

Abstract
Purpose
Prior studies show that intramuscular (IM) injection of xenogeneic orthologues of melanosomal antigens (tyrosinase, gp100) induces CD8+ T cell responses to the syngeneic protein. To further define the optimal vaccination strategy, we conducted a pilot clinical trial comparing IM injection with particle-mediated epidermal delivery (PMED).
Experimental Design
Human leukocyte antigen (HLA)-A*0201+ disease-free melanoma patients were randomized to the PMED or IM arm, receiving 8 vaccinations over 4 months. Patients received 4 μg or 2000 μg per injection, respectively, of mouse gp100 DNA. Peripheral blood mononuclear cells (PBMCs) were collected, cultured with gp100 peptides and analyzed by tetramer and intracellular cytokine staining (ICS) for responses to HLA-A*0201-restricted gp100 epitopes [gp100209-217 (ITDQVPFSV) and gp100280-288 (YLEPGPVTA)].
Results
Twenty seven patients with stage IIB-IV melanoma were analyzable for immune response. The only common toxicity was grade I injection site reaction in 9 patients with no intergroup difference, while one dose-limiting toxicity of acute hypersensitivity occurred in a PMED patient with undiagnosed gold allergy. Four of 27 patients produced gp100 tetramer+CD8+ T cells, all carrying the CCR7loCD45RAlo effector-memory phenotype. Five of 27 patients generated interferon-γ+ (IFN-γ) CD8+ T cells, one who was also tetramer-positive. Overall, vaccination induced a response in 30% of patients, which was not significantly associated with study arm or clinical outcome. However, the PMED group showed a trend toward increased IFN-γ+CD8+ T cell generation (p=0.07).
Conclusion
A comparable efficacy and safety profile was demonstrated between the IM and PMED arms, despite a significantly decreased dose of DNA used for PMED injection.
Keywords: gp100, DNA, vaccine, PMED, melanoma
Introduction
Currently, the only FDA approved adjuvant therapy for melanoma is high dose interferon alpha, which does not produce a significant prolongation in overall survival but does improve relapse-free survival.(1) Therefore, there is a substantial interest in alternative adjuvant therapy including tumor vaccines such as DNA vaccines. Evidence for immunotherapy efficacy continues to mount with immunomodulatory antibodies including anti-CTLA-4 antibodies.(2-5) Melanoma provides an ideal setting for evaluating tumor antigen-specific immune responses, given selective expression of differentiation antigens.
Gp100, a melanocytic differentiation protein present mainly in melanocytes and melanoma, provides a specific immunization target. In fact, pre-existing gp100 antigen-specific T cells have been demonstrated in melanoma patients. This supports the goal of expanding functional tumor antigen-specific T cell responses with anti-tumor immunity. Our group and others have compared gp100 vaccination utilizing the (HLA)-A *0201 gp100209-217 (210M) peptide (IMDQVPFSV) or gp100 cDNA, comparing different adjuvants and intramuscular gp100 DNA, with demonstration of enhanced antigen-specific T cell responses.(6-7) The prior work has defined the adjuvant criteria for peptide immunization. In addition, xenogeneic DNA vaccination has provided sufficient antigenic disparity to overcome immune tolerance or ignorance.(8) However, much of the pre-clinical DNA vaccination development in mouse models was done utilizing particle mediated epidermal delivery (PMED). Genetic immunization through PMED has demonstrated to be capable of generating potent immune responses.(9) We have previously shown that DNA vaccination by PMED can result in frequent tumor specific T cell responses in mice.(10) However, clinical utilization of intramuscular DNA vaccination has been less efficacious than would be suggested by murine studies.
Pre-clinical studies comparing IM and gene gun (PMED) methods of immunization have shown greater IFN-γ secreting CD8+ T cell responses with PMED, although with similar protective tumor immunity and therapeutic efficacy. However, a lesser amount of vaccination DNA was required for the gene gun technique suggesting it was the most potent method per unit of DNA.(11) Additional pre-clinical information suggested that antibody production was greatest with gene gun vaccination compared with intramuscular vaccination.(12) Furthermore, a prior clinical study of PMED vaccination with gp100 and GM-CSF resulted in few adverse events.(7) However, no comparison with IM was conducted. There is a critical need to determine optimal methods of plasmid DNA delivery to generate effective immune responses. Therefore, utilizing our platform of immunizing against xenogeneic differentiation antigens, we performed a study to prospectively compare gp100 plasmid DNA delivered by gold particle conjugates with standard intramuscular injection in patients with high risk melanoma after surgery.
Materials and Methods
Eligibility criteria
To be eligible to participate in this study, patients must have been diagnosed with American Joint Committee on Cancer (AJCC) stage IIB, IIC, III and IV malignant melanoma, histologically confirmed at the Memorial Sloan-Kettering Cancer Center. Those with stage IIB, IIC or III must have had already undergone initial standard therapy (surgery), and all patients free of disease after surgical resection were only eligible if they had refused interferon-alpha therapy or had a recurrence while on it. Patients with choroidal melanoma required a basal diameter >16mm, height >8mm or involvement of the ciliary body. Additional eligibility criteria included a Karnofsky performance status ≥ 80%, HLA-A *0201 positivity, the absence of detectable brain metastases, a negative anti-double-stranded DNA serum antibody screen, and adequate organ and marrow function as defined by prior standards. Exclusion criteria included prior chemotherapy, immunotherapy or radiotherapy within 4 weeks of participation in the study, previous immunization with a gp100-containing vaccine, preexisting choroidal eye disease, pregnancy or nursing, allergy to gold, and any comorbidity or medication (e.g., corticosteroids) that could interfere with the treatment course. The Institutional Review Board approved this study and consent was received by all patients (NCT00398073).
