

Abstract
Introduction: Dystrophinopathy is a rare, severe muscle disorder, and nonsense mutations are found in 13% of cases. Ataluren was developed to enable ribosomal readthrough of premature stop codons in nonsense mutation (nm) genetic disorders. Methods: Randomized, double-blind, placebo-controlled study; males ≥5 years with nm-dystrophinopathy received study drug orally 3 times daily, ataluren 10, 10, 20 mg/kg (N = 57); ataluren 20, 20, 40 mg/kg (N = 60); or placebo (N = 57) for 48 weeks. The primary endpoint was change in 6-Minute Walk Distance (6MWD) at Week 48. Results: Ataluren was generally well tolerated. The primary endpoint favored ataluren 10, 10, 20 mg/kg versus placebo; the week 48 6MWD Δ = 31.3 meters, post hoc P = 0.056. Secondary endpoints (timed function tests) showed meaningful differences between ataluren 10, 10, 20 mg/kg, and placebo. Conclusions: As the first investigational new drug targeting the underlying cause of nm-dystrophinopathy, ataluren offers promise as a treatment for this orphan genetic disorder with high unmet medical need. Muscle Nerve 50: 477–487, 2014
Keywords: Duchenne muscular dystrophy, genetic, pediatric, nonsense mutation, orphan
Approximately 13% of patients with dystrophinopathy have a nonsense mutation in the gene for dystrophin.1 A nonsense mutation results in a premature stop codon within the protein coding region of the corresponding messenger ribonucleic acid (mRNA) and causes premature termination of translation and generation of a truncated, unstable, nonfunctional protein. There are 3 different types of premature stop codons: opal, amber (uridine-adenosine-guanosine), and ochre (uridine-adenosine-adenosine). Nonsense-mediated mRNA decay (NMD) weakens mRNA, and readthrough of nonsense mutations allows for production of functional protein by altering the level of mRNA deterioration when readthrough activity is established.2 Readthrough of a premature stop codon is a novel approach to treat genetic disorders due to a nonsense mutation. Aminoglycoside antibiotics such as gentamicin have been investigated for their potential ability to promote premature stop codon readthrough in patients with nonsense mutation dystrophinopathy.2–5 The development of a safe, orally bioavailable drug that has readthrough activity would be beneficial to Duchenne muscular dystrophy (DMD) patients with nonsense mutations in their dystrophin gene.4 To treat genetic disorders due to a nonsense mutation, ataluren (PTC124) has been developed as a first-in-class, investigational new drug designed to enable ribosomal readthrough of premature stop codons.
Ataluren's activity has been demonstrated independently in a large number of peer-reviewed articles of multiple disease models spanning many different organ systems, including models of dystrophinopathy.2,3,6–21 A proof-of-concept Phase 2a trial demonstrated that ataluren produced dystrophin in patients with nonsense mutation dystrophinopathy.22 A second Phase 2a study demonstrated that ataluren produced cystic fibrosis transmembrane conductance regulator (cftr) protein in nonsense mutation cystic fibrosis patients.23,24
Based on the results of the proof-of-concept clinical trials, we conducted a Phase 2b registration-directed study. Before this study, very few large-scale, randomized, controlled trials had been performed in dystrophinopathy, and none had been performed using a new chemical entity targeting the underlying cause of DMD.25 This Phase 2b study is a randomized, double-blind, placebo-controlled international study that evaluated the efficacy and safety of 2 doses of ataluren in patients with nonsense mutation dystrophinopathy (referred to in this study as nonsense mutation DMD [nmDMD]). This trial in dystrophinopathy used the 6-Minute Walk Test (6MWT) as an outcome measure. As this was the first study for registration in DMD, there were no established primary or secondary endpoints from a regulatory perspective, and there was limited DMD natural history data available at the time the study was designed. Completion of this trial has provided a better understanding of the natural history of DMD using the 6MWT and has established the 6MWT as a validated primary endpoint in DMD clinical trials; in addition, the data from this trial have helped to identify the best secondary endpoints in DMD trials and lay the clinical trial groundwork for future therapies for this disease.26
MATERIALS AND METHODS
Participants
Patients were enrolled at 37 sites in 11 countries, which featured the following inclusion criteria: male, ≥5 years of age with a documented nonsense mutation in the dystrophin gene, onset of dystrophinopathy symptoms by age 9 years, elevated serum creatine kinase (CK), and difficulty ambulating but able to walk ≥75 meters unassisted during a 6MWT at screening. Stable use of concomitant glucocorticoids was allowed.
At each participating institution, institutional review boards/ethics committees and health authorities approved the study protocol. All parents/participants provided signed informed consent/assent before study initiation. The trial was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice and was registered (Identifier NCT00592553) at www.clinicaltrials.gov.
Procedures
Patients were stratified prospectively by age (<9 or ≥9 years), use of glucocorticoids (yes or no), and baseline 6-Minute Walk Distance (6MWD) (≥350 or <350 meters) and were randomized 1:1:1 to receive study drug orally 3 times daily for 48 weeks (ataluren 10, 10, 20 mg/kg, henceforth referred to as ataluren 40 mg/kg/day; or ataluren 20, 20, 40 mg/kg, referred to as ataluren 80 mg/kg/day; or placebo). Evaluations were performed at screening, baseline and every 6 weeks. The primary outcome measure was the 6MWD.27,28 Secondary outcome measures of physical functioning included timed function tests ([TFTs] - stand from supine, 4-stair ascend, 4-stair descend, and 10 meter run/walk), functional test method grading, at-home activity, myometry (knee flexion and extension, elbow flexion and extension, and shoulder abduction), patient/caregiver-reported accidental falls, and the Pediatric Quality of Life Inventory (PedsQL) physical functioning and psychosocial domains,29 Treatment Satisfaction Questionnaire for Medication (TSQM), verbal memory and attention, heart rate, and serum CK. Safety, study drug compliance, and ataluren plasma concentrations before (C0h) and 2 h (C2h) following the morning dose30 were evaluated.
Biceps muscle dystrophin expression was also assessed (fully detailed in Supplementary Appendix – available online). Biopsy of the biceps brachii was performed at baseline (pretreatment sample) and from the contralateral arm at Week 36 ± 14 days (posttreatment sample) to assess for production of dystrophin. Biopsies were performed at the 37 study sites in 11 countries (United States, United Kingdom, Italy, Australia, Germany, Canada, France, Sweden, Spain, Belgium, and Israel). Samples were shipped to Covance Central Lab and stored at −80°C.
Statistical Analysis
The study hypothesis was that mean change in 6MWD from baseline to 48 weeks would be 30 meters better in at least 1 ataluren arm versus placebo. Thirty meters was selected based on the 6MWD treatment effects seen in trials of drugs which have been approved for the treatment of other rare diseases with neuromuscular complications.31,32 Based primarily on earlier observational 6MWD data in DMD patients,27,28 as well data from other diseases,31,32 the standard deviation of the change in 6MWD was hypothesized in the protocol to be 50 meters, and this standard deviation was assumed for sample size determination. The prespecified intent-to-treat (ITT) population included all randomized subjects with a valid 6MWT available at baseline and ≥1 postbaseline visit. Mixed-model repeated-measures (MMRM) analyses of changes from baseline to Week 48 were performed. Terms in the model included treatment, visit, treatment*visit, and the stratification factors. Original data were to be analyzed, if the data were normally distributed; otherwise, log-transformed data were to be analyzed, if the log-transformed data were normally distributed; otherwise, rank-transformed data were to be analyzed. Shapiro-Wilk testing was used to determine if the data were distributed normally. The MMRM model fit was improved post hoc by the addition of a baseline*visit interaction term.33,34 The baseline values for 2 patients (1 placebo-dosed and 1 treated with ataluren 80 mg/kg) were replaced by their screening values, because their baseline 6MWDs were radically lower than their screening and Week 6 values due to lower-limb injuries before the baseline test. This is referred to as the corrected ITT (cITT) population. The post hoc analysis was performed on the untransformed data with deviations from assumptions addressed by means of a re-randomization test (10,000 iterations) using MMRM. Details of prespecified statistical methods and post hoc modifications are provided in the Supplementary Appendix.
