

Abstract
Objective:
Self-microemulsifying drug delivery system (SMEDDS) and solid-SMEDDS of telmisartan was aimed at overcoming the problems of poor solubility and bioavailability.
Methodology:
The formulation strategy included selection of oil phase based on saturated solubility studies and surfactant and co-surfactant screening on the basis of their emulsification ability. Ternary phase diagrams were constructed to identify the self-emulsifying region using a dilution method. The prepared formulations of SMEDDS were evaluated for their drug content, loading efficiency, morphology, globule size determination. Solid-SMEDDS were prepared by adsorption technique using microcrystalline cellulose (1% w/w) and were evaluated for micromeritic properties, scanning electron microscopy, differential scanning calorimetry, X-ray diffraction.
Results:
The formulation containing telmisartan (20 mg), castor oil (30% w/w), tween 20 (55% w/w), propylene glycol (15% w/w) was concluded to be optimized. The optimized SMEDDS and solid-SMEDDS exhibited 100% in vitro drug release up to 120 min, which was significantly higher (P < 0.05, t-test) than that of the pure drug. Solid-SMEDDS may be considered as a better solid dosage form as solidified formulations are more ideal than liquid ones in terms of its stability.
Conclusion:
These results suggest the potential use of SMEDDS and solid-SMEDDS to improve the dissolution and hence oral bioavailability of poorly water-soluble drugs like telmisartan through oral route.
Keywords: Bioavailability, phase diagram, solid-self-microemulsifying drug delivery system, telmisartan
INTRODUCTION
Telmisartan an angiotensin II receptor antagonist is used in the management of high blood pressure (hypertension). Telmisartan exerts potent and sustained antagonism of angiotensin types II-mediated presser responses in vivo, and effectively lowers blood pressure in animal models of hypertension as well as in humans.[1] Being a class II of biopharmaceutics classification system its poor solubility aqueous medium (0.078 mg/ml) low absolute bioavailability (42-58%) and stumpy stability are the challenging problems of conventional dosage of telmisartan. Currently, it is available as tablets dosage forms in the market. Thus increasing aqueous solubility and dissolution of telmisartan is of therapeutic meaning and foremost aim.[2,3]
Previous researchers have made attempts to improve the aqueous solubility of telmisartan by preparing solid dispersion[4] and solid lipid nanoparticles.[5] The preparation of solid dispersion is easy, but its limitations include stability of the drug and the difficulty of incorporating into solid dispersion in suitable dosage forms. For solid dispersion, the amount of carriers used is often large, and thus if the dose of the active ingredient is high, the tablets or capsules formed will be large in volume and difficult to swallow. Moreover, the carriers used are usually expensive and the freeze-drying or spray-drying method requires particular facilities and processes, leading to a high production cost. Though a traditional solvent method can be adopted instead, it is difficult to deal with co-precipitates with a high viscosity. One potential problem with solid lipid nanoparticles formulation is that the drug may favor a more thermodynamically stable state, which can result in the compound crystallizing in the polymer matrix. There is a necessity to develop a formulation that would offer rapid dissolution of temisartan and improve its bioavailability and finally therapeutic efficacy. Lipid-based formulation approaches, predominantly the self-microemulsifying drug delivery system (SMEDDS), illustrate their potential as alternative approaches for the delivery of hydrophobic drugs. Dosing of drug substances that exhibit poor water solubility, but sufficient lipophilic properties in a predissolved state are advantageous in view of the fact that the energy input allied with a solid-liquid phase transition is circumvented, thus overcoming the slow dissolution process after oral intake.[6]