Study design and treatment plan
In this study, patients were randomized to receive mouse gp100 DNA vaccine by means of either intramuscular (IM) injection or particle-mediated epidermal delivery (PMED). Patients receiving IM delivery were given 1000 μg plasmid DNA per injection using the Biojector2000 jet delivery device (Bioject, Tualatin, OR), while those in the PMED subgroup were vaccinated with 2 μg plasmid DNA coated on 1000 μg gold per actuation via the ND10 delivery system (PowderMed/Pfizer, Sandwich, UK). Regardless of delivery method, all patients were treated with 2 injections/day every two weeks for 4 months, with rotation of injection sites and avoidance of location with prior draining regional lymph node removal. Treatment was discontinued in the presence of a dose-limiting toxicity (DLT) or with development of progressive or recurrent disease requiring systemic treatment or radiation therapy. DLT was defined as any grade 3/4 toxicity or grade ≥2 allergic/immunologic toxicity, as per the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0.
DNA vaccine construct
Mouse gp100 plasmid DNA had been previously sequenced and introduced into the pING vector by our group (13), which has extensively utilized this conventional eukaryotic expression vector in both pre-clinical studies and clinical trials (14-15). This vector contains a cytomegalovirus promotor and a kanamycin resistence selection marker (KanR). KanR is the antibody selection gene, in accordance with the Food and Drug Administration's Point to Consider for DNA vaccination. The mouse gp100 plasmid DNA was subsequently tested for endotoxin, sterility and animal safety. Production of clinical-grade material was accomplished by Althea Technologies (San Diego, CA).
Evaluations at baseline and during therapy
Prior to initializing therapy, a complete history and physical examination was performed for all patients, along with a baseline ophthalmologic examination to evaluate for preexisting retinal or choroidal eye disease. Routine blood work, chest imaging (X-ray or CT) and a brain MRI was also completed, in addition to appropriate radiographic imaging of lesions in patients with measurable disease.
For immune function monitoring, blood samples were drawn at 1 week before and immediately prior to the first vaccination and at weeks 18 and 30, associated with 3 and 15 weeks post-vaccination completion, respectively. To ensure the acquisition of a sufficient quantity of peripheral blood mononuclear cells (PBMC), leukapheresis was performed at baseline and week 18, if possible. In addition, patients underwent clinical and radiologic monitoring as indicated to assess for disease recurrence and adverse events.
Immune function monitoring
Assessment of T-cell response was made using tetramer and intracellular cytokine staining (ICS) assays coupled with multiparameter flow cytometry. Frozen PBMC samples were utilized for such assays, originally obtained at baseline and weeks 18 and 30 as described above. In short, thawed PBMCs were incubated at a 1:30 ratio with HLA-A *0201-transfected K562 cells pulsed with either of the following peptides at 10 μg/ml each: HLA A2-restricted gp100209-217 (ITDQVPFSV) and gp100280-288 (YLEPGPVTA) (JPT Peptide Technologies, Berlin, Germany). Every 2 days, the cells were re-fed with complete media (10% pooled human serum and RPMI), 10 units/mL IL-2 and 10 ng/mL IL-15. The cells were harvested at day 10 and analyzed immediately by tetramer staining. The following tetramers and fluorochrome-labeled antibodies were used: HLA-A *0201-PE-labeled tetramers loaded with gp100209-217 (ITDQVPFSV) and gp100280-288 (YLEPGPVTA) (Tetramer Core, Lausanne Branch, Ludwig Institute of Cancer Research, Lausanne, Switzerland), APC-Cy7-CD8, FITC-Interferon (IFN)-γ, PE-Cy7-CD3, APC-CD3, PE-Macrophage inflammatory protein (MIP)-1β (BD Pharmingen, San Jose, CA), PE-Cy7-Tumor necrosis factor (TNF)-α (eBioscience, San Diego, CA), ECD-CD4, ECD-CD45RA (Beckman Coulter Inc., Fullerton, CA) and FITC-CCR7 (R&D Systems, Minneapolis, MN).
For ICS, the cells were further incubated for 20 minutes with PE-Cy5-CD107a (20 μl/ml, BD Pharmingen) and then re-stimulated with either of the preceding gp100 peptides for 2 hours. Brefeldin A and monensin (BD Biosciences, San Jose, CA) were then added at 5 μg/ml each and the cells were incubated overnight for no more than 16 hours.(16) Cells were analyzed using a CyAn flow cytometer with Summit software (Dako Cytomation California Inc., Carpinteria, CA). Analysis was performed using FlowJo software (version 8.8; TreeStar, Inc., Ashland, OR).