The P-values of the primary and secondary outcome measures were adjusted for comparisons of 2 dose levels against placebo. All analyses were 2-sided at the 0.05 level of significance. Where P-values are described as nominal, they are not adjusted for multiplicity.
RESULTS
Patient Disposition and Characteristics
The ITT population included all 174 randomized patients, of whom 57 were assigned to placebo, 57 to ataluren 40 mg/kg/day, and 60 to ataluren 80 mg/kg/day (Supplementary Appendix, which is available online). One patient discontinued at Week 6 due to noncompliance. The remaining 173 patients completed 48 weeks. Patients ranged in age from 5 to 20 years (Table1). All 3 premature stop codon types were represented. There was no significant difference among the 3 arms in any patient characteristic.
Table 1.
Patient characteristics.
| Characteristic | Treatment arm | ||
|---|---|---|---|
| Placebo N=57 | 40 mg/kg/day N=57 | 80 mg/kg/day N=60 | |
| Demographics | |||
| Age, years | |||
| Mean (SD) | 8.3 (2.33) | 8.8 (2.91) | 8.4 (2.53) |
| Median | 8.0 | 8.0 | 8.0 |
| Range | 5–15 | 5–20 | 5–16 |
| Race, n (%) | |||
| Caucasian | 54 (94.7) | 53 (93.0) | 50 (83.3) |
| Black | 0 (0.0) | 1 (1.8) | 1 (1.7) |
| Asian | 1 (1.8) | 1 (1.8) | 4 (6.7) |
| Hispanic | 1 (1.8) | 1 (1.8) | 2 (3.3) |
| Other | 1 (1.8) | 1 (1.8) | 3 (5.0) |
| Body height, cm | |||
| Mean (SD) | 123 (11.8) | 125 (15.3) | 126 (13.8) |
| Median | 122 | 121 | 126 |
| Range | 104–163 | 99–173 | 99–173 |
| Body weight, kg | |||
| Mean (SD) | 29 (9.1) | 31 (12.1) | 32 (12.8) |
| Median | 26 | 27 | 28 |
| Range | 16–55 | 16–76 | 17–84 |
| Stop codon type, n (%) | |||
| UGA | 31 (54.4) | 29 (50.9) | 23 (38.3) |
| UAG | 12 (21.1) | 17 (29.8) | 19 (31.7) |
| UAA | 14 (24.6) | 11 (19.3) | 18 (30.0) |
| Stratification factors | |||
| Age group, n (%) | |||
| <9 years | 32 (56) | 32 (56) | 34 (57) |
| ≥9 years | 25 (44) | 25 (44) | 26 (43) |
| Glucocorticoid use, n (%) | |||
| Yes | 40 (70) | 41 (72) | 43 (72) |
| No | 17 (30) | 16 (28) | 17 (28) |
| Baseline 6MWD, n (%) | |||
| ≥350 m | 35 (61) | 32 (56) | 33 (55) |
| <350 m | 22 (39) | 25 (44) | 27 (45) |
| Functional characteristics | |||
| 6MWD, m, mean (SD) | 361 (87.5) | 350 (97.6) | 361 (99.7) |
| %-predicted 6MWD, mean (SD) | 61.9 (16.26) | 59.6 (18.06) | 61.6 (17.78) |
| Climb 4 stairs, s, mean (SD) | 6.0 (5.67) | 6.9 (6.47) | 7.5 (7.46) |
| Descend 4 stairs, s, mean (SD) | 5.5 (5.75) | 6.1 (5.98) | 6.7 (7.21) |
| 10-m run/walk, s, mean (SD) | 6.7 (2.67) | 7.4 (4.37) | 7.4 (4.36) |
| Supine to stand, s, mean (SD) | 11.5 (11.44) | 10.8 (9.92) | 12.3 (11.19) |
| Falls/day*, mean (SD) | 0.5 (0.94) | 0.3 (0.48) | 0.4 (0.60) |
Baseline falls/day data were available for 48, 48, and 54 patients in the placebo, ataluren 40 mg/kg/day, and ataluren 80 mg/kg/day treatment arms, respectively.
SD, standard deviation; UAA, uridine-adenosine-adenosine; UAG, uridine-adenosine-guanosine; UGA, uridine-guanosine-adenosine
Median study drug compliance was >97%. Ataluren concentrations before and 2 h after the morning dose were dose-proportional and remained stable over time.
Seventy-one percent (124/174) of patients were receiving glucocorticoids, which was equal across treatment arms, and of these, 92 (74%) received glucocorticoids daily, 7 (6%) received glucocorticoids every other day, and 25 (20%) were on other regimens. Changes in glucocorticoid regimens were minimal during the study. No patients discontinued glucocorticoid use during the study.
Safety
Ataluren was generally well tolerated at both dose levels (Supplementary Appendix). There were no study discontinuations due to adverse events. No ataluren-related serious adverse events were reported. Most treatment-emergent adverse events were mild or moderate. Investigator attributions of drug-related adverse effects showed similar frequencies across the placebo- and ataluren-treated arms. Changes in laboratory and physical parameters generally were not significant clinically.
Efficacy
Primary Endpoint
In the ITT population, mean declines in 6MWD at Week 48 of 42.6 and 12.9 meters were observed for placebo and ataluren 40 mg/kg/day, respectively (Δ = 29.7 meters, nominal P = 0.149, MMRM on ranks). This difference was consistent with the targeted treatment effect size of 30 meters. In the corrected ITT population, mean declines in 6MWD at Week 48 of 44.1 and 12.8 meters were observed for placebo and ataluren 40 mg/kg/day, respectively (see Fig. 1; Δ = 31.3 meters, P = 0.056, re-randomization MMRM adjusted for multiplicity). The difference in the mean change in 6MWD from baseline to Week 48 between placebo and ataluren 80 mg/kg/day was negligible.
Figure 1.
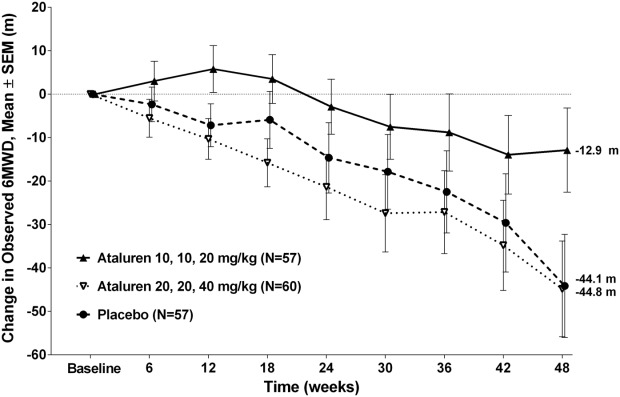
Change in 6MWD. Mean changes in the ataluren 10, 10, 20 mg/kg and placebo arms were −12.86 and −44.14 meters, respectively, resulting in a difference of 31.28 meters.
