

Abstract
The prion protein (PrP), widely recognized to misfold into the causative agent of the transmissible spongiform encephalopathies, has previously been shown to bind to lipid membranes with binding influenced by both membrane composition and pH. Aside from the misfolding events associated with prion pathogenesis, PrP can undergo various posttranslational modifications, including internal cleavage events. Alpha- and beta-cleavage of PrP produces two N-terminal fragments, N1 and N2, respectively, which interact specifically with negatively charged phospholipids at low pH. Our previous work probing N1 and N2 interactions with supported bilayers raised the possibility that the peptides could insert deeply with minimal disruption. In the current study we aimed to refine the binding parameters of these peptides with lipid bilayers. To this end, we used neutron reflectometry to define the structural details of this interaction in combination with quartz crystal microbalance interrogation. Neutron reflectometry confirmed that peptides equivalent to N1 and N2 insert into the interstitial space between the phospholipid headgroups but do not penetrate into the acyl tail region. In accord with our previous studies, interaction was stronger for the N1 fragment than for the N2, with more peptide bound per lipid. Neutron reflectometry analysis also detected lengthening of the lipid acyl tails, with a concurrent decrease in lipid area. This was most evident for the N1 peptide and suggests an induction of increased lipid order in the absence of phase transition. These observations stand in clear contrast to the findings of analogous studies of Ab and α-synuclein and thereby support the possibility of a functional role for such N-terminal fragment-membrane interactions.
Introduction
Transmissible spongiform encephalopathies (TSEs) or prion diseases are fatal neurodegenerative diseases affecting humans and animals. Misfolded conformers of the prion protein (PrPSc) are widely recognized as the causative agent of TSEs with expression of wildtype or cellular prion protein (PrPC) indispensable for disease transmission and pathogenesis (1,2). During disease the predominantly alpha-helical PrPC becomes misfolded into beta-sheet-rich conformers that can template further misfolding of PrPC resulting in propagation of pathogenic PrPSc.
The misfolding process has been replicated in vitro utilizing recombinant PrP (rPrP), and the development of a serial protein misfolding amplification assay (PMCA) has made production of misfolded rPrP highly efficient (3). However, initial experiments with misfolded rPrP could only generate a transmissible prion species if the PMCA reaction was “seeded” with infectious PrPSc taken from a disease source (4). Such observations raised questions over the potential role for cellular cofactors in PrPSc propagation. Various anionic species have been suggested as cofactors in the production of infectious prions; these have included nucleic acids, glycosaminoglycans, and lipids (reviewed in (5,6)). In the first study to report use of the PMCA to produce genuinely infectious (transmissible in animal bioassay) rPrP species in the absence of an infectious disease-derived seed, a combination of 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG) and nucleic acid cofactors were required (7). In addition to the inclusion of pure phospholipid species in misfolding reactions, synthetic membranes of mixed lipid composition have also been employed to mimic rPrP binding to cellular structures. These studies have demonstrated the affinity of rPrP for lipids, with binding invariably influenced by lipid composition, metal ion binding, and pH (8–10) and often resulting in structural changes to rPrP that might predispose it to or initiate misfolding (9–12).
Phospholipid-PrP binding interactions may have a wider significance beyond protein misfolding. Although a “normal” PrPC function is still undetermined, suggestions have included trafficking and signal transduction (reviewed in (13)), functions intimately linked with membranes. Mature PrPC is a glycosylphosphatidylinositol (GPI)-anchored membrane protein directed to cholesterol-rich membrane domains on the outer leaflet of the cell plasma-membrane (14). These membrane domains are often associated with cellular signaling reactions and the control of signal protein activation; such a localization may support a role for PrPC in these functions. Part of the reason that elucidating one individual function of PrPC has been challenging is likely to be the influence of its posttranslational modifications. Protein cleavage is a cellular mechanism of functional activation and deactivation. PrPC is known to undergo well-characterized alpha- and beta-cleavage events as well as secretory cleavage from the cell surface (14–17). Alpha-cleavage occurs either side of residue 111 (15) producing N1 and C1 fragments and the β-cleavage site is located around residue 90 (18) producing N2 and C2 fragments. The N1 and N2 fragments contain a far N-terminal polybasic region and an octameric repeat, metal ion binding domain. The fragments differ at their C terminus by one further polybasic region containing an additional metal ion binding site that is only present in the N1 fragment. PrPC, therefore, exists as a minimum of six different species before other posttranslational modifications (such as complex glycosylation) are considered, with the potential for differing functions of different fragments depending on cellular environment. As stated, posttranslational cleavage may be used for protein activation or deactivation and, therefore, the fragments produced by these cleavages could be functional and may alter the functional capacity of full-length PrP. Both the N1 and N2 cleavage fragments have been demonstrated to have neuroprotective functions (19,20), but the cleavage events that produce them are thought to occur at different cellular locations, with N1 produced in the Golgi (21) and N2 at the cell surface (16), indicating their production may be stimulated by different signals.
Our previous studies using synthetic N1 and N2 have shown that the peptides have a propensity to interact with anionic lipid membranes independent of the PrP globular C terminus region (22). These studies suggested that the liberated peptides could interact with the acyl chain core of the bilayer membrane without substantially disrupting the homogeneity of the lipid system and thus our previous work raised the possibility that the peptides may insert benignly through the bilayer. Our recent work aimed to expand these findings by directly probing the peptide-lipid interactions using neutron reflectometry (NR). NR directs a beam of neutrons into a sample, measuring the intensity of reflected neutrons as a function of momentum transfer. The pattern of reflection in the context of membranes provides information on the thickness and composition of the surface and from these the fluidity can be inferred by deriving the area per lipid molecule. This technique was employed specifically for its capacity to accurately probe the depth that a peptide penetrates into the bilayer, which can be measured using isotopic labeling. Furthermore, NR is nondestructive and nonperturbing, therefore giving a more valid insight into the interactions within the sample. Our findings show that both N1 and N2 interact with anionic membranes at pH 5 as originally proposed without pore-forming or lytic capacity but neither peptide penetrates deeply into the membrane.
Materials and Methods
Vesicle and peptide preparation
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG), 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-L-serine] (POPS), 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (DMPG), 1-palmitoyl(d31)-2-oleoyl-sn-glycero-3-phosphocholine (d31-POPC), 1-palmitoyl(d31)-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (d31-POPG), and 1-palmitoyl(d31)-2-oleoyl-sn-glycero-3-[phospho-L-serine] (d31-POPS) were purchased from Avanti Polar Lipids (Alabaster, AL) and used without further purification. Lipids were dissolved in chloroform/methanol (4:1 by volume) in glass vials to give POPC only, POPC/POPG (2:1 molar ratio), POPC/POPS (2:1 molar ratio), and DMPC/DMPG (2:1 molar ratio) lipid mixtures. The same lipid ratios were used for preparing vesicles with lipids containing the d31-palmitoyl chain. The solvent was evaporated first by blowing nitrogen over the lipid solutions and then by storing under vacuum overnight to remove any residual solvent. The dried lipid thin films were hydrated with a buffered solution of 10 mM MOPS, 150 mM NaCl pH 7.0 (in 100% D2O for NR experiments) with vortex mixing to a concentration of 0.5 mg/mL. The hydrated lipid solutions were incubated at 25°C for PO-containing lipids and 30°C for DM-containing lipids for 1 h before small unilamellar vesicles were prepared by sonicating the lipid solution until clear.