Statistical methods
In order to determine positive T-cell responses, we calculated the standard deviation of the prevaccination replicate values taken at two timepoints (one week prior to vaccination and the value immediately before vaccination). A T-cell response at any post-vaccination time-point was considered positive if it had a value ≥3 standard deviations greater than the mean value at baseline and having an absolute value >0.1%. Differences between groups were analyzed using Fisher's exact test. Progression-free survival (PFS) and overall survival were estimated using the Kaplan-Meier method.
Results
Patient Demographics
Thirty four patients were enrolled with equal numbers in both the IM and PMED arms, all evaluable for survival. Twenty nine patients received all 8 vaccinations, 27 of whom were assessable for immune function (15 IM, 12 PMED). Reasons for termination of vaccination or lack of immune assessment included progression of disease during vaccination course (1 IM, 4 PMED), development of multiple myeloma (1 IM) and an acute hypersensitivity reaction following the initial actuation (1 PMED). Post-vaccine PBMC samples were not collected on these 7 patients. Patient demographics are listed in Table 1.
Table 1.
Patient Demographics
| Characteristic | Overall | IM | PMED | |||
|---|---|---|---|---|---|---|
| n | % | n | % | n | % | |
| Number | 34 | 17 | 50% | 17 | 50% | |
| Age | ||||||
| Range | 26-79 | 37-79 | 26-78 | |||
| Median | 52.5 | 55 | 47 | |||
| Sex | ||||||
| Male | 26 | 76% | 11 | 65% | 15 | 88% |
| Female | 8 | 24% | 6 | 35% | 2 | 12% |
| Stage | ||||||
| IIB | 2 | 6% | 1 | 6% | 1 | 6% |
| IIC | 2 | 6% | 2 | 12% | 0 | 0% |
| III | 25 | 74% | 10 | 59% | 15 | 88% |
| IV | 5 | 15% | 4 | 24% | 1 | 6% |
| Karnofsky performance status | ||||||
| 90% | 14 | 41% | 7 | 41% | 7 | 41% |
| 100% | 20 | 59% | 10 | 59% | 10 | 59% |
| Prior therapy | ||||||
| None | 22 | 65% | 8 | 47% | 14 | 82% |
| Temozolomide | 2 | 6% | 2 | 12% | 0 | 0% |
| Radiation alone | 3 | 9% | 2 | 12% | 1 | 6% |
| Interferon alone | 4 | 12% | 3 | 18% | 1 | 6% |
| Multiple therapies | 3 | 9% | 2 | 12% | 1 | 6% |
| Radiation + IFN-α | 2 | 6% | 1 | 6% | 1 | 6% |
| IFN-α + IL-2 | 1 | 3% | 1 | 6% | 0 | 0% |
The median age of the patients was 55 (37-79) in the IM arm and 47 (26-78) in the PMED arm, with an overall male predominance of 74%. All patients had a Karnofsky performance status of 90% or greater at the time of initial vaccination. Eighty-eight percent (30/34) of the patients were Stage III or IV, with all patients having no evidence of disease. Twelve patients had received prior therapy, including temozolomide (2), radiation alone (3), interferon-α alone (4), or combination therapy of radiation plus interferon-α (2) or interferon-α plus interleukin-2 (1).
Toxicity and Survival
Toxicity was assessed in all patients receiving at least 1 vaccination. Therapy was generally well-tolerated with 10 patients (59%) in the IM arm versus 6 patients (35%) in the PMED arm having no adverse effects. There was 1 incident of dose-limiting toxicity, with 1 patient in the PMED arm developing an acute hypersensitivity reaction after the first injection. This was evaluated by a dermatologist who felt this was most consistent with gold sensitivity. The most common toxicity otherwise was grade 1 local injection site reactions in 4 patients in the IM arm and 5 patients in the PMED arm. Other potentially vaccine-associated toxicities included fatigue, watery eyes and abdominal pain in 2 patients each (6%), and nausea, arthralgia, myalgia, pruritus, and rash in 1 patient each (3%). In addition, one patient with a prior history of gout had an exacerbation after the third injection, and another previously-irradiated patient experienced radiation recall after the first injection. The median follow-up was 24 months with a median PFS of 17 months. Median survival was not yet reached.
Post-immunization increase in gp100-specific tetramer-reactive CD8+ cells
Following a 10-day culture, patient PBMC samples were stained with HLA *A0201 restricted gp100209-217 and gp100280-288 peptides at two baseline measurements and at weeks 18 and 30, again associated with 3 and 15 weeks after the final vaccination. We have found this 10-day culture necessary to detect low frequency self-antigen specific T cells, and it is also known that this relatively short in vitro culture does not cause in vitro priming.(16) Multi-parameter flow cytometry was then performed to analyze these samples.
Overall, four of the 27 assessed patients experienced an increase in gp100 tetramer-reactive CD8+ cells post-vaccination when compared to baseline, corresponding to 1 and 3 patients in the IM and PMED groups, respectively. Three patients were positive for gp100280-288 alone, and 1 patient was positive for both gp100209-217 and gp100280-288. Figure 1 demonstrates dot plots for a representative positive patient, while Figure 2 shows all patients’ changes in frequency of tetramer-reactive CD8+ cells from baseline to both post-vaccination time-points.