Progression of 6MWD was defined a priori based on time to persistent 10% 6MWD worsening relative to baseline. In the ITT population, 26% of patients in the ataluren 40 mg/kg/day arm, as compared with 44% of patients in the placebo arm, had experienced persistent 10% 6MWD worsening by Week 48. This corresponds to a 48% reduction in the risk of 10% 6MWD worsening (Figure 2; hazard ratio of ataluren 40 mg/kg/day vs. placebo of 0.52, nominal P = 0.039). Similar results were observed for the corrected ITT population (hazard ratio = 0.51, nominal P = 0.033). The proportion of patients with persistent 10% 6MWD worsening at Week 48 in the ataluren 80 mg/kg/day arm was similar to placebo.
Figure 2.
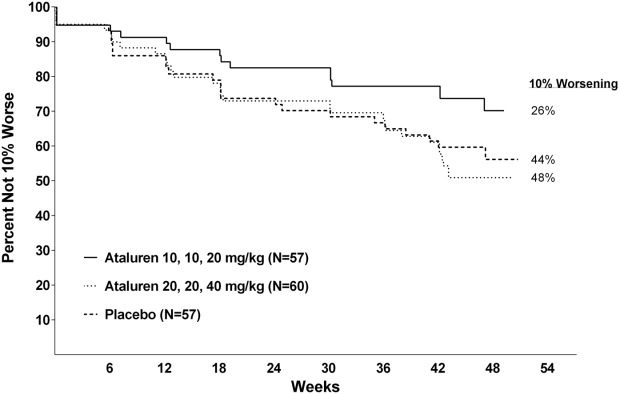
Time to persistent ≥10% worsening in 6MWD.
Recently, an age- and height-based equation35 has been used to convert 6MWD to %-predicted 6MWD in patients with DMD,36 which accounts for maturational differences in 6MWD. This equation was applied to 6MWD data in the corrected ITT population. At baseline, this DMD cohort performed the 6MWT at approximately 60% predicted. Mean declines in %-predicted 6MWD at Week 48 of 7.6% and 2.7% were observed for placebo and ataluren 40 mg/kg/day, respectively (Δ = 4.9%, P = 0.055, re-randomization MMRM adjusted for multiplicity). The mean decline for ataluren 80 mg/kg/day was 7.7%. The significance level of this %-predicted analysis is consistent with the results of the analysis of the 6MWD in meters, in which a P-value of 0.056 was observed.
Age, glucocorticoid use, and baseline 6MWD were prespecified as stratification factors, because these variables were likely to have prognostic significance. Analyses of mean changes in 6MWD within the 6 subgroups defined by the 3 stratification factors showed that all ataluren 40 mg/kg/day subgroups performed better relative to the corresponding placebo subgroup. The largest mean differences between ataluren 40 mg/kg/day and placebo were observed in patients <9 years old, patients receiving glucocorticoids, and patients with baseline 6MWD <350 meters.
The natural history of changes in ambulation as measured by the 6MWT,26 indicates that patients greater than 350 meters at baseline generally do not demonstrate substantial changes in their 6MWDs in 48 weeks while those less than 350 meters tend to show large declines. Based on this, and the fact that a baseline value of 350 meters was a prespecified stratification factor, an analysis was performed on the prespecified group with baseline 6MWD < 350 meters. In this prespecified subgroup with baseline 6MWD < 350 meters in patients treated with ataluren 40 mg/kg/day the mean change of 6MWD from baseline to Week 48 was 68.2 meters better than placebo-dosed patients (nominal P = 0.0053, see Figure 3).
Figure 3.
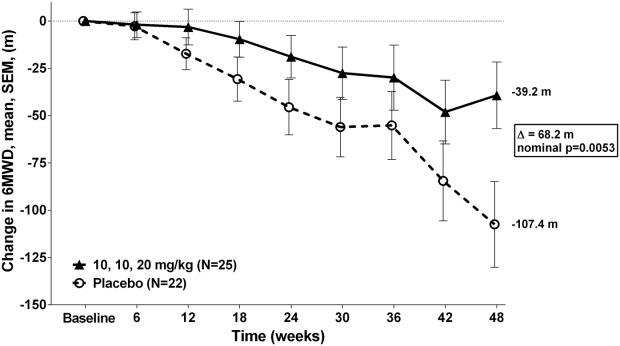
Mean change in 6MWD from baseline to week 48 in the < 350 meters 6MWD subgroup.
Further analysis was performed on patients likely to be in the ambulatory decline phase of the disease subsequent to age 7. This subgroup consisted of nmDMD patients aged 7 to 16 with a baseline %-predicted 6MWD ≤80% and, to minimize heterogeneity, who were taking corticosteroids and had a baseline 6MWD ≥150 meters. In this decline-phase subgroup, in patients treated with ataluren 40 mg/kg/day, the mean change of 6MWD from baseline to Week 48 was 49.9 meters better than placebo-dosed patients (nominal P = 0.0096; see Fig. 4). Separation between ataluren 40 mg/kg/day and placebo in the decline-phase subgroup occurred early (6 weeks), and by 24 weeks there was a 30 meter treatment difference between ataluren 40 mg/kg/day in comparison to the placebo group (nominal P = 0.047).
Figure 4.
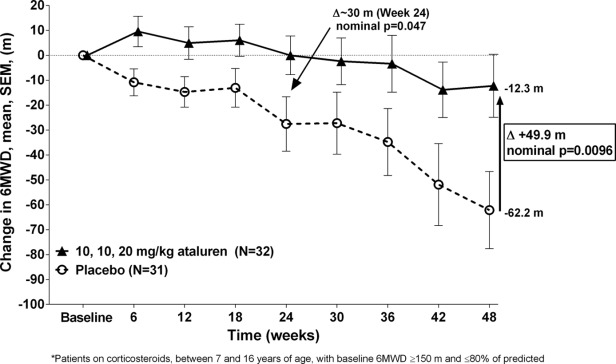
Mean change in 6MWD from baseline to week 48 in the decline-phase subgroup.
The baseline characteristics for the decline-phase subgroup and the < 350 meters subgroup were balanced across the 3 treatment arms (see Table2).
Table 2.
Frequency of patients within levels of stratification factors.
| Parameter | Placebo | Ataluren 40 mg/kg/day | Ataluren 80 mg/kg/day |
|---|---|---|---|
| Decline phase subgroup | |||
| N=31 | N=32 | N=33 | |
| < 9 years | 39% | 41% | 36% |
| ≥ 9 years | 61% | 59% | 64% |
| < 350 meters baseline 6MWD | 42% | 44% | 42% |
| ≥ 350 meters baseline 6MWD | 58% | 56% | 58% |
| Corticosteroid use | 100% | 100% | 100% |
| < 350 meters 6MWD subgroup | |||
| N=22 | N=25 | N=27 | |
| < 9 years | 36% | 44% | 48% |
| ≥ 9 years | 64% | 56% | 52% |
| < 350 meters baseline 6MWD | 100% | 100% | 100% |
| Corticosteroid use | 64% | 68% | 59% |
| No corticosteroid use | 36% | 32% | 41% |
Although the effect of ataluren was most evident in patients in the ambulatory decline-phase, the activity of ataluren was seen across the disease spectrum. As shown in Figure 5, the Phase 2b patients can be categorized based on %-predicted 6MWD at baseline. All patients, including milder patients (>70% predicted), showed a favorable effect for ataluren, and the overall results were not driven by milder patients.
Figure 5.
