mutagenesis of the extracellular juxtamembrane and transmembrane domains. J. Biol. Chem. 2006;281:7002–7011. doi: 10.1074/jbc.M512638200. [DOI] [PubMed] [Google Scholar]
- 14.Kubatzky K.F., Ruan W., Klingmüller U. Self assembly of the transmembrane domain promotes signal transduction through the erythropoietin receptor. Curr. Biol. 2001;11:110–115. doi: 10.1016/s0960-9822(01)00018-5. [DOI] [PubMed] [Google Scholar]
- 15.Li Q., Wong Y.L., Kang C. Solution structure of the transmembrane domain of the insulin receptor in detergent micelles. Biochim. Biophys. Acta. 2014;1838:1313–1321. doi: 10.1016/j.bbamem.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Vinogradova O., Badola P., Sanders C.R., 2nd Escherichia coli diacylglycerol kinase: a case study in the application of solution NMR methods to an integral membrane protein. Biophys. J. 1997;72:2688–2701. doi: 10.1016/S0006-3495(97)78912-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 18.Johnson B.A. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 19.Wishart D.S., Sykes B.D., Richards F.M. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 20.Shen Y., Delaglio F., Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Güntert P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 22.Veglia G., Zeri A.C., Opella S.J. Deuterium/hydrogen exchange factors measured by solution nuclear magnetic resonance spectroscopy as indicators of the structure and topology of membrane proteins. Biophys. J. 2002;82:2176–2183. doi: 10.1016/s0006-3495(02)75564-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kay L.E., Torchia D.A., Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 24.Gayen S., Li Q., Kang C. An NMR study of the N-terminal domain of wild-type hERG and a T65P trafficking deficient hERG mutant. Proteins. 2011;79:2557–2565. doi: 10.1002/prot.23089. [DOI] [PubMed] [Google Scholar]
- 25.Schwieters C.D., Kuszewski J.J., Clore G.M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 26.Banci L., Bertini I., Parigi G. Paramagnetism-based restraints for Xplor-NIH. J. Biomol. NMR. 2004;28:249–261. doi: 10.1023/B:JNMR.0000013703.30623.f7. [DOI] [PubMed] [Google Scholar]
- 27.Schwieters C.D., Kuszewski J.J., M. Clore G. Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2006;48:47–62. [Google Scholar]
- 28.Kang C., Tian C., Sanders C.R. Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry. 2008;47:7999–8006. doi: 10.1021/bi800875q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conner M., Hicks M.R., Conner A.C. Functional and biophysical analysis of the C-terminus of the CGRP-receptor; a family B GPCR. Biochemistry. 2008;47:8434–8444. doi: 10.1021/bi8004126. [DOI] [PubMed] [Google Scholar]
- 30.Gayen S., Li Q., Kang C. Structure of the C-terminal region of the Frizzled receptor 1 in detergent micelles. Molecules. 2013;18:8579–8590. doi: 10.3390/molecules18078579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang C., Li Q. Solution NMR study of integral membrane proteins. Curr. Opin. Chem. Biol. 2011;15:560–569. doi: 10.1016/j.cbpa.2011.05.025. [DOI] [PubMed] [Google Scholar]
- 32.Gayen S., Li Q., Kang C. Solution NMR study of the transmembrane domain of single-span membrane proteins: opportunities and strategies. Curr. Protein Pept. Sci. 2012;13:585–600. doi: 10.2174/138920312803582979. [DOI] [PubMed] [Google Scholar]
- 33.Constantinescu S.N., Huang L.J., Lodish H.F. The erythropoietin receptor cytosolic juxtamembrane domain contains an essential, precisely oriented, hydrophobic motif. Mol. Cell. 2001;7:377–385. doi: 10.1016/s1097-2765(01)00185-x. [DOI] [PubMed] [Google Scholar]
- 34.Laskowski R.A., Rullmannn J.A., Thornton J.M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 35.Hilty C., Wider G., Wüthrich K. Membrane protein-lipid interactions in mixed micelles studied by NMR spectroscopy with the use of paramagnetic reagents. ChemBioChem. 2004;5:467–473. doi: 10.1002/cbic.200300815. [DOI] [PubMed] [Google Scholar]
- 36.Clark E.H., East J.M., Lee A.G. The role of tryptophan residues in an integral membrane protein: diacylglycerol kinase. Biochemistry. 2003;42:11065–11073. doi: 10.1021/bi034607e. [DOI] [PubMed] [Google Scholar]
- 37.Livnah O., Johnson D.L., Wilson I.A. An antagonist peptide-EPO receptor complex suggests that receptor dimerization is not sufficient for activation. Nat. Struct. Biol. 1998;5:993–1004. doi: 10.1038/2965. [DOI] [PubMed] [Google Scholar]
- 38.Syed R.S., Reid S.W., Stroud R.M. Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature. 1998;395:511–516. doi: 10.1038/26773. [DOI] [PubMed] [Google Scholar]
- 39.Muthukumaran G., Kotenko S., Pestka S. Chimeric erythropoietin-interferon gamma receptors reveal differences in functional architecture of intracellular domains for signal transduction. J. Biol. Chem. 1997;272:4993–4999. doi: 10.1074/jbc.272.8.4993. [DOI] [PubMed] [Google Scholar]
- 40.Mackenzie K.R. Folding and stability of alpha-helical integral membrane proteins. Chem. Rev. 2006;106:1931–1977. doi: 10.1021/cr0404388. [DOI] [PubMed] [Google Scholar]
- 41.Moore D.T., Berger B.W., DeGrado W.F. Protein-protein interactions in the membrane: sequence, structural, and biological motifs. Structure. 2008;16:991–1001. doi: 10.1016/j.str.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruan W., Becker V., Langosch D. The interface between self-assembling erythropoietin receptor transmembrane segments corresponds to a membrane-spanning leucine zipper. J. Biol. Chem. 2004;279:3273–3279. doi: 10.1074/jbc.M309311200. [DOI] [PubMed] [Google Scholar]
- 43.Zhuang T., Jap B.K., Sanders C.R. Solution NMR approaches for establishing specificity of weak heterodimerization of membrane proteins. J. Am. Chem. Soc. 2011;133:20571–20580. doi: 10.1021/ja208972h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matthews E.E., Thévenin D., Engelman D.M. Thrombopoietin receptor activation: transmembrane helix dimerization, rotation, and allosteric modulation. FASEB J. 2011;25:2234–2244. doi: 10.1096/fj.10-178673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.MacKenzie K.R., Prestegard J.H., Engelman D.M. A transmembrane helix dimer: structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 46.Bocharov E.V., Mayzel M.L., Arseniev A.S. Left-handed dimer of EphA2 transmembrane domain: Helix packing diversity among receptor tyrosine kinases. Biophys. J. 2010;98:881–889. doi: 10.1016/j.bpj.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diller A., Loudet C., Dufourc E.J. Bicelles: a natural ‘molecular goniometer’ for structural, dynamical and topological studies of molecules in membranes. Biochimie. 2009;91:744–751. doi: 10.1016/j.biochi.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanders C.R., Prosser R.S. Bicelles: a model membrane system for all seasons? Structure. 1998;6:1227–1234. doi: 10.1016/s0969-2126(98)00123-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.