Figure 1. Representative gp100209-217 HLA-*A201-restricted tetramer analysis on CD8+ T cells following mouse gp100 DNA vaccination.
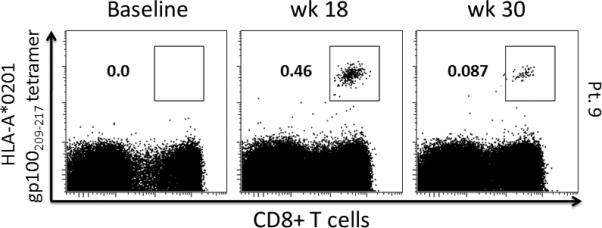
Gp100209-217 and gp100280-288 tetramer staining was performed at baseline, week 18 and week 30, and analyzed via multi-parameter flow cytometry. Tetramer positive cells, defined as greater than 3 times the standard deviation above the mean baseline value and an absolute value above 0.1%, were gated on CD3+CD8+ T cells. The above representative dot-plots demonstrate this gating within a positive patient's CD3+ lymphocyte population.
Figure 2. Changes in (A) gp100209-217 and (B) gp100280-288 HLA-*A201-restricted tetramer CD8+ T cells following mouse gp100 DNA vaccination.
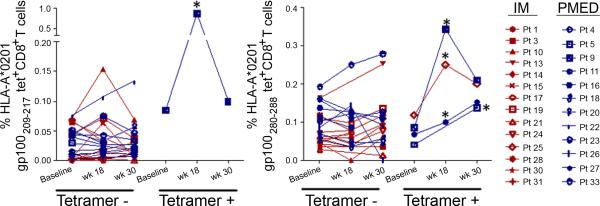
This figure shows the mean values of tetramer-positive CD8+ cells at each time-point, with positive patients presented on the right of each graph and positive time points marked by asterisks. Patients in the IM arm are shown in red, while those in the PMED arm are in blue.
Phenotypic analysis of the tetramer-positive CD8+ cells: indicative of an effector cell population
Chemokine receptor 7 (CCR7) and CD45RA are often used to subtype CD8+ T-cell populations, providing the following phenotypes: naïve cells (CCR7+ CD45RA+), central memory cells (CCR7+ CD45RA-), effector memory cells (CCR7- CD45RA-) and effector cells (CCR7- CD45RA +). We therefore further characterized the gp100 tetramer-positive CD8+ cells based on analyzing the expression of the above markers. Every tetramer-positive sample carried the phenotype of an effector memory cell, exhibiting the above-described CCR7lo CD45RAlo expression. Specifically, the tetramer+ T cells were, on average, 80.2% CCR7lo (range 60.2-96.1) and 76.3% CD45RAlo (range 67.7-94.9). See Figure 3 for representative dot-plots.
Figure 3. Phenotypic characterization of gp100209-217 HLA-*A201-restricted tetramer-positive CD8+ cells.
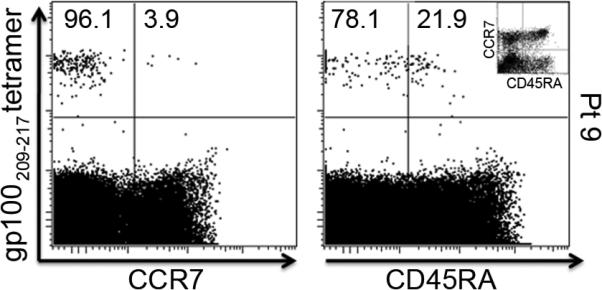
All gp100209-217 and gp100280-288 tetramer-positive CD8+ cells were further analyzed for the expression of CCR7 and CD45RA. This figure shows representative dot-plots for a tetramer-positive patient, demonstrating the CCR7loCD45RAlo effector memory phenotype. The plot in the upper-right corner demonstrates the gating strategy.
Post-immunization increases in CD8+ IFN-γ+ cells and polyfunctionality
Intracellular cytokine staining (ICS) was also performed on all samples and time-points to assess the effect of vaccination on the intracellular cytokine constitution. Five of the 27 analyzed patients showed an increase in CD8+IFN-γ+ cells post-vaccination when compared to baseline, with 1 and 4 in the IM and PMED groups, respectively. All 5 of these patient samples were restimulated with the gp100280-288 peptide, while none of the samples restimulated with the gp100209-217 peptide showed such a response. These 5 patients were further analyzed for the intracellular presence of macrophage inhibitory protein (MIP)-1β, tumor necrosis factor (TNF)-α and CD107a. These are common cytokines used in vaccine response assessment and formed the basis of polyfunctionality analysis in our prior study.(17) An increase in polyfunctionality (2 or more intracellular cytokines) was observed in 4 of the 5 patients, with the positive values corresponding to the time points of IFN-γ positivity. Figure 5 demonstrates representative dot-plots. Figure 6 shows the trends in each intracellular cytokine of the 5 patients who generated CD8+IFN-γ+ cells.
Figure 5. Representative polyfunctional intracellular cytokine analysis following mouse gp100 DNA vaccination.