Fluorescent large unilamellar vesicles (LUVs) for calcein release assays were prepared as follows. Chloroform solutions (10 mg/mL) of 1-palmitoyl-2-oleoyl-sn-Glycero-3-phosphcholine (POPC) and 1-palmitoyl-2-oleoyl-sn-Glycero-3-[phospho-rac-(1-glycerol)] (POPG) were combined (2:1 w/w) in a round-bottom flask and the solvent removed by rotary evaporation. The lipid film was hydrated in 10 mM Tris-HCl pH 7.4 (150 mM NaCl) containing 70 mM calcein (Sigma-Aldrich Pty Ltd, Sydney, Australia) for 1 h at 40°C with gentle agitation. The multilamellar dispersion was freeze-thawed in liquid nitrogen five times, then extruded through a 100 nm polycarbonate membrane 11 times. The resultant calcein-loaded LUVs were passed through a column of Sephadex G25 resin (Sigma) preequilibrated with 50 mM Na/K acetate pH 5.2 (130 mM NaCl) to remove free calcein from the solution. Calcein-loaded LUVs were diluted to a concentration of ca. 50 μM in acetate buffer immediately before use.
The mouse PrP amino acid sequence was used to generate peptide fragments corresponding to N1 (23-111) and N2 (23-90) cleavage fragments. The Peptide Technology Laboratory (Bio21 Institute, University of Melbourne) used microwave synthesis to prepare the peptides as previously described (23). Verification of the peptide sequence and purity was performed by HPLC and mass spectrometry. Freeze-dried peptides were dissolved in buffer (17 mM sodium acetate, 150 mM NaCl pH 5.0) to a concentration of 10 μM immediately before use.
Neutron reflectometry experiments and data analysis
Polished n-type circular silicon wafers 100 mm in diam. and 10 mm thick (El-Cat Inc., Ridgefield Park, NJ) were cleaned for 1 h at 85°C in a strongly acidic “piranha” solution of H2O/H2SO4/H2O2 (4:3:1 by volume) to remove surface impurities. After cleaning in the corrosive acid solution, the silicon wafers were rinsed in Milli-Q water and dried before being UV-ozone cleaned for 20 min. A final rinse with Milli-Q water and propan-2-ol was carried out before drying under a stream of nitrogen. The silicon wafers were assembled in aluminum cells with a silicon backing plate, which had a 50 μm deep solvent reservoir and inlet and outlet tubes to allow for solvent/sample exchange.
Neutron reflectivity data were measured using the Platypus time-of-flight neutron reflectometer and a cold neutron spectrum (2.8 Å ≤ λ ≤ 18.0 Å) at the OPAL 20 MW research reactor (Sydney, Australia) (24,25). Neutron pulses of 20 Hz were generated using a disc chopper system (EADS Astrium GmbH, Munich, Germany) in the low resolution mode (Δλ/λ = 8%), and recorded on a two-dimensional 3He neutron detector (Denex GmbH, Lüneburg, Germany). Reflected beam spectra were collected for each of the surfaces at 0.45° for 15 min (0.72 mm slits), 1.6° for 45 min (2.56 mm slits), and 4.5° for 2 h (7.2 mm slits), respectively. Direct beam measurements were collected under the same collimation conditions for 1 h each. The data were reduced using the Slim reduction package, which stitches the three data sets together at the appropriate overlap region, re-bins the data at instrument resolution and corrects for background and detector efficiency (26). The final scaled reflectivity, R, is presented as a function of momentum transfer, Q, defined as follows:
where θ is the angle of incidence and λ is the neutron wavelength. The reflectivity data was multiplied by Q4 to remove the Fresnel reflectivity and thus final reflectivity data presented as RQ4 versus Q.
Structural parameters for the native oxide layer on the silicon blocks, lipid bilayer, and peptide layers were refined using the MOTOFIT reflectivity analysis software (27). In the fitting routines, the genetic algorithm was selected to minimize χ2 values by varying the thickness (τ), roughness (σ), and neutron scattering length density (nSLD) of each layer. Model fitting of the resulting reflectivity profiles yields information on the real space neutron scattering length density profile normal to the surface, from which the structure of a lipid bilayer may be deduced. The nSLD, ρ, can be considered as a neutron refractive index and is a function of the chemical composition of the material according to the following:
where NA is Avogadro’s number, pi is the mass density, Ai is the atomic mass, and bi is the nuclear scattering length of component i. The errors generated in the analysis software are ±1 standard deviation of the parameter value from which the percentage error was calculated. The advantage of using neutrons, particularly for soft matter and biological systems, is the difference in scattering length between hydrogen (bH = -3.74 ×10−5 Å) and its isotope deuterium (bD = +6.67 ×10−5 Å). By selective deuteration of molecules (in this case the palmitoyl tails) different segments of the lipid bilayer can be probed by choosing a suitable solvent contrast. We used either a pure D2O solvent contrast (ρ = 6.35 ×10−6 Å-2) to highlight the protonated peptide or pure H2O (ρ = -0.56 ×10−6 Å-2) to highlight the partially deuterated lipid chains. When changing between different subphase contrasts, the physical structure of the system is assumed not to change. This means that the thickness and roughness parameters in the models are constrained to be the same between each subphase while only letting the scattering length density vary. This form of simultaneous fitting provides for a unique solution to the model. The nSLD on each component in a layer contributes to the final nSLD of the layer, ρlayer, such that
where φ is the volume fraction of each component. Once the volume fraction has been determined the surface excess, Γ, of each component (in mol m−2) can be calculated from the following:
where V is the molecular volume. From the surface of excess the lipid to peptide ratio can be deduced.
For consideration of changes in lipid order, the area per lipid molecule, Alipid, is determined from the following:
where V is the molecular volume of the lipid (1230 Å3 for POPC/POPG bilayers and 1226 Å3 for POPC/POPS bilayers (28–30), φ is the volume fraction, and τ is half the total bilayer thickness. The values used to calculate parameters can be found in Table S1 in the Supporting Material.
Quartz crystal microbalance (QCM) experiments and analysis
The Q-senseE4 instrument (Q-Sense, Gothenburg, Sweden) fitted with a peristaltic pump (Ismatec SA, Glattbrugg, Switzerland) used a flow rate of 100 μL min−1, a constant temperature of 22°C, and silicon dioxide-coated sensor crystals (QSX-303, Q-sense). The sensor crystals were cleaned by UV-ozone for 20 min, rinsing with 2% (v/v) Hellmanex followed by copious rinsing with > 18 MΩ water and drying under nitrogen. The sensors underwent a final clean using UV-ozone for 20 min immediately before use. The piezoelectric quartz crystal was excited at its fundamental frequency (5 MHz) and the change in frequency (Δf) was observed for the third, fifth, seventh, ninth, eleventh, and thirteenth overtones. Data was collected using QSoft401 software and processed into frequency and dissipation versus time data using Q-Tools 301 v2.1. A decrease in frequency corresponds to an increased mass on the surface of the sensor. For rigid films with little water content that have minimal changes in dissipation (< 1), the Sauerbrey equation can be used to relate mass (Δm) and frequency (Δf) as follows:
where ρq is the density of quartz (2648 kg m−3), vq is the speed of sound through quartz (3340 m s−1), F is the fundamental frequency (5 MHz), and n is the overtone number. For each overtone the change in dissipation (ΔD) was also measured. The dissipation is the proportion of energy dissipated during one cycle of the frequency oscillation and provides information on the viscoelastic properties of the materials deposited on the sensor surface. For films that are nonrigid and have a dissipation that is large (> 1), the Sauerbrey equation is no longer valid.
Vesicles prepared in buffer (50 mM sodium acetate/acetic acid pH 5.2, 130 mM NaCl) were deposited onto the silicon dioxide surface. Once the vesicles had ruptured and the bilayer formed, the excess lipid was removed with a buffer wash. Peptide at 10 μM prepared in the same buffer was added and incubated on the bilayer for 60 min after which the excess peptide was removed with a buffer wash.