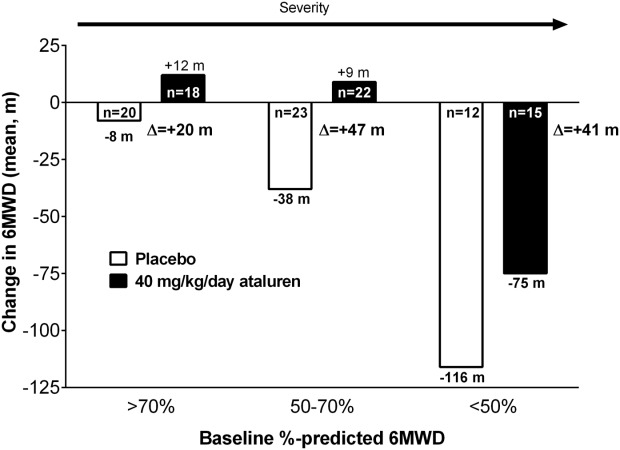
Mean change in 6MWD by disease severity.
Secondary Endpoints
In TFTs, ataluren-treated patients demonstrated smaller increases in the time it takes to climb 4 steps, descend 4 steps, and run/walk 10 meters relative to placebo (Table3). The log of the threshold for the clinically meaningful difference in TFTs was estimated to be 0.4 s,37 which corresponds to ∼1.5 s on the untransformed scale. These trends were more prominent at the 40 mg/kg/day dose, which meets the threshold (∼1.5 s) and suggests clinically meaningful differences in TFTs.37 Positive trends for ataluren 40 mg/kg/day were also seen in functional method grading (Table3); see detailed explanation in Supplementary Appendix. Differences between ataluren versus placebo for mean changes in supine to stand were small for both dose levels, and at baseline 23% of patients were unable to stand from supine within the predefined maximum of 30 seconds (vs. ≤3% patients in other TFTs), limiting the ability to demonstrate a treatment effect with this test.
Table 3.
Timed function test scores (corrected ITT population).
| Mean time and change from baseline to week 48 (Δ) in seconds | ||||||
|---|---|---|---|---|---|---|
| Placebo | 40 mg/kg/day ataluren | 80 mg/kg/day ataluren | ||||
| Endpoint* | Baseline | Week 48 | Baseline | Week 48 | Baseline | Week 48 |
| Climb 4 stairs, s | 6.0 | 10.8 | 6.9 | 9.3 | 7.7 | 11.2 |
| Δ=4.8 | Δ=2.4 | Δ=3.5 | ||||
| Descend 4 stairs | 5.5 | 9.6 | 6.1 | 8.5 | 6.8 | 9.7 |
| Δ=4.0 | Δ=2.4 | Δ=3.0 | ||||
| Run/walk 10 m | 6.8 | 9.8 | 7.4 | 9.1 | 7.8 | 10.2 |
| Δ=3.0 | Δ=1.7 | Δ=2.4 | ||||
| Supine to stand | 11.4 | 14.6 | 10.8 | 14.0 | 12.4 | 15.4 |
| Δ=3.2 | Δ=3.2 | Δ=3.0 | ||||
| Comparison of change from baseline to week 48 between ataluren and placebo, mean (95% CI) | ||
|---|---|---|
| 40 mg/kg/day ataluren vs Placebo | 80 mg/kg/day ataluren vs Placebo | |
| Climb 4 stairs | −2.4 (−4.8, 0.0) | −1.3 (−4.0, 1.4) |
| Descend 4 stairs | −1.6 (−4.2, 1.0) | −1.1 (−3.9, 1.7) |
| Run/walk 10 m | −1.4 (−3.7, 0.9) | −0.7 (−3.0, 1.7) |
| Supine to stand | −0.0 (−2.5, 2.4) | −0.2 (−2.6, 2.2) |
For timed function tests, negative differences between ataluren and placebo represent better outcomes in ataluren-treated patients.
CI, confidence interval; Corrected ITT, corrected intent-to-treat population.
Compared with the results of the overall population, in the prespecified baseline 6MWD < 350 meters subgroup, the results favoring ataluren were even greater in values of 10 meter run/walk (3.5 s), the time to climb 4 steps (6.4 s), and the time to descend 4 steps (5.0 s). Also similar to these results, the ambulatory decline-phase subgroup exhibited greater differences in the TFTs of 4-stair climb, 4-stair descend, and 10-meter run/walk of ataluren over placebo compared with the overall study population. In this subgroup, the results favoring ataluren were nearly twice the 1.5 s clinically meaningful threshold in the 10-meter run/walk (2.8 s), the time to climb 4 steps (2.9 s), and the time to descend 4 steps (2.9 s) (see Fig. 6).
Figure 6.
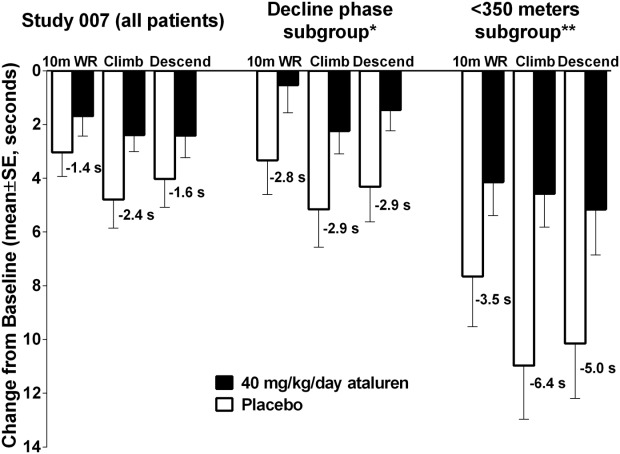
Timed function tests change from baseline to week 48 in Study 007 overall population versus decline-phase subgroup.
Positive trends favoring ataluren 40 mg/kg/day versus placebo were seen in patient-reported physical functioning, as measured by the PedsQL. The difference in the mean change in physical functioning score was 3.4 at Week 48. This was more pronounced in the ambulatory decline-phase subgroup with a difference of 6.1 in the mean change in physical functioning score, favoring ataluren 40 mg/kg/day over placebo at Week 48.
Accidental falling is the most common cause of limb fractures in boys with DMD,38 and ∼35 to 40% of lower-limb fractures result in permanent loss of ambulation.39 This patient-reported outcome was monitored by patient/caregiver diaries. The results show reductions in accidental falling for ataluren versus placebo; the relative ratios of the estimated fall rates at Week 48 were 0.38 (95% CI = 0.16, 0.94) for ataluren 40 mg/kg/day versus placebo.
Additional positive trends favoring ataluren 40 mg/kg/day versus placebo were seen across the other secondary outcome measures of physical functioning, including activity and wheelchair use in the community setting, as well as myometric evaluation of muscle strength, (see Supplementary Appendix). In 5- to 6-year-old patients treated with ataluren 40 mg/kg/day, a 30 meter treatment benefit in 6MWD versus placebo was seen. In addition, 5- to 6-year-old patients treated with ataluren 40 mg/kg/day showed stable or improved TFTs, whereas patients treated with ataluren 80 mg/kg/day or dosed with placebo showed worsening over 48 weeks. These younger patients treated with ataluren 40 mg/kg/day also showed an improvement across all myometry measures compared with patients dosed with placebo (For further 5 to 6 year old data, see Supplementary Appendix). Secondary outcome measures unrelated to physical functioning did not show a difference between ataluren and placebo.