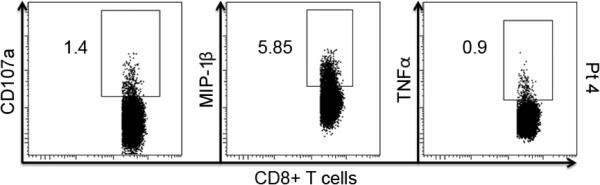
Intracellular cytokine staining for MIP-1β, TNF-α and CD107a was also performed at baseline, week 18 and week 30 for those patients who scored positive for producing IFN-γ in CD8+ T cells. This figure shows representative dot-plots of a patient who was positive for all measured intracellular cytokines after vaccination.
Figure 6. Polyfunctional intracellular cytokine changes following mouse gp100 DNA vaccination.
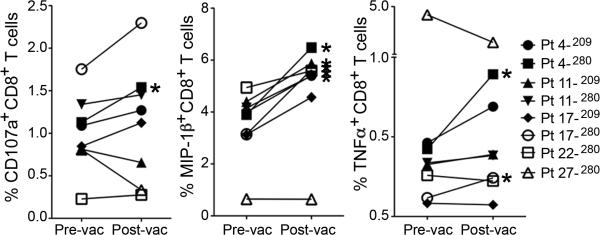
This figure shows the mean values of CD107a+, MIP-1β+, and TNF-α+ cells within the CD8+ T cell population before and after vaccination for all time points when the patient produced significant IFN-γ+CD8+ T cells. Positive time points are marked by asterisks.
Correlation between immune responses, vaccination method and clinical outcome
All demographic characteristics as well as treatment arm were analyzed for association with immune response and clinical outcome. There were no associations between any demographics and tetramer response or clinical outcome. However, 2 borderline associations became evident when assessing the ICS data which failed to reach statistical significance. First, all of the patients producing IFN-γ+CD8+ cells had no previous treatment (p=0.06). In addition, this response may have been associated with treatment arm, as 80% of these responders were in the PMED group (p=0.07). Nevertheless, neither demographic information nor study arm was associated with overall immune response or clinical outcome, nor was the presence of an immune response associated with clinical outcome. An overall summary of specific immune response and clinical outcomes by study arm can be seen in Table 2.
Table 2.
Immune monitoring summary and clinical outcomes of patients who produced a measureable immune response.
| Arm | Patient No. | Increase in gp100 Tetramer-reactive Cells | Increase in CD8+ IFN-γ+ Cells | Clinical Status1 |
|---|---|---|---|---|
| Intramuscular | 17 | + | POD | |
| 25 | gp100280-288 + | POD | ||
| Particle-Mediated Epidermal Delivery | 4 | + | POD | |
| 5 | gp100280-288 + | NED | ||
| 9 | gp100209-217 +; gp100280-288 + | POD | ||
| 11 | + | POD | ||
| 22 | + | NED | ||
| 27 | gp100280-288 + | + | NED |
Abbreviations: NED, no evidence of disease; POD, progression of disease.
Discussion
This randomized clinical trial was designed to validate and expand on the understanding of methods for DNA vaccination, both in comparing intramuscular (IM) injection to particle-mediated epidermal delivery (PMED) and in assessing the safety and feasibility of using PMED as an administration technique. With these endpoints in mind, we showed the ability of PMED to provide an equal or even enhanced tumor antigen-specific immune response at a significantly lower DNA dose. Furthermore, we showed that using PMED is a safe tool for DNA vaccination, providing minimal toxicity, aside from 1 patient with a presumed unknown gold allergy, and no autoimmune manifestations.
Previous research from our laboratory and others has explored utilizing xenogeneic vaccination to overcome poor immunogenicity of self antigens. Pre-clinical models have shown the effectiveness of inducing tumor immunity using homologous DNA from other species (18), which has been further demonstrated with a number of different antigens (18-21). This hypothesis has been validated by the USDA licensure of the first approved therapeutic cancer vaccine in the US, a human tyrosinase DNA vaccine for treatment of melanoma-afflicted dogs (14, 22). Currently, there are also ongoing DNA vaccination trials in multiple human malignancies including prostate, breast, lymphoma and melanoma.
This is the first clinical comparison of PMED as a vaccination method compared with IM injection as far as we are aware. At first, it was shown that genetic vaccination using PMED generated immune responses (9). We then observed that DNA vaccination using PMED induced tumor-specific T-cell responses (10). There are a number of theories as to why PMED could have a superior potency to IM injection. First, it allows for direct transfection of resident antigen presenting cells (APCs) in the epidermis, including dendritic and Langerhans cells, by the gold particles. Furthermore, the particulate nature of this type of vaccine generates a stronger inflammatory response compared to that which would be produced from liquid IM injection. This increased inflammation may induce dendritic cell migration to local draining lymph nodes and enhance cytokine and chemokine production (23). Finally, by vaccinating directly into the epidermis and dermis, a rich supply of resident APCs are available, optimizing the opportunity for direct APC transfection. Pre-clinical models have explored this comparison, demonstrating an increased transgene expression with PMED, producing protective immunity in a variety of animal models of infection and cancer. (9, 24)
Consistent with our previous gp100 DNA vaccination trial, there was an increased immune response compared to baseline. Four of 27 patients (15%) generated a gp100-specific tetramer-reactive CD8+ T-cell response. This was comparable to results in our tyrosinase367-377 peptide vaccine trial, where 3 of 18 patients (17%) produce a positive CD8+ tetramer+ response (15). Further analysis in this study revealed that these tetramer-positive CD8+ T cells carried an effector memory phenotype. In addition, 5 of 27 patients showed an increased post-vaccination CD8+ IFN-γ+ cell production, 4 of which were not tetramer positive. Therefore, 30% (8/27) of our patients produced at least one marker of gp100 immune response after gp100 DNA vaccination.