Calcein Release Measurements
For each condition, 95 μL of calcein-loaded LUVs was added to a black 96-narrow-well plate and the initial fluorescence (f0) monitored using a POLARstar OPTIMA microplate reader (BMG LABTECH, Ortenberg, Germany) using 480 ± 10 nm excitation and 520 ± 20 nm emission filters. Following addition of 5 μL peptide (100 μM N1, N2, or melittin dissolved in MQ grade water), the solution briefly mixed by pipetting up and down the fluorescence signal was further monitored as a function of time. Finally, 5 μL Triton X-100 (10% v/v) was added to determine the maximum fluorescence response (f1). The normalized fluorescence (f') was obtained from the raw fluorescence signal (f) using the following expression:
Results
NR is an established technique for studying the structure of solid-supported phospholipid bilayers along the axis perpendicular to the plane of the bilayer (the z-axis) (31,32). The bilayers were generated using the vesicle deposition technique (33) onto silicon oxide surfaces typically 15 ± 1 Å thick and with an interfacial roughness of 3 Å. In these experiments we used mono-unsaturated diacyl phospholipids where the palmitoyl chain was deuterated so that there was contrast against the hydrogenous peptides. For modeling the bilayer data, the bilayers were separated into three discrete layers: headgroup one (closest to the silicon oxide surface), acyl tails, and headgroup two (closest to the bulk solvent). A 3 to 7 Å solvent layer between the silicon oxide surface and headgroup one was included in the model for each bilayer as previously described (34,35). The experiments were conducted at room temperature, which is above the phase transition temperature of POPC, POPG, and POPS (Tm of -2°C, -2°C, and 14°C, respectively) and thus the bilayers were in the fluid (Lα) phase. The thickness of the bilayers ranged from 38 ± 3 Å to 56 ± 3 Å (Table 1 and Tables S2 and S3) and the area per lipid molecule ranged from 35 to 47 Å2 in the tail region. These observations are consistent with molecular dynamics simulation data of POPC bilayers in the fluid phase (28).
Table 1.
Thickness and derived properties of the lipid bilayers with and without N1 and N2 peptide bound from the fitting of the neutron reflectometry data
| Layer | Thickness (no peptide) / Å | φlipid (no peptide) | Thickness (peptide bound) / Å | φlipid (peptide bound) | φpeptide | Γlipid / μmol m−2 | Γpeptide / μmol m−2 |
|---|---|---|---|---|---|---|---|
| d31-POPC/d31-POPG (2:1) bilayer + N1 peptide | |||||||
| Headgroup 1 | 15 ± 1 | 0.94 ± 0.02 | 18 ± 1 | 0.75 ± 0.01 | - | 7.46 ± 0.12 | - |
| Tails | 20 ± 1 | 1.00 ± 0.02 | 23 ± 1 | 1.00 ± 0.01 | - | 4.11 ± 0.01 | - |
| Headgroup 2 | 10 ± 1 | 0.41 ± 0.02 | 7 ± 1 | 0.10 ± 0.01 | 0.47 ± 0.01 | 0.39 ± 0.01 | 0.07 ± 0.01 |
| Peptide | - | - | 21 ± 2 | - | 0.15 ± 0.01 | - | 0.08 ± 0.01 |
| d31-POPC/d31-POPS (2:1) bilayer + N1 peptide | |||||||
| Headgroup 1 | 7 ± 1 | 0.94 ± 0.06 | 8 ± 1 | 0.99 ± 0.03 | - | 4.58 ± 0.16 | - |
| Tails | 20 ± 1 | 1.00 ± 0.02 | 29 ± 1 | 0.97 ± 0.01 | - | 4.85 ± 0.05 | - |
| Headgroup 2 | 11 ± 1 | 0.94 ± 0.03 | 9 ± 1 | 0.08 ± 0.01 | 0.47 ± 0.01 | 0.42 ± 0.01 | 0.07 ± 0.01 |
| Peptide | - | - | 21 ± 1 | - | 0.25 ± 0.01 | - | 0.08 ± 0.01 |
| d31-POPC/d31-POPG (2:1) bilayer + N2 peptide | |||||||
| Headgroup 1 | 7 ± 1 | 0.56 ± 0.01 | 7 ± 1 | 0.56 ± 0.02 | - | 2.30 ± 0.09 | - |
| Tails | 27 ± 1 | 1.00 ± 0.02 | 31 ± 1 | 1.00 ± 0.02 | - | 5.38 ± 0.12 | - |
| Headgroup 2 | 11 ± 1 | 0.86 ± 0.03 | 12 ± 1 | 0.14 ± 0.05 | 0.33 ± 0.01 | 0.90 ± 0.03 | 0.08 ± 0.01 |
| Peptide | - | - | 27 ± 3 | - | 0.20 ± 0.03 | - | 0.12 ± 0.01 |
| d31-POPC/d31-POPS (2:1) bilayer + N2 peptide | |||||||
| Headgroup 1 | 13 ± 1 | 0.89 ± 0.02 | 12 ± 1 | 0.78 ± 0.02 | - | 5.44 ± 0.11 | - |
| Tails | 26 ± 1 | 0.98 ± 0.01 | 27 ± 1 | 0.89 ± 0.01 | - | 4.17 ± 0.05 | - |
| Headgroup 2 | 18 ± 1 | 0.24 ± 0.02 | 16 ± 1 | 0.20 ± 0.01 | 0.53 ± 0.01 | 1.90 ± 0.03 | 0.18 ± 0.01 |
| Peptide | - | - | 20 ± 2 | 0 | 0.28 ± 0.01 | - | 0.12 ± 0.01 |
For all NR experiments a peptide concentration of 10 μM in a pH 5.0 buffer was employed, which has been previously shown to be effective for binding to anionic bilayers (18). The approach used was bilayer formation and characterization after which peptide was incubated on the surface and then the excess removed with a pH 5.0 buffer wash in either D2O or H2O. When both the N1 and N2 peptides were added to zwitterionic d31-POPC bilayers, no change in reflectivity was observed (Fig. S1) and no presence of peptide is observed in the corresponding real space nSLD profile. This corresponds with previous data that the N1 and N2 peptides do not bind to zwitterionic bilayers (22).
When the N1 peptide was added to the negatively charged d31-POPC/d31-POPG (2:1 molar ratio) bilayer, a reduction in the intensity of the fringe at Q = 0.101 Å-1 was observed along with a slight shift in fringe position to a lower value of Q by 0.01 Å-1 (compare black and red reflectivity profiles in Fig. 1 A). These changes indicate that the N1 peptide bound to the d31-POPC/d31-POPG bilayer without dramatic structural changes to the bilayer. A similar effect was observed when the N1 peptide was added to a d31-POPC/d31-POPS (2:1 molar ratio) bilayer; however, the reduction in the intensity was less than for d31-POPG-containing bilayers suggesting that less peptide was bound to the bilayer (Fig. 1 B). The modeling of the data to produce the real-space nSLD profiles provided more details on the membrane-bound peptide (Fig. 1 C and D). There was no change in the nSLD of the acyl tails region before peptide addition or when peptide was bound to both the d31-POPG- and d31-POPS-containing bilayers in the D2O contrast. This shows that the N1 peptide does not penetrate into the bilayer acyl tail layer. Additionally, changing the isotopic contrast to H2O in the presence of the N1 peptide also showed no isotopic-dependent change in the nSLD of the bilayer tails region, which further confirms the acyl tails layer is devoid of peptide and that there is no detectable pore-formation by the peptide. The calculated volume fractions of the lipid tails before and after peptide addition showed that there was no loss of lipid from the surface (Table 1), also suggesting no detectable peptide action through the membrane lysis process known as the “carpet mechanism” (36). The most profound effect that the N1 peptide had on both bilayers was in the outer headgroups of the phospholipids. The N1 peptide was present in this region along with solvent. Using the volume fractions of lipid and peptide, the surface excess and lipid-to-peptide ratio for the outer headgroup was calculated. From the lipid-to-peptide ratios in Fig. 2 it can be seen that a higher amount of peptide is bound to the d31-POPG-containing bilayer than the d31-POPS-containing bilayer, indicating a stronger interaction between d31-POPG and the N1 peptide than for d31-POPS. Both bilayers have a 21 Å layer of peptide above membrane. The peptide layer is not dense; having a peptide volume fraction of 0.15 and therefore a high solvent content (Table 1). A further observation was that the thickness of the acyl tail zone was increased on peptide binding, indicating a potential increase in lipid ordering (Table 1). This was also evident as a contraction of the area per lipid molecule of the membrane following peptide addition (Table 2), but no phase transition of the lipids was seen.