Based on an analysis of patients with pre- and posttreatment muscle biopsy samples, a mean change from pretreatment to posttreatment of 2.8% in dystrophin/spectrin ratio was observed in the ataluren 40 mg/kg/day dose group: 1.3% in the ataluren 80 mg/kg/day dose group and 0.09% in the placebo group. The method used and results are further detailed in the Supplementary Appendix.
Exposure-Response
The inverse dose-response observed with ataluren was evaluated further by means of assessment of exposure-response using ataluren C2h (plasma concentration 2 h postmorning dose), which correlates with ataluren area under the concentration-time curve (Supplementary Appendix). In patients who received ataluren 40 mg/kg/day, mean C2h across all visits ranged between 3.4 and 19.2 µg/ml. In patients who received ataluren 80 mg/kg/day, mean C2h ranged between 6.5 and 42.1 µg/ml. Approximately 40% of 80 mg/kg/day patients had a mean C2h that overlapped with the range observed in 40 mg/kg/day patients (<19.3 µg/ml). This cutoff point was used to analyze changes in 6MWD and timed function tests by dividing the higher-dose arm into low-concentration (<19.3 µg/ml) and high-concentration (≥19.3 µg/ml) groups. This allowed assessment of whether patients who received 80 mg/kg/day, but with exposure in the range of 40 mg/kg/day, performed better than patients with higher exposure. Consistent with the inverse dose-response relationship, less mean decline in 6MWD and better performance of timed function tests was seen in the group of high-dose patients with mean C2h <19.3 µg/ml (Figure 7).
Figure 7.
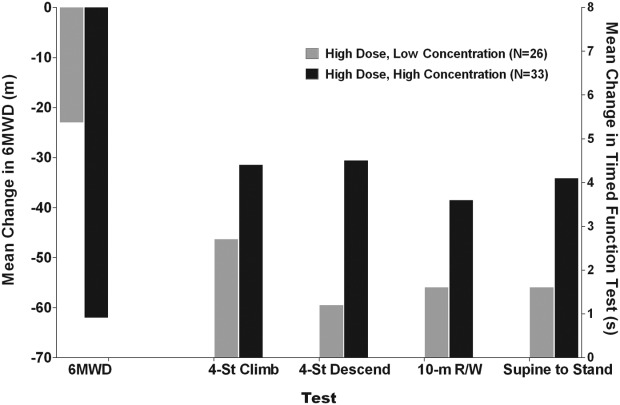
Mean change in 6MWD and timed function tests by concentration.
We also performed an exposure-response analysis by performing a step-wise scan across the entire ataluren plasma concentration range (see Fig. 8). P-values for the concentration cuts within the range of exposure for the 40 mg/kg/day dose are significant statistically, whereas the P-values for exposures outside that range (i.e., the high exposure range of the 80 mg/kg/day dose) are not.
Figure 8.
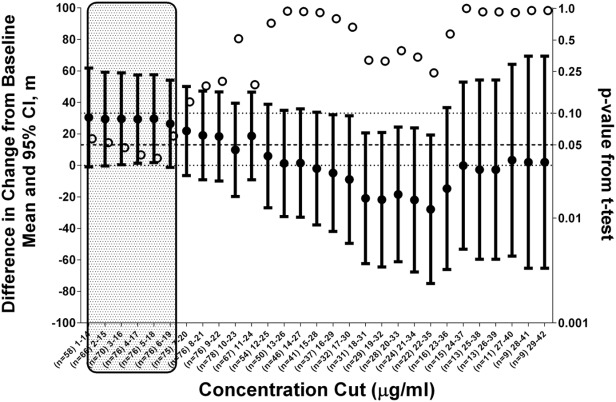
Difference in change from baseline and statistical significance by concentration. Note: Shaded area represents exposure range for 40 mg/kg/day ataluren.
DISCUSSION
Before this study, there was no accepted primary endpoint identified as suitable for evaluating efficacy in clinical trials of patients with dystrophinopathy. The major goal of intervention during the ambulatory phase of dystrophinopathy is to maintain walking ability for as long as possible.40 The 6MWT, a standardized assessment of ambulation,41 had been used to evaluate efficacy in various diseases,31,32 including myotonic dystrophy type 1.42 An observational study showed that the 6MWT is feasible and reliable in dystrophinopathy.27 Subsequent studies have further established the 6MWT as a clinically meaningful outcome measure in dystrophinopathy, and natural history studies show that patients increase in walking ability in the early years, stabilize, then enter a decline phase, which leads, potentially rapidly, to complete loss of ambulation.28,43,44
The results from this Phase 2b study showed that ataluren at a dose of 40 mg/kg/day demonstrated clinical benefit and a favorable clinical benefit/risk profile in ambulatory patients ≥5 years old with dystrophinopathy due to a nonsense mutation. This study was planned in collaboration with regulatory authorities and academic investigators, and the design and results of this trial lay the groundwork for future clinical trials in dystrophinopathy.
The results showed that ataluren 40 mg/kg/day slowed the rate of decline of walking ability and achieved the targeted mean 30-meter difference between ataluren and placebo in 6MWD over 48 weeks. No effect was observed in the 80 mg/kg/day dose. The 30-meter target was based upon 6MWD data from registration-directed studies for other drugs in other diseases with neuromuscular manifestations.31,32,42 Differences in mean change in 6MWD between active and placebo ranged from 28 to 44 meters in these previous studies.
Several lines of evidence support the clinical meaningfulness of a 30-meter difference in 6MWT. These include: (1) 2 distribution-based methods show that a 28.5 to 31.7 meters difference in 6MWD should be considered the minimal clinically important difference (MCID), and it is clinically relevant for nmDMD patients.26 Although not statistically significant, patients treated with ataluren 40 mg/kg/day demonstrated a 31.3-meter difference in change in 6MWD relative to placebo. (2) A 30-meter improvement over placebo in the 6MWT is in the range in which other drugs have been approved in multiple inherited conditions, including mucopolysaccharidosis and Pompe disease. (3) Evidence of the clinical relevance of these results comes from a recent report which showed that a 30-meter change in 6MWD over 48 weeks was considered a clinically meaningful change based on the patient/parent-reported Pediatric Outcomes Data Collection Instruction (PODCI), a quality of life measure, in DMD patients with disease status45 similar to this study. (4) Recent results of longitudinal 6MWT natural history data in DMD demonstrate that each 30-meter decrease in baseline 6MWD predicts increasing risk of loss of ambulation over the following 2 years (Eugenio Mercuri, MD, unpublished data adapted from a longitudinal multicentric cohort study46).
Consistent with ataluren's activity in DMD patients, a time-to-event analysis of the 6MWT showed that ataluren-treated patients were less likely to lose walking ability, defined as a 10% reduction in 6MWD from baseline. This analysis showed that ataluren 40 mg/kg/day substantially slowed disease progression in nmDMD patients. The separation between ataluren and placebo occurred early in the study and continued to the end of the study; by Week 48, 74% of patients who received ataluren 40 mg/kg/day did not experience disease progression versus 56% of patients who received placebo (P = 0.0386; see Fig. 2).
Delaying ambulatory decline provides the direct clinical benefit of affording boys with nmDMD a longer period of self-sufficiency. Importantly, slowing the loss of walking ability may also have beneficial effects that could not be measured within a 48-week timeframe. For example, maintenance of ambulatory capacity has been associated with prevention or delay of onset and reduced severity of scoliosis and the need for major surgery.47,48
The variability of the 6MWT over 48 weeks in this disease was unknown at the time the study was designed. By Week 48, however, it was evident that there was considerable heterogeneity in the rate of disease progression in nmDMD. This contributed to the higher-than-anticipated standard deviation ranging from 72–90 meters (see Supplementary Table1).