As opposed to the prior study, one patient generated gp100209-217 tetramer-reactive cells, compared to none previously. This remained less than the 4 patients who generated gp100280-288 tetramer-reactive cells, and interestingly one of which was positive for both gp100 peptides. This supports the theory that whole antigen vaccination may produce a tiered peptide-specific response with dominance of a specific epitope which, however, does not prevent co-development of alternate epitopes such as gp100209-217. Also remarkable was that 1 patient in the IM injection arm produced a CD8+ IFN-γ+ response, equal to that of the previous trial. However, 4 patients in the PMED arm induced such a response, potentially indicative of a higher level of immunogenicity related to the mechanism of delivery. Within these groups, however, there did not appear to be significant difference in inducing a polyfunctional cytokine response. In addition, the polyfunctionality of the cytokine response was not as robust as that which was seen in our prior DNA vaccine trials, including gp100 and GM-CSF DNA vaccines.(6, 25)
There was also no progression-free survival advantage when comparing both arms of the study, nor when comparing who had a detectable immune response to treatment. It should be noted, however, that this is a pilot trial and was not designed to analyze survival. Nevertheless, it can be presumed that an immunologic response to vaccination is required for an effect on clinical outcome, but it is not enough.
While the immune response following DNA vaccination may not be sufficient for clinical response, it provides a backbone for enhancement with immunomodulatory antibodies. In fact, much research has been developing showing detrimental outcomes of multiple vaccinations as a result of immunosuppression. A recent study using a mouse model demonstrated that this immune suppression is often mediated by regulatory T-cells, which must be eliminated in order for vaccines to retain their efficacy (26). Potential adjuvants which may accomplish this task include the anti-OX40, anti-4-1BB, anti-CD25, anti-PD-1 and anti-CTLA-4 antibodies (27-28). Perhaps the most studied of these therapeutics is the antibody against CTLA-4 (ipilimumab/tremelimumab), a cell surface molecule which normally serves to control immune responses and avoid autoimmunity (29-30). As monotherapy, it has been shown to be an effective treatment in advanced melanoma patients, producing beneficial and lasting clinical responses in phase II (31-32). Anti-CTLA-4 therapy also can produce or augment both antigen-specific CD8+ responses both quantitatively and qualitatively with respect to polyfunctional cytokine production (17). Using immunomodulatory antibody therapy as an adjuvant in conjunction with DNA vaccination may augment immunologic potency.
Moreover, the above vaccine strategies have to be compared at different dosing levels. For instance, this trial only utilized intramuscular injection at a dose of 2000μg, while our prior trial comparing different doses of gp100 DNA vaccine administered intramuscularly found that the lowest dose, 100μg, was potentially more immunogenic than the highest dose, 1500μg/injection, although no definitive dose-response was found. A consensus in the field is that one reason for drop-off in immunity to DNA immunization seen going from mouse studies to human studies could be a dose scaling effect. Therefore, we used the highest practical dose for both IM and PMED administration. For IM, this was based on volume and number of injections and for PMED it was based on the manufacturing methods developed by PowderMed. Determination of an optimal vaccination strategy may prove critical in effective anti-tumor immunization and treatment.
In summary, this pilot trial compared 2 methods of delivery for our gp100 DNA vaccine, demonstrating comparable efficacy in both study arms in terms of immune response and clinical outcome. Both mechanisms shared a good safety profile, aside from a hypersensitivity reaction following PMED injection in a patient with previously undiagnosed gold allergy. Both techniques induced a measurable immune response, via either HLA-A*0201 restricted gp100 tetramer+ CD8+ T cells carrying an effector memory phenotype or by producing IFN-γ+ CD8+ T cells, with variable effects on polyfunctionality within the intracellular cytokine profile. Therefore, with the PMED delivery method's ability to generate immune response (with a possible trend towards enhanced immunogenicity compared to IM injection) combined with its small quantity of DNA required to generate such a response, this study supports additional exploration in the use of this approach in further human studies, including in combination with other adjuvants.
Statement of Translational Relevance.
This paper, “Immunologic Response to Xenogeneic gp100 DNA in Melanoma Patients: Comparison of Particle Mediated Epidermal Delivery with Intramuscular Injection”, presents the results of a pilot clinical trial directly comparing 2 vaccination methods in terms of safety and immunogenicity. From a laboratory and scientific perspective, it provides insight into the human immune response to xenogeneic plasmid DNA, and through comprehensive monitoring of antigen-specific CD8+ T cell response, provides a better understanding of the mechanism of action. Designed as a randomized clinical trial in resected AJCC stage IIB-IV melanoma patients, it has clear translational applications with respect to the safety and efficacy of using DNA vaccination as a means of clinical management of metastatic melanoma. Perhaps most importantly, this is the first cancer vaccine clinical trial to directly compare the standard technique of intramuscular injection with another such as particle-mediated epidermal delivery, providing a foundation for the optimization of vaccination strategies.