Figure 1.
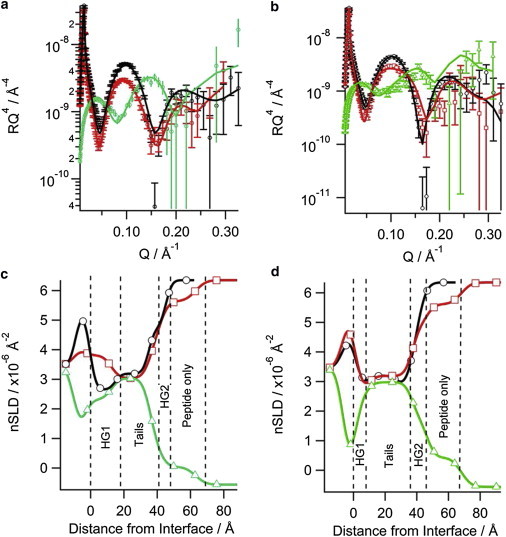
Neutron reflectometry of the N1 peptide binding to solid-supported phospholipid bilayers on silicon: (a) d31-POPC/d31-POPG (2:1) bilayer, (b) d31-POPC/d31-POPS (2:1) bilayer, the symbols with error bars are the data and the solid lines the fit. The corresponding real-space nSLD profile from the fits is also shown: (c) d31-POPC/d31-POPG (2:1) bilayer, (d) d31-POPC/d31-POPS (2:1) bilayer. The dashed vertical lines delineate the different layers in the peptide bound state. In all panels black (○) is bilayer before N1 peptide addition in D2O, red (□) is bilayer with N1 peptide bound in D2O, and green (▵) is bilayer with peptide bound in H2O. To see this figure in color, go online.
Figure 2.
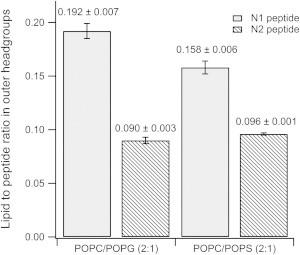
The lipid-to-peptide ratio of N1 and N2 peptides in the outer headgroups in anionic phospholipid bilayers as determined from neutron reflectometry data.
Table 2.
The change in area per lipid molecule upon binding N1 and N2 peptides
| Bilayer | Peptide | Alipid before peptide addition / Å2 | Alipid after peptide addition / Å2 |
|---|---|---|---|
| POPC / POPG (2:1) | N1 | 55 ± 4 | 51 ± 4 |
| POPC / POPS (2:1) | N1 | 65 ± 7 | 54 ± 4 |
| POPC / POPG (2:1) | N2 | 55 ± 5 | 49 ± 4 |
| POPC / POPS (2:1) | N2 | 44 ± 3 | 50 ± 3 |
NR showed the N2 peptide displayed similar behavior to the N1 peptide when binding to negatively charged phospholipid bilayers. As with the N1 peptide, when N2 was bound to solid-supported bilayers the intensity of the fringe at Q ∼ 0.1 Å-1 decreased suggesting that the N2 peptide bound to the bilayer (Fig. 3 A and B). For the H2O contrast in the d31-POPC/d31-POPS bilayer with N2 bound (Fig.3 B) the background of the data points did not match the fitted curve; however, the fit is still within a reasonable χ2 value of 1.24. When the background was constrained, a poorer fit that was not biologically credible resulted. The real-space nSLD profile showed no presence of peptide in the acyl tail region for both d31-POPC/d31-POPG bilayer and d31-POPC/d31-POPS bilayers (Fig. 3 C and D). The d31-POPS-containing bilayer showed an isotopic dependent change in the tail layer when changing between the D2O and H2O contrasts, highlighting the presence of solvent in the tail region (Fig. 3 D). The volume fraction of the solvent in the tail region of the d31-POPC/d31-POPS bilayer was 0.11 with the lipid volume fraction reducing down to 0.89 indicting some loss of lipid material from the surface. This marginal loss of lipid material over the period that the peptide was bound to the bilayer (∼ 7 h) makes it unlikely that the N2 peptide has any lytic or pore-forming mechanism. The predominant binding of the N2 peptide was at the outer headgroups forming a peptide layer above the bilayer in both negatively charged membranes. The lipid-to-peptide ratio of the N2 peptide in the outer headgroup region for each bilayer was calculated. Fig. 2 shows that there was essentially no difference in the lipid-to-peptide ratio between d31-POPG- or d31-POPS-containing bilayers. However, the lipid-to-peptide ratio of N1 was higher than N2 in both cases indicating less N2 bound to the bilayer than N1.
Figure 3.
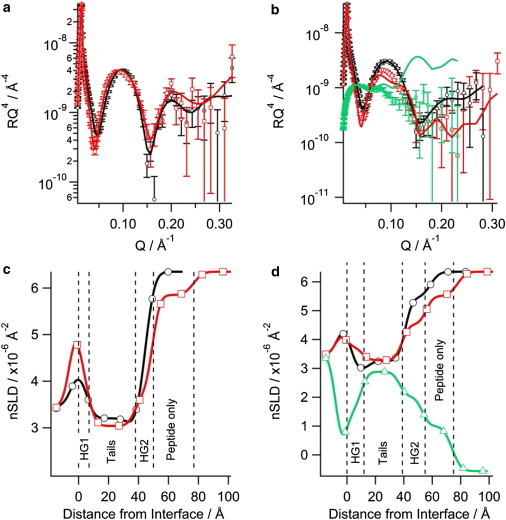
Neutron reflectometry of the N2 peptide binding to solid-supported phospholipid bilayers on silicon: (a) d31-POPC/d31-POPG (2:1) bilayer, (b) d31-POPC/d31-POPS (2:1) bilayer, the symbols with error bars are the data and the solid lines the fit. The corresponding real-space nSLD profile from the fits is also shown: (c) d31-POPC/d31-POPG (2:1) bilayer, (d) d31-POPC/d31-POPS (2:1) bilayer. The dashed vertical lines delineate the different layers in the peptide bound state. In all panels black (○) is bilayer before peptide addition in D2O, red (□) is bilayer with N2 peptide bound in D2O, and green (▵) is bilayer with N2 peptide bound in H2O. To see this figure in color, go online.