Within all subgroups created by the stratification factors, ataluren 40 mg/kg/day treated patients performed better than placebo patients. This was true for subgroups based on age, baseline 6MWD, and glucocorticoid use. Clinically meaningful differences in disease progression due to ataluren treatment were also found in both the decline-phase subgroup, which included patients aged >7, on steroids, and baseline values of 6MWD from 150 meters to 80% predicted (Δ 6MWD = 49.9 meters), and in the prespecified subgroup with baseline 6MWD < 350 meters (Δ 6MWD = 68.2 meters). These treatment effects over a 48-week study duration in a placebo-controlled trial of DMD are substantial, clinically meaningful, and unprecedented in a corticosteroid-treated population of boys with DMD. The minimal decline in 6MWD among placebo-treated patients with baseline 6MWD ≥350 meters (Δ 6MWD = −9 meters) indicates that these patients are in a more stable phase of the disease.
Timed function tests (TFTs) are well-established and sensitive to changes in disease status.43,44,49,50 Over 48 weeks, ataluren-treated patients showed less decline in TFTs than placebo. More recent data have shown that TFTs are important endpoints, and, like the 6MWT, are predictive of the time for a patient to become nonambulatory.26 Natural history data from the Cooperative International Neuromuscular Group (CINRG) and from Study 007 show that a >6-s time to climb 4 stairs is predictive of a greater likelihood of 10% progression in the 6MWD, whereas a >8-s stair climb predicts greater likelihood of loss of ambulation over 1 year.26 The methods used by patients to perform these tests were evaluated by means of functional method grading, which was a novel assessment introduced in this study. It consisted of a 6-point scale that assessed functional ability independently for each timed function test (the complete methodology is detailed in the Supplementary Appendix).
Patients treated with ataluren 40 mg/kg/day trended toward less decline in muscle function compared with placebo as measured by TFTs. The results were not statistically significant, but they met the threshold for clinically meaningful differences, supporting the primary endpoint results. Among the TFTs, the largest effect for ataluren 40 mg/kg/day was seen in stair-climbing, which is one of the most difficult activities of daily living for patients with DMD.51 These data are particularly important, given that Mazzone et al. have reported that the 6MWT and TFTs provide complementary information and should be used in combination in dystrophinopathy clinical trials.43 Positive trends favoring ataluren 40 mg/kg/day versus placebo were also seen for accidental fall frequency, functional method grading, activity and wheelchair use in the community setting, myometry, and patient-reported physical functioning. The consistency of these findings supports an ataluren 40 mg/kg/day treatment effect on physical functioning in ambulatory patients with nmDMD.
The lack of effect on 6MWD at 80 mg/kg/day is consistent with nonclinical data and the exposure-response analysis. A bell-shaped concentration-response curve for production of dystrophin has been observed in cultured myotubes isolated from mdx mice2 and from patients with nmDMD when they were exposed to ataluren.22 Consistent with these findings, recently published results showed that ataluren promoted readthrough of a nonsense mutation in the dystrophin gene of a zebrafish DMD model.52 A bell-shaped dose-response relationship of ataluren activity in promoting dystrophin expression was also observed, consistent with the bell-shaped dose response curve observed in human and mouse myotubes.
In the current study, an analysis of 6MWD and timed function tests by ataluren C2h showed that ataluren 80 mg/kg/day patients with lower concentrations (i.e., those in the range observed with the 40 mg/kg/day dose) experienced better outcomes than those patients with higher concentrations (Fig. 7; see Supplementary Appendix for further details). A bell-shaped exposure-response relationship has also been seen with aminoglycoside antibiotics (e.g., gentamicin) and other compounds evaluated for their ability to promote readthrough of premature stop codons.53–55
The dystrophin expression results were difficult to interpret due to generally poor sample quality as determined by the central laboratory pathologist, including freezing artifact, orientation, and fibrotic replacement (See Supplementary Appendix). Furthermore, a sensitive and reliable method for quantifying dystrophin is not currently available.56 This issue has been recognized in the DMD research community, where an initiative to develop and validate a reliable dystrophin quantification protocol is ongoing.57
Even though the samples were of poor quality, all tissue obtained was immunostained for dystrophin and spectrin (used as the control) in a dual label protocol. A quantitative method for assessing the ratio of dystrophin/spectrin intensity values (similar to that used in the Phase 2a proof of concept study22) was used whereby 4 readings per sample were generated and used to produce mean pre- and posttreatment dystrophin and spectrin intensity values. A small positive trend was observed for ataluren compared with placebo.
Ataluren was generally well tolerated at both dose levels over 48 weeks. Adverse event profiles were similar in ataluren- and placebo-treated patients. No patients discontinued from the study due to an adverse event, and no ataluren-related serious adverse events were reported.
A lesson learned from this trial is that the study was underpowered, given the unexpectedly large standard deviation of the 6MWD scores over 48 weeks. However, the encouraging results of this double blind, placebo-controlled, long-term study suggest that ataluren may have a clinically meaningful effect in patients with nonsense mutation dystrophinopathy. Because dystrophin stabilizes muscle function but does not build strength, a dystrophin restoration therapy for DMD patients would be anticipated to preserve muscle function and delay disease progression. For this reason, in a 48-week trial, the efficacy of ataluren should be expected to be more notable in patients who have marked disease progression. The study results confirm this post hoc, as it was demonstrated that ataluren's effect is most evident in DMD patients with advanced disease, i.e., patients who have begun a phase of decline in their ambulatory ability.
Collectively, these data indicate that ataluren has clinical activity and a favorable safety profile. There is currently a lack of other disease-modifying treatment options for patients with nonsense mutations. As the first investigational new drug to address the underlying cause of dystrophinopathy, ataluren represents an important advance in personalized, genetic-based treatment of nonsense mutation disease.
Acknowledgments
This study was sponsored by PTC Therapeutics, funded in part by a grant from the FDA Office of Orphan Products. We thank the patients and their families for their participation in this study; individuals who were instrumental in the conduct of this study and the collection of data, particularly Principal Investigators (see Supplementary Appendix), supporting investigators, clinical coordinators, the clinical evaluator trainers (Ted Abresch, Kim Coleman, Michelle Eagle, Julaine Florence, Ed Gappmaier, Allan Glanzman, and Erik Henricson), clinical evaluators, and study coordinators; Laura Taylor of Nationwide Children's Hospital for assistance with dystrophin quantification; Zejiang Yang of INC Research for statistical programming support. We also thank the patient advocacy organizations (including Valerie Cwik, MD and the Muscular Dystrophy Association and Giovanna Spinella, MD and the Parent Project Muscular Dystrophy) for the collaboration and support which made this trial possible.
Steven Moore was partially supported by the Iowa Wellstone MDCRC NS053672.
We gratefully acknowledge the support of the Clinical Translational Research Center at the Children's Hospital of Philadelphia (UL1-RR-024134). The study in Newcastle was supported by the staff of the MRC Translational Centre for neuromuscular diseases, especially Mr. Geoffrey Bell, the Muscular Dystrophy Campaign, and the TREAT-NMD network. The Newcastle team acknowledges the support of the National Institute for Health Research through the Newcastle Clinical Research Facility and across the UK by means of the support for the Medicines for Children Research Network. We gratefully acknowledge the support of Stefania Corti, Francesca Magri, Valeria Lucchini at the Milan, Italy study site.