Figure 4. Representative analysis of IFN-γ+ CD8+ T cells following mouse gp100 DNA vaccination.
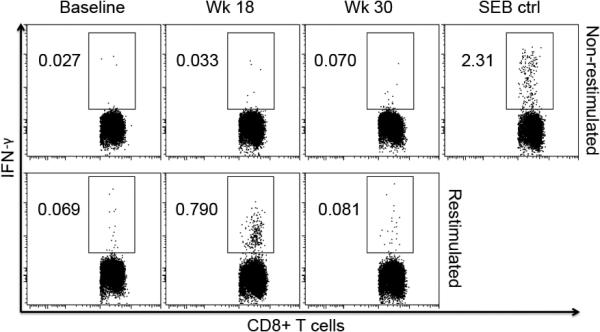
Intracellular cytokine staining was also performed at baseline, week 18 and week 30, and analyzed via multi-parameter flow cytometry, with the same criteria for positivity as for tetramer analysis. This figure shows representative dot-plots of Pt 27, who was positive following vaccination, but returned to baseline by week 30, along with the associated “no restimulation” negative controls and SEB positive control.
Acknowledgements
This work was supported by NIH P01CA33049, Swim Across America, the Experimental Therapeutics Center of MSKCC and the Ludwig Trust. JDW was supported by a Damon Runyon-Lilly Clinical Investigator Award and a Melanoma Research Foundation/Live4Life award . We would also like to acknowledge Peter Loudon and John Beadle from Pfizer for their support.
Abbreviations
- CTL
cytotoxic T lymphocyte
- PBMC
peripheral blood mononuclear cell
- ICS
intracellular cytokine staining
- (TNF)-α
Tumor necrosis factor-α
- PFS
Progression-free survival
- PMED
particle mediated epidermal delivery
- IM
intramuscular
Footnotes
Author contributions: B.A.G., H.F.G., A.N.H, J.Y. and J.D.W. designed research. B.A.G., H.F.G., T.S.R., M.A., Z.M., S.T., B.B.B. R.A. R., P. C., G. S., R.C., K.S.P. S.L.T. and J.Y. performed research. B.A.G. K.S.P. and J.Y. analyzed data; and B.A.G. J.Y and J.D.W. wrote paper.
References
- 1.Treisman J, Garlie N. Systemic therapy for cutaneous melanoma. Clin Plast Surg. 2010;37:127–46. doi: 10.1016/j.cps.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Weber JS, O'Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol. 2008;26:5950–6. doi: 10.1200/JCO.2008.16.1927. [DOI] [PubMed] [Google Scholar]
- 3.Camacho LH, Antonia S, Sosman J, et al. Phase I/II trial of tremelimumab in patients with metastatic melanoma. J Clin Oncol. 2009;27:1075–81. doi: 10.1200/JCO.2008.19.2435. [DOI] [PubMed] [Google Scholar]
- 4.Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the Evaluation of Immune Therapy Activity in Solid Tumors: Immune-Related Response Criteria. Clin Cancer Res. 2009;15:7412–20. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 5.Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15:5591–8. doi: 10.1158/1078-0432.CCR-09-1024. [DOI] [PubMed] [Google Scholar]
- 6.Yuan J, Ku GY, Gallardo HF, et al. Safety and immunogenicity of a human and mouse gp100 DNA vaccine in a phase I trial of patients with melanoma. Cancer Immun. 2009;9:5. [PMC free article] [PubMed] [Google Scholar]
- 7.Cassaday RD, Sondel PM, King DM, et al. A phase I study of immunization using particle-mediated epidermal delivery of genes for gp100 and GM-CSF into uninvolved skin of melanoma patients. Clin Cancer Res. 2007;13:540–9. doi: 10.1158/1078-0432.CCR-06-2039. [DOI] [PubMed] [Google Scholar]
- 8.Dyall R, Bowne WB, Weber LW, et al. Heteroclitic immunization induces tumor immunity. J Exp Med. 1998;188:1553–61. doi: 10.1084/jem.188.9.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang DC, DeVit M, Johnston SA. Genetic immunization is a simple method for eliciting an immune response. Nature. 1992;356:152–4. doi: 10.1038/356152a0. [DOI] [PubMed] [Google Scholar]
- 10.Gnjatic S, Altorki NK, Tang DN, et al. NY-ESO-1 DNA vaccine induces T-cell responses that are suppressed by regulatory T cells. Clin Cancer Res. 2009;15:2130–9. doi: 10.1158/1078-0432.CCR-08-2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trimble C, Lin CT, Hung CF, et al. Comparison of the CD8+ T cell responses and antitumor effects generated by DNA vaccine administered through gene gun, biojector, and syringe. Vaccine. 2003;21:4036–42. doi: 10.1016/s0264-410x(03)00275-5. [DOI] [PubMed] [Google Scholar]
- 12.Best SR, Peng S, Juang CM, et al. Administration of HPV DNA vaccine via electroporation elicits the strongest CD8+ T cell immune responses compared to intramuscular injection and intradermal gene gun delivery. Vaccine. 2009;27:5450–9. doi: 10.1016/j.vaccine.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldberg SM, Bartido SM, Gardner JP, et al. Comparison of two cancer vaccines targeting tyrosinase: plasmid DNA and recombinant alphavirus replicon particles. Clin Cancer Res. 2005;11:8114–21. doi: 10.1158/1078-0432.CCR-05-1410. [DOI] [PubMed] [Google Scholar]