The NR data clearly indicates that the N1 and N2 peptides do not insert into the hydrophobic tail region of a phospholipid bilayer. To further confirm the absence of tail insertion by the N1 and N2 peptides (and to ensure the current results were not because of batch variation in peptides), we conducted QCM-D experiments comparing the N1 and N2 fragments with melittin from bee venom, a peptide known to insert into the hydrophobic tails of anionic phospholipid bilayers (37). Fig. S2 shows that when melittin is added to a bilayer of POPC/POPG (2:1) or POPC/POPS (2:1) the dissipation increases, the overtones no longer overlap, and the Sauerbrey equation is no longer valid as is consistent with recent publications (38). This is indicative of the vibration of the bilayer no longer being in concert with the vibrations of the oscillating quartz crystal because of the increased hydration of the bilayer. Although the mass of the bound melittin cannot be determined through the use of the Sauerbrey equation, the frequency of each overtone remains more negative than the frequency of the bilayer. This shows that there is an overall mass gain suggesting the increased hydration of the bilayer rather than loss of lipid material from the surface (which would result in a less negative frequency than the initial bilayer). As can be seen, the QCM-D trace for the addition of melittin was very different from that of either the N1 or N2 peptides binding to anionic bilayers (Fig. 4). The QCM-D traces for the N1 and N2 peptides (N2 trace presented in Fig. 4 and tabulated results shown in Table S4) show an overall mass gain on the bilayer indicating no lytic action by the peptides and all the overtones overlap with a low dissipation indicating that the bilayer was intact and had not changed its hydration state. Control QCM-D experiments were also conducted with DM lipids with no differences found from the PO lipids (Table S4). By plotting Δf versus ΔD, time is excluded as a parameter and the plot shows how the lipid bilayer structure changes per unit of peptide mass added. This provides a “finger print” of the mechanistic action of peptides with lipid bilayers (39). Fig. 5 shows the Δf-ΔD plots for N1 and N2 binding to both the POPG and POPS-containing bilayers. The origin of the each graph in Fig. 5 is the point of peptide addition (the point marked PA in Fig. 4 A). In all cases there is a linear increase of ΔD with increasing mass (i.e., a more negative Δf) as the peptide is binding to the bilayer until the lipid bilayers are saturated with peptide and there is no further change in Δf or ΔD resulting in the large cluster of data points on the right-hand of each graph. The shape of data in Fig. 5 is indicative of peptides that have minimal structural impact on lipid bilayers and is similar to the nonlytic peptides apidaecin (40) and oncocin (41). Finally, to further ensure minimal membrane perturbation and no lytic activity of the peptides calcein release from LUVs in response to the N1 and N2 peptides was compared with the action of melittin in POPC and POPC/POPG (2:1) vesicles (Fig. 6). No change from baseline fluorescence was seen for the POPC LUVs and minimal fluctuation was measured for the POPC/POPG LUVs when exposed to the N1 and N2 peptides compared with melittin, which demonstrated rapid and almost complete release. These results further exclude pore-formation by N1 or N2 and confirm our previous findings.
Figure 4.
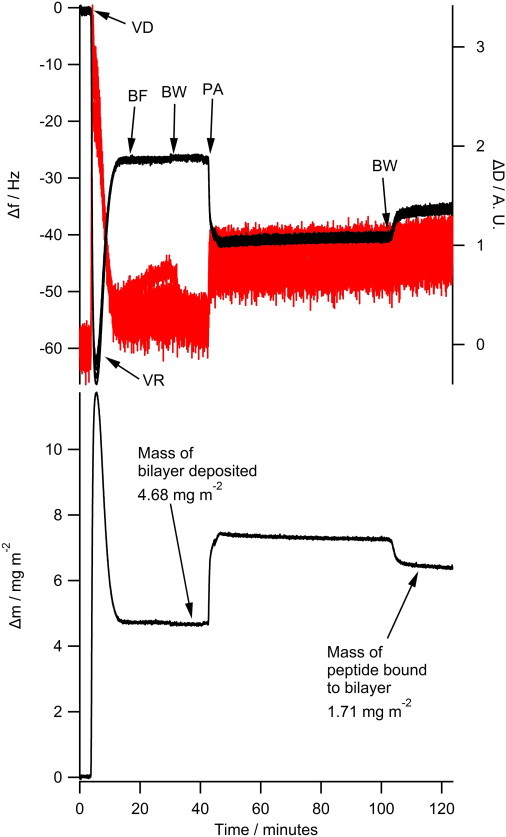
The QCM-D of 10 μM N2 peptide binding to a phospholipid bilayer of POPC/POPS (2:1). The upper panel shows the Δf trace on the left (black trace) and the ΔD trace is shown on the right (red trace). The 3rd, 5th, 7th, 9th, 11th, and 13th overtones are shown but are indistinguishable as all overtones overlap. The lower panel shows the corresponding Sauerbrey mass at each step of the measurement with final masses deposited indicated. VD—vesicle deposition, VR—vesicle rupture, BF—bilayer formation, BW—buffer wash, PA—peptide addition. To see this figure in color, go online.
Figure 5.
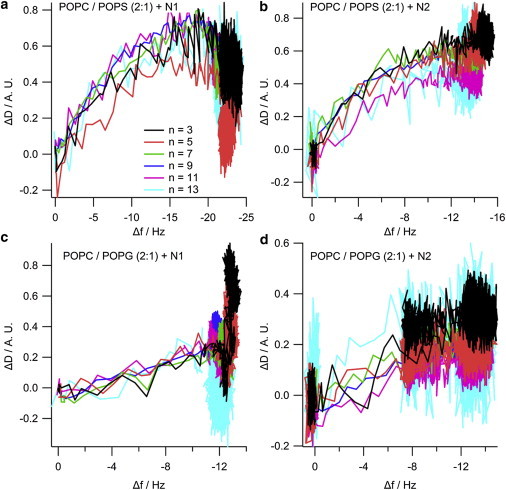
Δf-ΔD plots from the QCM-D data of (a) 10 μM N1 or (b) N2 binding to POPC/POPS (2:1) bilayers, or binding to (c and d) POPC/POPG (2:1) bilayers. The data shown is from the 3rd (black), 5th (red), 7th (green), 9th (blue), 11th (magenta), and 13th (cyan) overtones. To see this figure in color, go online.
Figure 6.
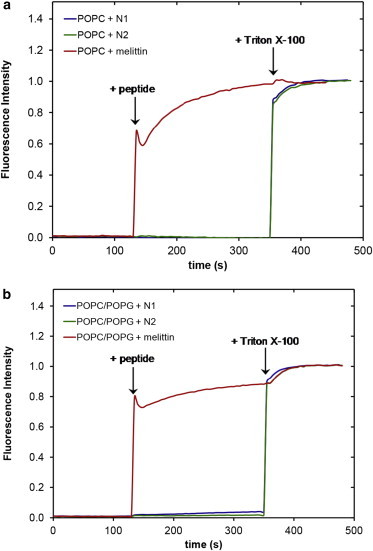
Calcein release from (a) POPC or (b) POPC/POPG (2:1) LUVs when exposed to N1 (blue), N2 (green), or melittin (red) peptides. Triton X-100 was added to demonstrate maximum release. To see this figure in color, go online.
Discussion
The NR data presented in the current study confirm the binding capabilities of the N1 and N2 endogenous cleavage fragments to phospholipid model membranes and further defines the specific engagement and changes to the lipid environment upon peptide binding. Our previous work showed interactions of the N1 and N2 peptides with anionic lipids that were consistent with either benign uniform insertion of the peptide across the bilayer or no insertion with peptide bound to the surface of the bilayer causing lipid ordering (22). Previous studies using 2H-solid-state NMR with only the palmitoyl chain of the POPC lipid labeled with deuterium in a 2:1 mixture of POPC:POPS were only able to report on the ordering effect on 33% of the tails in the lipid mixture. The 2H chemical shift anisotropy increased from 23.2 kHz for lipid only to 25.6 and 24.4 kHz in the presence of N1 and N2, respectively, indicating an ordering of the palmitoyl tails in POPC. This modest increase in ordering could reflect peptides inserting into the lipid tails; however, the increases did not account for a substantial perturbation in the freedom of movement of the palmitoyl tails associated with bilayer disruption and could suggest that the N1 and N2 peptides do not interact with the bulk lipid bilayer (22). In the study all parts of the lipid bilayer and the peptide were observed providing a complete picture and strong support that the latter situation is true, finding that binding was primarily in the outer headgroup region with no penetration into the acyl tails, and also confirmed that the binding affinity is greater for N1 than N2 on both the POPS/POPC and POPG/POPC bilayers, most likely because of the additional polybasic region at the C terminus of the N1 peptide (15). Therefore, these data continue to support a nonperturbing PrP N1/N2-lipid interaction and are not consistent with the peptide causing bilayer damage by other lipid disruptions such as the carpet mechanism (36).