Glossary
- 6MWD
six-minute walk distance
- 6MWT
six-minute walk test
- C0h
plasma concentration before the morning dose
- C2h
plasma concentration 2 hours after the morning dose
- CINRG
Cooperative International Neuromuscular Group
- cITT
corrected intent-to-treat
- CK
creatine kinase
- DMD
Duchenne muscular dystrophy
- ITT
intent-to-treat
- MCID
minimal clinically important difference
- MMRM
mixed-model repeated-measures
- mRNA
messenger ribonucleic acid
- nm
nonsense mutation
- nmDMD
nonsense mutation Duchenne muscular dystrophy
- PedsQL
pediatric quality of life inventory
- PODCI
pediatric outcomes data collection instruction
- TFTs
timed function tests
- TSQM
treatment satisfaction questionnaire for medication.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
REFERENCES
- 1.Dent KM, Dunn DM, von Niederhausern AC, Aoyagi AT, Kerr L, Bromberg MB. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am J Med Genet A. 2005;134:295–298. doi: 10.1002/ajmg.a.30617. [DOI] [PubMed] [Google Scholar]
- 2.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 3.Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci U S A. 2008;105:2064–2069. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D, Shukla C, Liu X, Schoeb TR, Clarke LA, Bedwell DM. Characterization of an MPS I-H knock-in mouse that carries a nonsense mutation analogous to the human IDUA-W402X mutation. Mol Genet Metab. 2010;99:62–71. doi: 10.1016/j.ymgme.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, Al-Dahhak R. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol. 2010;67:771–780. doi: 10.1002/ana.22024. [DOI] [PubMed] [Google Scholar]
- 6.Goldmann T, Overlack N, Wolfrum U, Nagel-Wolfrum K. PTC124-mediated translational readthrough of a nonsense mutation causing Usher syndrome type 1C. Hum Gene Ther. 2011;22:537–547. doi: 10.1089/hum.2010.067. [DOI] [PubMed] [Google Scholar]
- 7.Sarkar C, Zhang Z, Mukherjee AB. Stop codon read-through with PTC124 induces palmitoyl-protein thioesterase-1 activity, reduces thioester load and suppresses apoptosis in cultured cells from INCL patients. Mol Genet Metab. 2011;104:338–345. doi: 10.1016/j.ymgme.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan L, Narayan SB, Chen J, Meyers GD, Bennett MJ. PTC124 improves readthrough and increases enzymatic activity of the CPT1A R160X nonsense mutation. J Inherit Metab Dis. 2011;34:443–447. doi: 10.1007/s10545-010-9265-5. [DOI] [PubMed] [Google Scholar]
- 9.Buck NE, Wood LR, Hamilton NJ, Bennett MJ, Peters HL. Treatment of a methylmalonyl-CoA mutase stopcodon mutation. Biochem Biophys Res Commun. 2012;427:753–757. doi: 10.1016/j.bbrc.2012.09.133. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Hilarion S, Beghyn T, Jia J, Debreuck N, Berte G, Mamchaoui K. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J Rare Dis. 2012;7:58. doi: 10.1186/1750-1172-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kayali R, Ku JM, Khitrov G, Jung ME, Prikhodko O, Bertoni C. Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum Molec Genet. 2012;21:4007–4020. doi: 10.1093/hmg/dds223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanchez-Alcudia R, Perez B, Ugarte M, Desviat LR. Feasibility of nonsense mutation readthrough as a novel therapeutical approach in propionic acidemia. Hum Mutat. 2012;33:973–980. doi: 10.1002/humu.22047. [DOI] [PubMed] [Google Scholar]
- 13.Goldmann T, Overlack N, Möller F, Belakhov V, van Wyk M, Baasov T. A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol Med. 2012;11:1186–1199. doi: 10.1002/emmm.201201438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartolomeo R, Polishchuk EV, Volpi N, Polishchuk RS, Auricchio A. Pharmacological read-through of nonsense ARSB mutations as a potential therapeutic approach for mucopolysaccharidosis VI. J Inherit Metab Dis. 2013;36:363–371. doi: 10.1007/s10545-012-9521-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drake KM, Dunmore BJ, McNelly LN, Morrell NW, Aldred MA. Correction of nonsense BMPR2 and SMAD9 mutations by Ataluren in Pulmonary Arterial Hypertension. Am J Respir Cell Mol Biol. 2013;49:403–409. doi: 10.1165/rcmb.2013-0100OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du L, Jung ME, Damoiseaux R, Completo G, Fike F, Ku JM. A new series of novel small molecular weight compounds induce readthrough of all three types of nonsense mutations in the ATM gene. Mol Ther. 2013;21:1653–1660. doi: 10.1038/mt.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Y, Jiang Q, Takahagi S, Shao C, Uitto J. Premature termination codon read-through in the ABCC6 gene: potential treatment for Pseudoxanthoma elasticum. J Invest Dermatol. 2013;133:2672–2677. doi: 10.1038/jid.2013.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller JN, Chan CH, Pearce DA. The role of nonsense-mediated decay in neuronal ceroid lipofuscinosis. Hum Mol Genet. 2013;22:2723–2734. doi: 10.1093/hmg/ddt120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregory-Evans CY, Wang X, Wasan KM, Zhao J, Metcalfe AL, Gregory-Evans K. Postnatal manipulation of Pax6 dosage reverses congenital tissue malformation defects. J Clin Invest. 2014;124:111–116. doi: 10.1172/JCI70462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu H, Liu X, Huang J, Zhang Y, Hu R, Pu J. Comparison of read-through effects of aminoglycosides and PTC124 on rescuing nonsense mutations of HERG gene associated with long QT syndrome. Int J Mol Med. 2014;33:729–735. doi: 10.3892/ijmm.2013.1601. [DOI] [PubMed] [Google Scholar]
- 21.Lojewski X, Staropoli JF, Biswas-Legrand S, Simas AM, Haliw L, Selig MK. Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum Mol Genet. 2014;23:2005–2022. doi: 10.1093/hmg/ddt596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finkel R, Flanigan K, Wong B, Bönnemann C, Sampson J, Sweeney HL. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation muscular dystrophy. Plos One. 2013;8:e81302. doi: 10.1371/journal.pone.0081302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372:719–727. doi: 10.1016/S0140-6736(08)61168-X. [DOI] [PubMed] [Google Scholar]
- 24.Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med. 2010;182:1262–1272. doi: 10.1164/rccm.201001-0137OC. [DOI] [PubMed] [Google Scholar]
- 25.Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 26.McDonald CM, Henricson EK, Abresch RT, Florence JM, Eagle M, Gappmaier E. The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve. 2013;48:343–356. doi: 10.1002/mus.23902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDonald CM, Henricson EK, Han JJ, Abresch RT, Nicorici A, Elfring GL. The 6-minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve. 2010;41:500–510. doi: 10.1002/mus.21544. [DOI] [PubMed] [Google Scholar]
- 28.McDonald CM, Henricson EK, Han JJ, Abresch RT, Nicorici A, Atkinson L. The 6-minute walk test in Duchenne/Becker muscular dystrophy: longitudinal observations. Muscle Nerve. 2010;42:966–974. doi: 10.1002/mus.21808. [DOI] [PubMed] [Google Scholar]
- 29.Varni JW. The PedsQL™ measurement model for the pediatric quality of life inventory. 2007. http://www.pedsql.org/about_pedsql.html. [DOI] [PubMed]