- 14.Bergman PJ, Camps-Palau MA, McKnight JA, et al. Development of a xenogeneic DNA vaccine program for canine malignant melanoma at the Animal Medical Center. Vaccine. 2006;24:4582–5. doi: 10.1016/j.vaccine.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 15.Wolchok JD, Yuan J, Houghton AN, et al. Safety and immunogenicity of tyrosinase DNA vaccines in patients with melanoma. Mol Ther. 2007;15:2044–50. doi: 10.1038/sj.mt.6300290. [DOI] [PubMed] [Google Scholar]
- 16.Yuan J, Gallardo HF, Rasalan T, et al. In vitro expansion of Ag-specific T cells by HLA-A*0201-transfected K562 cells for immune monitoring. Cytotherapy. 2006;8:498–508. doi: 10.1080/14653240600868262. [DOI] [PubMed] [Google Scholar]
- 17.Yuan J, Gnjatic S, Li H, et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc Natl Acad Sci U S A. 2008;105:20410–5. doi: 10.1073/pnas.0810114105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weber LW, Bowne WB, Wolchok JD, et al. Tumor immunity and autoimmunity induced by immunization with homologous DNA. J Clin Invest. 1998;102:1258–64. doi: 10.1172/JCI4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bowne WB, Srinivasan R, Wolchok JD, et al. Coupling and uncoupling of tumor immunity and autoimmunity. J Exp Med. 1999;190:1717–22. doi: 10.1084/jem.190.11.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawkins WG, Gold JS, Blachere NE, et al. Xenogeneic DNA immunization in melanoma models for minimal residual disease. J Surg Res. 2002;102:137–43. doi: 10.1006/jsre.2001.6302. [DOI] [PubMed] [Google Scholar]
- 21.Hawkins WG, Gold JS, Dyall R, et al. Immunization with DNA coding for gp100 results in CD4 T-cell independent antitumor immunity. Surgery. 2000;128:273–80. doi: 10.1067/msy.2000.107421. [DOI] [PubMed] [Google Scholar]
- 22.Liao JC, Gregor P, Wolchok JD, et al. Vaccination with human tyrosinase DNA induces antibody responses in dogs with advanced melanoma. Cancer Immun. 2006;6:8. [PMC free article] [PubMed] [Google Scholar]
- 23.Bowne WB, Wolchok JD, Hawkins WG, et al. Injection of DNA encoding granulocyte-macrophage colony-stimulating factor recruits dendritic cells for immune adjuvant effects. Cytokines Cell Mol Ther. 1999;5:217–25. [PubMed] [Google Scholar]
- 24.Cheng L, Ziegelhoffer PR, Yang NS. In vivo promoter activity and transgene expression in mammalian somatic tissues evaluated by using particle bombardment. Proc Natl Acad Sci U S A. 1993;90:4455–9. doi: 10.1073/pnas.90.10.4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perales MA, Yuan J, Powel S, et al. Phase I/II study of GM-CSF DNA as an adjuvant for a multipeptide cancer vaccine in patients with advanced melanoma. Mol Ther. 2008;16:2022–9. doi: 10.1038/mt.2008.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lacelle MG, Jensen SM, Fox BA. Partial CD4 depletion reduces regulatory T cells induced by multiple vaccinations and restores therapeutic efficacy. Clin Cancer Res. 2009;15:6881–90. doi: 10.1158/1078-0432.CCR-09-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007;7:95–106. doi: 10.1038/nrc2051. [DOI] [PubMed] [Google Scholar]
- 28.Wong RM, Scotland RR, Lau RL, et al. Programmed death-1 blockade enhances expansion and functional capacity of human melanoma antigen-specific CTLs. Int Immunol. 2007;19:1223–34. doi: 10.1093/intimm/dxm091. [DOI] [PubMed] [Google Scholar]
- 29.Brunner MC, Chambers CA, Chan FK, Hanke J, Winoto A, Allison JP. CTLA-4-Mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813–20. [PubMed] [Google Scholar]
- 30.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–65. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ribas A. Overcoming immunologic tolerance to melanoma: targeting CTLA-4 with tremelimumab (CP-675,206). Oncologist. 2008;13(Suppl 4):10–5. doi: 10.1634/theoncologist.13-S4-10. [DOI] [PubMed] [Google Scholar]
- 32.Wolchok JD, Saenger Y. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation. Oncologist. 2008;13(Suppl 4):2–9. doi: 10.1634/theoncologist.13-S4-2. [DOI] [PubMed] [Google Scholar]