Our findings contrast with NR studies of the neurotoxic peptide amyloid-beta (Aβ) found in Alzheimer’s disease that, in combination with atomic force microscopy, shows globular aggregates of freshly prepared Aβ1-40, Aβ1-42, or both are adsorbed to the membrane and disruptively penetrate into the core increasing hydration (42). Further, once the Aβ has initially inserted into the hydrophobic core of negatively charged lipids, causing disordering of the lipids, an induction of folding and template assembly of Aβ fibrils occurs (43). As with the N1 and N2 peptides, Aβ forms a peptide layer on the surface of the lipid bilayer. However, with Aβ, a thicker (∼ 40 Å), more crystalline layer is formed than observed with N1 and N2 (43). The binding parameters of the N1 and N2 peptides also differ from those of α-synuclein, which forms intracellular aggregates called Lewy bodies in Parkinson’s disease. Similar to N1 and N2, α-synuclein mainly embeds in the outer headgroups of mixed lipid bilayers containing anionic lipids, with a greater binding affinity seen at lower pH (44), but its membrane interaction causes thinning of the bilayer rather than the thickening effect seen for the PrP fragments. Also, binding to lipid membranes is known to induce a helical secondary structure of α-synuclein (45), although we previously observed the formation of increased beta-sheet secondary structure on binding of N2 to PC/PS (22). The structural change of α-synuclein and its lipid interactions is thought to be involved in disease pathogenesis (46). A similar situation is unlikely to be true for N1 and N2 given these fragments do not contain the amyloidogenic region of PrP and the data presented indicate that the membrane binding events of these N-terminal fragments of PrP appear minimally perturbing and would not be expected to be directly toxic to cells. Indeed viability studies in cells have shown that these peptides do not cause lysis and are not toxic but instead demonstrate cytoprotective properties (19,20). Considering these properties, and that the N1/N2 binding events show no evidence of being acutely damaging to membranes, these interactions may be relevant to the cellular functions of these fragments and/or full-length PrP.
Analysis of the NR data of N1 and N2 peptide binding to POPG and N1 binding to POPS containing membranes showed that there was a thickening of the lipid bilayer, reducing the area per lipid. Such changes are indicative of an ordering of the lipids in the absence of a phase transition. Changes in lipid order are required for the formation of scaffolds for protein binding and the assembly of multimeric protein complexes, such as those associated with Annexin A6 (47), which through structuring membrane domains and linking with actin were found to be involved in membrane repair. Interestingly, PrPC has been reported to bind to a number of proteins, with the N-terminal region (residues 23–32) also containing a tubulin binding site (48), indicating that full length PrPC and the N1 and N2 fragments could link a protein scaffold to lipid membrane domains and result in membrane ordering events under specific circumstances. The membrane ordering changes were not observed for the N2 peptide with the POPS-containing membrane. This may have a technical basis, most likely because of the small loss of lipid from the bilayer over the time of the experiment. We do not believe that this necessarily represents a biologically relevant event as the NR experiments were run over a period of hours but we have previously shown that the half-life of this peptide in cell systems is very short, approximately minutes (20), and therefore it is unlikely to persist for sufficiently long times to cause such an effect in vivo.
The N-terminal domain of PrPC has been shown to control PrPC membrane relocalization from lipid rafts and subsequent clathrin mediated internalization (49). The formation of protein scaffolds supporting tubule formation for budding of trafficking vesicles requires organization of the lipid membrane domain to which the protein scaffold attaches. The ability to engage a lipid scaffold may be highly important for the trafficking of full-length PrPC and potentially the N-terminal cleavage fragments. However, the absolute dependence on low pH for the observed membrane interactions suggests that any physiological role of N1 or N2 interaction with anionic lipids is likely to occur inside the cell in organelles of increased acidity. N-terminal interactions may also direct trafficking once inside the cell. A pH of 5 is representative of the pH within late endosomes, from which PrPC might undergo retrograde transportation to the Golgi (50), be transferred to recycling endosomes or multivesicular bodies, or it may be targeted to lysosomes for destruction. N-terminal engagement with intracellular membranes may therefore be as part of an endosomal sorting signal and direct the final destination of PrPC and/or its N-terminal cleavage fragments. A role in membrane interaction that directs intracellular trafficking might also account for intricate changes that alter exosomal membrane structure during prion disease (51). Exosomes are formed through membrane fission from multivesicular bodies (52) to which PrPC may become directed by endosomal trafficking. Notably, N-terminal processing of PrPC is altered in exosomes from prion-infected cells (53).
Ordering of lipids is also seen in the formation of membrane signaling complexes. An alternative hypothesis to N1 and N2 being involved in dictating intracellular trafficking is that their endocytosis is required to permit the formation of membrane signaling complexes. Endosomes are known to have a role in cellular signaling, controlling the “off” and “on” of some pathways (54). The pathways activated or deactivated are influenced by their type, maturity and cellular location (55). We have found that the protective signaling of N2 is transduced through MEK1 only after correct engagement of its internalization pathways (C. L. Haigh, unpublished observations). Therefore, endocytosis of N1 and/or N2 may instigate a lipid-protein engagement that facilitates intracellular signaling.
Although the observed membrane interactions of the N-terminal peptides did not indicate that such contact could be responsible for toxicity during disease, they may still be relevant to pathogenesis in the context of PrP misfolding. The involvement of POPG in the formation of genuinely infectious misfolded rPrP (7,56) suggests that the ability of PrP to bind these lipids as a full-length protein could be important for the generation of prions. Misfolding during prion disease is significantly more efficient if the N-terminal regions of PrP are present (57), and it has been shown that POPG binding to rPrP results in a structural change that exposes a cryptic binding site for the N-terminal polybasic region that may enhance further rPrP recruitment and templated misfolding (58). In cell cultures, high levels of PrP α-cleavage confer resistance to prion infection (59), most likely because the C1 fragment serves as a suboptimal substrate for misfolding; however, β-cleavage is seen to be increased during disease (18). In a cellular context, a greater ratio of C2 to full length PrP could result in aberrant membrane binding of C2, permitting conformational change and thus enhanced production of misfolded PrP during disease. β-cleavage leaves the core of misfolded PrP intact and C2 retains the second polybasic site that might substitute for 23-28. Studies considering rPrP fragments equivalent to the C2 region of PrPC have shown that the ability to bind lipids and misfold is maintained (60). Additionally, the central domain has a significant role in lipid binding in full-length PrPC and is influenced by disease-associated mutations of the proline residues at 102 and 105 (mouse sequence) to leucine residues (61). This supports that the N1 peptide should have a greater capacity to bind lipids than the N2 fragment that lacks this central domain, although disruption of lipid membranes is only observed for misfolded and aggregated rPrP, not for monomeric rPrP (60), supporting that N1 or N2 are unlikely to be membrane disruptive in vivo. When the N2 peptide was added to cells in culture at high concentrations (10 μM), a very small amount of dimer could be visualized by western blotting but no higher molecular weight oligomers, which were observed for a peptide with the prolines of the 23-28 polybasic region mutated to alanines (20). This indicates that the propensity for the N-terminal fragments to oligomerise and for any putative oligomers to cause membrane damage is also low.
Much previous work looking at PrPC and lipid binding has focused on full-length recombinant PrP, thus remaining open to the criticism that, because rPrP is not GPI-anchored or complex glycosylated, it may not be accurately representative. The peptides used in the current study are representative of genuine cleavage fragments that occur in the brain (18), separating the N-terminus from the structured, glycosylated, and membrane-anchored C terminus, and the data therefore present a stronger argument for how these cleavage fragments interact with cell membranes. Both N-terminal cleavage fragments have been shown to have neuroprotective functions and therefore lipid binding interactions could be crucial for these peptides to effect protection. The binding parameters of full length PrPC, especially in the context of membrane anchoring, remain to be determined; however, the data presented herein suggest that specific lipid binding interactions could potentially be corrupted during prion disease, when the cleavage events shift toward greater β-cleavage.