Self-microemulsifying drug delivery system formulations are isotropic mixtures of an oil, a surfactant, a co-surfactant (or solubilizer) and a drug. The basic principle of this system is its ability to form fine oil in water (o/w) microemulsions under gentle agitation following dilution by aqueous phases that is, the digestive motility of the stomach and intestine provide the agitation required for self-emulsification in vivo in the lumen of the gut.[7] This spontaneous formation of an emulsion in the gastrointestinal tract presents the drug in a solubilized form, and the small size of the formed droplet provides a large interfacial surface area for drug absorption.[8,9] Apart from solubilization, the presence of lipid in the formulation further helps improve bioavailability by affecting the drug absorption.[10]
Telmisartan with its low daily oral dose (10-80 mg) and high log P (octanol/water) of 6.6 providing strong justification to develop SMEDDS and solid-SMEDDS of telmisartan.[11] SMEDDS can be converted into solid-SMEDDS by using methods such as met granulation, spray-drying, adsorption etc. Furthermore, solid-SMEDDS have better prospects for the development of solid dosage forms such as tablets, capsules, dry emulsion, pellets etc. Solid-SMEDDS also combines the advantages of both SMEDDS and solid dosage form. Further, solid-SMEDDS are more superior in terms of stability when compared to SMEDDS. SMEDDS of telmisartan are also studied by Bhagwat and Dsouza 2012. They optimized SMEDDS formulation for telmisartan using boxbehnken design.[3] However, in this study, SMEDDS and solid-SMEDDS have been prepared by dilution method and adsorption technique respectively and compared for the development of more bioavailable dosage form. The aim of the present investigation was to develop the SMEDDS and solid-SMEDDS of drug telmisartan to enhance its solubility as well as its bioavailability, which can lead to reduction in dose and side-effects of the drug.
MATERIALS AND METHODS
Material
Telmisartan was obtained as a gift sample from Glenmark Pharmaceuticals Pvt. Ltd., Baddi, India. Castor oil was purchased from Himalaya Agro Company, Ludhiana, India. Tween 20 (Polyoxyethylene sorbitan monolaurate, HLB 16.7) was purchased from Gattefosse, Mumbai, India. Other chemicals used were of analytical grade.
Methods
Phosphate buffer saline 7.4
The most common composition of phosphate buffer saline (PBS): Salt concentration (g/L) NaCl 8.0, KCl 0.2, Na2HPO4 1.44, KH2PO4 0.24.
Solubility of telmisartan
The solubility of telmisartan in various oils (castor oil, oleic acid, olive oil, cod liver oil, arachis oil), surfactants (tween 20, tween 80, span 20, span 80) and co-surfactants (propylene glycol, polyethylene glycol [PEG] 200, PEG 400, glycol) was determined by dissolving an excess amount of telmisartan in 500 mg of each of selected oils, surfactants and co-surfactants in stoppered vials. The mixtures were continuously stirred using vortex mixer for 10 min and kept at 37°C ± 0.5°C in water bath shaker for 72 h to attain equilibrium. The equilibrated samples were centrifuged (3000 rpm for 15 min) and supernatant was filtered through 0.45 μm membrane filter and diluted with mobile phase. Drug content was quantified by using ultraviolet-visible (UV-VIS) spectrophotometer (Shimadzu-1700, Japan) at 296 nm.[12]
Screening of components
Screening of surfactant and co-surfactant was done on the basis of percent transmittance. Emulsification ability of surfactants (tween 20, tween 80, span 20, span 80) was assessed by adding each; (300 mg) to selected oil (300 mg). The mixture was gently heated at 40-45°C for 30 s to achieve homogenization. Out of this mixture, 50 mg was weighed and diluted up to 50 ml with double distilled water to yield fine emulsion. The resulting mixture was observed visually for the relative turbidity. The emulsions were allowed to stand for 2 h and transmittance was assessed by UV-VIS spectrophotometer (Shimadzu-1700, Japan) at 638 nm, using double distilled water as blank.