- 30.Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S. Safety, tolerability, and pharmacokinetics of PTC124, a non-aminoglycoside, nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. Clin Pharmacol. 2007;47:430–444. doi: 10.1177/0091270006297140. [DOI] [PubMed] [Google Scholar]
- 31.Wraith JE, Clarke LA, Beck M, Kolodny EH, Pastores GM, Muenzer J. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase) J Pediatr. 2004;144:581–588. doi: 10.1016/j.jpeds.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 32.Muenzer J, Wraith JE, Beck M, Giugliani R, Harmatz P, Eng CM. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome) Genet Med. 2006;8:465–473. doi: 10.1097/01.gim.0000232477.37660.fb. Erratum in: Genet Med 2006;8:599. [DOI] [PubMed] [Google Scholar]
- 33.Mallinckrodt CH. Recommendations for the primary analysis of continuous endpoints in longitudinal clinical trials. Drug Info J. 2008;42:303–319. [Google Scholar]
- 34.Mallinckrodt CH, Kenward MG. Conceptual considerations regarding endpoints, hypotheses, and analyses for incomplete longitudinal clinical trial data. Drug Info J. 2009;43:449–458. [Google Scholar]
- 35.Geiger R, Strasak A, Treml B, K Gasser, A Kleinsasser, V Fischer. Six-minute walk test in children and adolescents. J Pediatr. 2007;150:395–399. doi: 10.1016/j.jpeds.2006.12.052. 399.e1–2. [DOI] [PubMed] [Google Scholar]
- 36.Henricson E, Abresch R, Han JJ, A Nicorici, E GoudeKeller, G Elfring. Percent-predicted 6-minute walk distance in Duchenne muscular dystrophy to account for maturational influences. Version 2. PLoS Curr. 2012;4:RRN1297. doi: 10.1371/currents.RRN1297. [revised 2012 Feb 2]; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Escolar DM, Hache LP, Clemens PR, Cnaan A, McDonald CM, Viswanathan V. Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy. Neurology. 2011;77:444–452. doi: 10.1212/WNL.0b013e318227b164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonald CM. Physical activity, health impairments, and disability in neuromuscular disease. Am J Phys Med Rehabil. 2002;81((Suppl)):S108–S120. doi: 10.1097/00002060-200211001-00012. [DOI] [PubMed] [Google Scholar]
- 39.Vestergaard P, Glerup H, Steffensen BF, Rejnmark L, Rahbek J, Moseklide L. Fracture risk in patients with muscular dystrophy and spinal muscular atrophy. J Rehabil Med. 2001;33:150–155. [PubMed] [Google Scholar]
- 40.Worton RG, Molnar MJ, Brais B, Karpati G. The muscular dystrophies. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. 8. Vol. 4. New York: McGraw-Hill; 2001. pp. 5493–5523. [Google Scholar]
- 41.ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166:111–117. doi: 10.1164/ajrccm.166.1.at1102. [DOI] [PubMed] [Google Scholar]
- 42.Kierkegaard M, Tollback A. Reliability and feasibility of the six minute walk test in subjects with myotonic dystrophy. Neuromuscul Disord. 2007;17:943–949. doi: 10.1016/j.nmd.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 43.Mazzone E, Martinelli D, Berardinelli A, Messina S, D'Amico A, Vasco G. North Star Ambulatory Assessment, 6-minute walk test and timed items in ambulant boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2010;20:712–716. doi: 10.1016/j.nmd.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 44.Mazzone E, Vasco G, Sormani MP, Torrente Y, Berardinelli A, Messina S. Functional changes in Duchenne muscular dystrophy: a 12-month longitudinal cohort study. Neurology. 2011;77:250–256. doi: 10.1212/WNL.0b013e318225ab2e. [DOI] [PubMed] [Google Scholar]
- 45.Henricson E, Abresch R, Han JJ, Nicorici A, Goude Keller E, de Bie E. The 6-minute walk test and person-reported outcomes in boys with Duchenne muscular dystrophy and typically developing controls: longitudinal comparisons and clinically-meaningful changes over one year. Plos Curr. 2013;5 doi: 10.1371/currents.md.9e17658b007eb79fcd6f723089f79e06. pii: ecurrents md.9e17658b007eb79fcd6f723089f79e06. doi: 10.1371/currents.md.9e17658b007eb79fcd6f723089f79e06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mazzone ES, Pane M, Sormani MP, Scalise R, Berardinelli A, Messina S. 24 month longitudinal data in ambulant boys with Duchenne muscular dystrophy. PLoS One. 2013;8:e52512. doi: 10.1371/journal.pone.0052512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yilmaz O, Karaduman A, Topaloglu H. Prednisolone therapy in Duchenne muscular dystrophy prolongs ambulation and prevents scoliosis. Eur J Neurol. 2004;11:541–544. doi: 10.1111/j.1468-1331.2004.00866.x. [DOI] [PubMed] [Google Scholar]
- 48.Kinali M, Main M, Eliahoo J, Messina S, Knight RK, Lehovsky J. Predictive factors for the development of scoliosis in Duchenne muscular dystrophy. Eur J Paediatr Neurol. 2007;11:160–166. doi: 10.1016/j.ejpn.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 49.McDonald CM, Abresch RT, Carter GT, Fowler WM Jr, Johnson ER, Kilmer DD. Profiles of neuromuscular diseases. Duchenne muscular dystrophy. Am J Phys Med Rehabil. 1995;74((Suppl)):S70–S92. doi: 10.1097/00002060-199509001-00003. [DOI] [PubMed] [Google Scholar]
- 50.Beenakker EAC, Maurits NM, Fock JM, Brouwer OF, van der Hoeven JH. Functional ability and muscle force in healthy children and ambulant Duchenne muscular dystrophy patients. Eur J Paediatr Neurol. 2005;9:387–393. doi: 10.1016/j.ejpn.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 51.Uchikawa K, Liu M, Hanayama K, Tsuji T, Fujiwara T, Chino N. Functional status and muscle strength in people with Duchenne muscular dystrophy living in the community. J Rehabil Med. 2004;36:124–129. doi: 10.1080/16501970410023461. [DOI] [PubMed] [Google Scholar]
- 52.Li M, Andersson-Lendahl M, Sejersen T, Arner A. Muscle dysfunction and structural defects of dystrophin-null sapje mutant zebrafish larvae are rescued by ataluren treatment. FASEB J. 2014;28:1593–1599. doi: 10.1096/fj.13-240044. [DOI] [PubMed] [Google Scholar]
- 53.Burke JF, Mogg AE. Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G-418 and paromomycin. Nucleic Acids Res. 1985;13:6265–6272. doi: 10.1093/nar/13.17.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lai CH, Chun HH, Nahas SA, Mitui M, Gamo KM, Du L. Correction of ATM gene function by aminoglycoside-induced read-through of premature termination codons. Proc Natl Acad Sci U S A. 2004;101:15676–15681. doi: 10.1073/pnas.0405155101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bellais S, Le Goff C, Dagoneau N, Munnich A, Cormier-Daire V. In vitro readthrough of termination codons by gentamycin in the Stüve-Wiedemann Syndrome. Eur J Hum Genet. 2010;18:130–132. doi: 10.1038/ejhg.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown KJ, Marathi R, Fiorillo AA, Ciccimaro EF, Sharma S, Rowlands DS. Accurate quantitation of dystrophin protein in human skeletal muscle using mass spectrometry. J Bioanal Biomed. 2012;S7 doi: 10.4172/1948-593X.S7-001. pii: 001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Treat-NMD Neuromuscular Network. Treat-NMD Newsletter. Web site. Treat-NMD. 2010;87:1–3. http://www.treat-nmd.eu/downloads/file/newsletter/archive/2010/treat-nmd_newsletter_no87.pdf. Published 2010. Updated October 2010. Accessed December 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information