Conclusion
The new data generated by NR support our previous findings that endogenous PrP N-terminal cleavage fragments can interact with negatively charged supported phospholipid bilayers at low physiological pH. We further show that this interaction lies predominantly at the level of the phospholipid headgroups with no clear penetration into the acyl tail plane and without damage to the structural integrity of the membrane but with induction of a domain ordering effect. These observations stand in clear contrast to the findings of analogous studies of Aβ and α-synuclein and thereby support the possibility of a functional role for such N-terminal fragment-membrane interactions.
Acknowledgments
This work was supported by the Australian Nuclear Science and Technology Organisation (ANSTO), proposal ID2211, Australian Institute for Nuclear Science and Engineering (AINSE Ltd.) for travel support, and a National Health and Medical Research Council (NHMRC) program grant (No. 628946). S. J. C. is funded by an NHMRC Practitioner Fellowship (No. APP100581). S. C. D. is funded by an Australian Research Council Future Fellowship (FT110100199). A. P. L. B. is funded by an Australian Research Council Discovery Early Career Research Award (DE140101788). The authors declare that they have no conflicts of interest.
Footnotes
Anton P. Le Brun and Cathryn L. Haigh contributed equally to this work.
Martin P. Boland’s present address is School of Psychological and Clinical Sciences, Charles Darwin University, Darwin, Northern Territory, 0815, Australia.
Michael James’ present address is Australian Synchrotron, 800 Blackburn Road, Clayton, Victoria, 3168, Australia.
Supporting Material
References
- 1.Prusiner S.B. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Weissmann C., Bueler H., Aguet M. PrP-deficient mice are resistant to scapie. In: Bjornsson J., Carp R.I., Love A., Wisniewski H.M., editors. Slow Infections of the Central Nervous System: The Legacy of Dr Bjorn Sigurdsson. New York Academic Sciences; New York: 1994. pp. 235–240. [DOI] [PubMed] [Google Scholar]
- 3.Saborio G.P., Permanne B., Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 4.Kim J.I., Cali I., Surewicz W.K. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J. Biol. Chem. 2010;285:14083–14087. doi: 10.1074/jbc.C110.113464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welton J.M., Lawson V.A. The site and host dependent requirements for prion propagation. In: Collins S.J., Lawson V.A., editors. The Cellular and Molecular Biology of Prion Disease. Research Signpost; Scarborough, Canada: 2011. pp. 189–206. [Google Scholar]
- 6.Zhou Z., Xiao G. Conformational conversion of prion protein in prion diseases. Acta Biochim. Biophys. Sin. (Shanghai) 2013;45:465–476. doi: 10.1093/abbs/gmt027. [DOI] [PubMed] [Google Scholar]
- 7.Wang F., Wang X., Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–1135. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Critchley P., Kazlauskaite J., Pinheiro T.J.T. Binding of prion proteins to lipid membranes. Biochem. Biophys. Res. Commun. 2004;313:559–567. doi: 10.1016/j.bbrc.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Re F., Sesana S., Masserini M. Prion protein structure is affected by pH-dependent interaction with membranes: a study in a model system. FEBS Lett. 2008;582:215–220. doi: 10.1016/j.febslet.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Dong S.L., Cadamuro S.A., Renner C. Copper binding and conformation of the N-terminal octarepeats of the prion protein in the presence of DPC micelles as membrane mimetic. Biopolymers. 2007;88:840–847. doi: 10.1002/bip.20860. [DOI] [PubMed] [Google Scholar]
- 11.Robinson P.J., Pinheiro T.J.T. Phospholipid composition of membranes directs prions down alternative aggregation pathways. Biophys. J. 2010;98:1520–1528. doi: 10.1016/j.bpj.2009.12.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steunou S., Chich J.F., Vidic J. Biosensing of lipid-prion interactions: insights on charge effect, Cu(II)-ions binding and prion oligomerization. Biosens. Bioelectron. 2010;26:1399–1406. doi: 10.1016/j.bios.2010.07.064. [DOI] [PubMed] [Google Scholar]
- 13.Haigh C.L., Marom S.Y., Collins S.J. Copper, endoproteolytic processing of the prion protein and cell signalling. Front. Biosci. 2010;15:1086–1104. doi: 10.2741/3663. (Landmark ed.) [DOI] [PubMed] [Google Scholar]
- 14.Borchelt D.R., Rogers M., Prusiner S.B. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology. 1993;3:319–329. doi: 10.1093/glycob/3.4.319. [DOI] [PubMed] [Google Scholar]
- 15.Harris D.A., Huber M.T., Wang R. Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry. 1993;32:1009–1016. doi: 10.1021/bi00055a003. [DOI] [PubMed] [Google Scholar]
- 16.McMahon H.E.M., Mangé A., Lehmann S. Cleavage of the amino terminus of the prion protein by reactive oxygen species. J. Biol. Chem. 2001;276:2286–2291. doi: 10.1074/jbc.M007243200. [DOI] [PubMed] [Google Scholar]
- 17.Mangé A., Béranger F., Lehmann S. Alpha- and beta-cleavages of the amino-terminus of the cellular prion protein. Biol. Cell. 2004;96:125–132. doi: 10.1016/j.biolcel.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Chen S.G., Teplow D.B., Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 1995;270:19173–19180. doi: 10.1074/jbc.270.32.19173. [DOI] [PubMed] [Google Scholar]
- 19.Guillot-Sestier M.V., Sunyach C., Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J. Biol. Chem. 2009;284:35973–35986. doi: 10.1074/jbc.M109.051086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haigh C.L., Drew S.C., Collins S.J. Dominant roles of the polybasic proline motif and copper in the PrP23-89-mediated stress protection response. J. Cell Sci. 2009;122:1518–1528. doi: 10.1242/jcs.043604. [DOI] [PubMed] [Google Scholar]
- 21.Walmsley A.R., Watt N.T., Hooper N.M. Alpha-cleavage of the prion protein occurs in a late compartment of the secretory pathway and is independent of lipid rafts. Mol. Cell. Neurosci. 2009;40:242–248. doi: 10.1016/j.mcn.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 22.Boland M.P., Hatty C.R., Collins S.J. Anionic phospholipid interactions of the prion protein N terminus are minimally perturbing and not driven solely by the octapeptide repeat domain. J. Biol. Chem. 2010;285:32282–32292. doi: 10.1074/jbc.M110.123398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karas J.A., Boland M., Scanlon D. Microwave synthesis of prion protein fragments up to 111 amino acids in length generates biologically active peptides. Int. J. Pept. Res. Ther. 2012;18:21–29. [Google Scholar]
- 24.James M., Nelson A., Klose F. The multipurpose time-of-flight neutron reflectometer “Platypus” at Australia’s OPAL reactor. Nucl. Instrum. Methods Phys. Res. A. 2011;632:112–123. [Google Scholar]
- 25.Saerbeck T., Klose F., James M. Invited article: polarization “down under”: the polarized time-of-flight neutron reflectometer PLATYPUS. Rev. Sci. Instrum. 2012;83:081301–081312. doi: 10.1063/1.4738579. [DOI] [PubMed] [Google Scholar]
- 26.Nelson A. Motofit—integrating neutron reflectometry acquisition, reduction and analysis into one, easy to use, package. J. Phys. Conf. Ser. 2010;251:012094. [Google Scholar]
- 27.Nelson A. Co-refinement of multiple-contrast neutron/x-ray reflectivity data using MOTOFIT. J. Appl. Crystallogr. 2006;39:273–276. [Google Scholar]
- 28.Chiu S.W., Jakobsson E., Scott H.L. Combined Monte Carlo and molecular dynamics simulation of fully hydrated dioleyl and palmitoyl-oleyl phosphatidylcholine lipid bilayers. Biophys. J. 1999;77:2462–2469. doi: 10.1016/S0006-3495(99)77082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pabst G., Danner S., Raghunathan V.A. On the propensity of phosphatidylglycerols to form interdigitated phases. Biophys. J. 2007;93:513–525. doi: 10.1529/biophysj.106.101592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrache H.I., Tristram-Nagle S., Nagle J.F. Structure and fluctuations of charged phosphatidylserine bilayers in the absence of salt. Biophys. J. 2004;86:1574–1586. doi: 10.1016/S0006-3495(04)74225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wacklin H.P. Neutron reflection from supported lipid membranes. Curr. Opin. Colloid Interface Sci. 2010;15:445–454. [Google Scholar]
- 32.Le Brun A.P., Darwish T.A., James M. Studies of biomimetic cellular membranes using neutron reflection. J. Chem. Biol. Interfaces. 2013;1:3–24. [Google Scholar]
- 33.Kalb E., Frey S., Tamm L.K. Formation of supported planar bilayers by fusion of vesicles to supported phospholipid monolayers. Biochim. Biophys. Acta—Biomembr. 1992;1103:307–316. doi: 10.1016/0005-2736(92)90101-q. [DOI] [PubMed] [Google Scholar]
- 34.Johnson S.J., Bayerl T.M., Sackmann E. Structure of an adsorbed dimyristoylphosphatidylcholine bilayer measured with specular reflection of neutrons. Biophys. J. 1991;59:289–294. doi: 10.1016/S0006-3495(91)82222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiessling V., Tamm L.K. Measuring distances in supported bilayers by fluorescence interference-contrast microscopy: polymer supports and SNARE proteins. Biophys. J. 2003;84:408–418. doi: 10.1016/S0006-3495(03)74861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandez D.I., Le Brun A.P., Separovic F. The antimicrobial peptide aurein 1.2 disrupts model membranes via the carpet mechanism. Phys. Chem. Chem. Phys. 2012;14:15739–15751. doi: 10.1039/c2cp43099a. [DOI] [PubMed] [Google Scholar]
- 37.Krueger S., Meuse C.W., Plant A.L. Investigation of hybrid bilayer membranes with neutron reflectometry: probing the interactions of melittin. Langmuir. 2001;17:511–521. [Google Scholar]
- 38.Lu N.Y., Yang K., Li J.L., Yuan B., Ma Y.Q. Vesicle deposition and subsequent membrane-melittin interactions on different substrates: a QCM-D experiment. Biochim. Biophys. Acta—Biomembr. 2013;1828:1918–1925. doi: 10.1016/j.bbamem.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 39.McCubbin G.A., Praporski S., Martin L.L. QCM-D fingerprinting of membrane-active peptides. Eur. Biophys. J. 2011;40:437–446. doi: 10.1007/s00249-010-0652-5. [DOI] [PubMed] [Google Scholar]
- 40.Piantavigna S., Czihal P., Martin L.L. Cell penetrating apidaecin peptide interactions with biomimetic phospholipid membranes. Int. J. Pept. Res. Ther. 2009;15:139–146. [Google Scholar]
- 41.Knappe D., Piantavigna S., Hoffmann R. Oncocin (VDKPPYLPRPRPPRRIYNR-NH2): a novel antibacterial peptide optimized against gram-negative human pathogens. J. Med. Chem. 2010;53:5240–5247. doi: 10.1021/jm100378b. [DOI] [PubMed] [Google Scholar]
- 42.Dante S., Hauss T., Dencher N.A. Nanoscale structural and mechanical effects of beta-amyloid (1–42) on polymer cushioned membranes: a combined study by neutron reflectometry and AFM Force Spectroscopy. Biochim. Biophys. Acta—Biomembr. 2011;1808:2646–2655. doi: 10.1016/j.bbamem.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 43.Chi E.Y., Ege C., Lee K.Y.C. Lipid membrane templates the ordering and induces the fibrillogenesis of Alzheimer’s disease amyloid-beta peptide. Proteins. 2008;72:1–24. doi: 10.1002/prot.21887. [DOI] [PubMed] [Google Scholar]
- 44.Hellstrand E., Grey M., Sparr E. Adsorption of α-synuclein to supported lipid bilayers: positioning and role of electrostatics. ACS Chem. Neurosci. 2013;4:1339–1351. doi: 10.1021/cn400066t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jao C.C., Hegde B.G., Langen R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bisaglia M., Mammi S., Bubacco L. Structural insights on physiological functions and pathological effects of alpha-synuclein. FASEB J. 2009;23:329–340. doi: 10.1096/fj.08-119784. [DOI] [PubMed] [Google Scholar]
- 47.Cornely R., Rentero C., Gaus K. Annexin A6 is an organizer of membrane microdomains to regulate receptor localization and signalling. IUBMB Life. 2011;63:1009–1017. doi: 10.1002/iub.540. [DOI] [PubMed] [Google Scholar]
- 48.Osiecka K.M., Nieznanska H., Nieznanski K. Prion protein region 23-32 interacts with tubulin and inhibits microtubule assembly. Proteins. 2009;77:279–296. doi: 10.1002/prot.22435. [DOI] [PubMed] [Google Scholar]
- 49.Taylor D.R., Watt N.T., Hooper N.M. Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J. Cell Sci. 2005;118:5141–5153. doi: 10.1242/jcs.02627. [DOI] [PubMed] [Google Scholar]
- 50.Lu L., Hong W. From endosomes to the trans-Golgi network. Semin. Cell Dev. Biol. 2014;31:30–39. doi: 10.1016/j.semcdb.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 51.Coleman B.M., Hanssen E., Hill A.F. Prion-infected cells regulate the release of exosomes with distinct ultrastructural features. FASEB J. 2012;26:4160–4173. doi: 10.1096/fj.11-202077. [DOI] [PubMed] [Google Scholar]
- 52.Von Bartheld C.S., Altick A.L. Multivesicular bodies in neurons: distribution, protein content, and trafficking functions. Prog. Neurobiol. 2011;93:313–340. doi: 10.1016/j.pneurobio.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vella L.J., Sharples R.A., Hill A.F. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 2007;211:582–590. doi: 10.1002/path.2145. [DOI] [PubMed] [Google Scholar]
- 54.von Zastrow M., Sorkin A. Signaling on the endocytic pathway. Curr. Opin. Cell Biol. 2007;19:436–445. doi: 10.1016/j.ceb.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taub N., Teis D., Huber L.A. Late endosomal traffic of the epidermal growth factor receptor ensures spatial and temporal fidelity of mitogen-activated protein kinase signaling. Mol. Biol. Cell. 2007;18:4698–4710. doi: 10.1091/mbc.E07-02-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Z., Zhang Y., Ma J. De novo generation of infectious prions with bacterially expressed recombinant prion protein. FASEB J. 2013;27:4768–4775. doi: 10.1096/fj.13-233965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lawson V.A., Priola S.A., Chesebro B. Flexible N-terminal region of prion protein influences conformation of protease-resistant prion protein isoforms associated with cross-species scrapie infection in vivo and in vitro. J. Biol. Chem. 2004;279:13689–13695. doi: 10.1074/jbc.M303697200. [DOI] [PubMed] [Google Scholar]
- 58.Zurawel A.A., Walsh D.J., Supattapone S. Prion nucleation site unmasked by transient interaction with phospholipid cofactor. Biochemistry. 2014;53:68–76. doi: 10.1021/bi4014825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lewis V., Hill A.F., Collins S.J. Increased proportions of C1 truncated prion protein protect against cellular M1000 prion infection. J. Neuropathol. Exp. Neurol. 2009;68:1125–1135. doi: 10.1097/NEN.0b013e3181b96981. [DOI] [PubMed] [Google Scholar]
- 60.Chich J.F., Chapuis C., Noinville S. Vesicle permeabilization by purified soluble oligomers of prion protein: a comparative study of the interaction of oligomers and monomers with lipid membranes. J. Mol. Biol. 2010;397:1017–1030. doi: 10.1016/j.jmb.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 61.Wang F., Yin S., Ma J. Role of the highly conserved middle region of prion protein (PrP) in PrP-lipid interaction. Biochemistry. 2010;49:8169–8176. doi: 10.1021/bi101146v. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.