Various co-surfactants (propylene glycol, PEG 200, PEG 400, glycol) were screened for formulation of SMEDDS. Mixtures of co-surfactant (100 mg), selected surfactant (200 mg) and selected oil (300 mg) were prepared and evaluated in same manner as described in the procedure of surfactant screening.[13]
Construction of ternary phase diagrams
Ternary phase diagram was constructed by dilution method.[14] The mixtures of oil, surfactant and co-surfactant were prepared in which concentration of oil varied from 30% w/w to 70% w/w, surfactant from 30% w/w to 70% w/w and co-surfactant varied from 0% w/w to 30% w/w. However, the total concentration of the mixture containing oil, surfactant and co-surfactant was always added to 100%.[15] First mixture consisted of 30% of oil, 70% of surfactant and 0% of co-surfactant. Subsequently, in further mixtures, oil concentration was kept constant, co-surfactant concentration was increased by 5% for each composition and the surfactant concentration was adjusted to obtain a total of 100%.
A total of 50 mg of each of the compositions was then diluted to 50 ml with double distilled water to evaluate the composition for microemulsion formulation by determining the % transparency, globule size and polydispersity index (PDI) of the resulting dispersion by dynamic light scattering with zetasizer. Dispersions having particle size in the range of SMEDDS were considered desirable for the construction of ternary diagram as well as for drug loading. The corresponding compositions of the dispersions were plotted to obtain the area of microemulsion formation for the respective system in which microemulsion with desired globule size was obtained.
Preparation of self-microemulsifying drug delivery system
The amount of oil, surfactant and co-surfactant to be taken was decided on the basis of microemulsification region in the ternary phase diagram. Telmisartan was accurately weighed into screw-capped glass vials and dissolved in oil. The mixture was warmed in a water bath at 37°C. Surfactant and co-surfactant were added to the mixture and stirred for 10 min using a magnetic bar. The formulations were further sonicated at 45°C for 15 min. Thirteen formulations (F1-F13) with different concentrations of oil, surfactant and co-surfactant, each containing telmisartan at a final loading of 20 mg drug were prepared.[16]
Physicochemical characterization of self-microemulsifying drug delivery system
Drug content
Self-microemulsifying drug delivery system containing telmisartan was added in 50 mL volumetric flask containing methanol and mixed well with shaking and was sonicated for 10-15 min. 0.1 mL of this solution was diluted with 25 mL fresh methanol and drug content was determined using UV-spectrophotometer at λmax 296 nm.[17]
Morphological studies
It was done using transmission electron microscopy (TEM): (Hitachi H7500, Japan). SMEDDS formulations were diluted with water (1:10). A drop of diluted SMEDDS was then directly deposited on the holey film grid, stained by 1% aqueous solution of phosphotungestic acid and observed after drying.
Globule size determination
Analysis of globule size and PDI measurement was carried out by dynamic light scattering with Zeta sizer HSA 3000 (Malvern Instruments Ltd, UK). All samples were subjected to sonication prior to globule size and PDI determination (Tenjarla, 1999). Zeta potential was determined using Zetasizer (Malvern Instrument Ltd, UK). The formulations (F1-F13) were subjected to sonication diluted with excess (100 times) double distilled water and then analyzed.[18]
Viscosity determination
20 g of each of formulation of SMEDDS was weighed and transferred to beaker and the viscosity of formulation was determined with the help of Brookfield Viscometer DV-E model, spindle no 6, at 10 rpm for 5 min and the corresponding dial reading on the viscometer was noted.[19]
Cloud point measurement
Self-microemulsifying drug delivery system was diluted with distilled water in the ratio of 1:250, placed in a water bath and its temperature was increased gradually. Cloud point was measured as the temperature at which there was a sudden appearance of cloudiness visually.
Robustness to dilution for self-microemulsifying drug delivery system
Robustness to dilution was studied by diluting SMEDDS to 50, 100 and 1000 times with water, phosphate buffer pH 6.8 and PBS 7.4. The diluted SMEDDS were stored for 12 h and observed for any signs of phase separation or drug precipitation.
Thermodynamic stability studies
It was determined by carrying heating cooling cycle, centrifugation test and freeze thaw cycle.[20]
Heating cooling cycle
Six cycles between refrigerator temperatures 4°C and 45°C with storage at each temperature for not <48 h was studied. If SMEDDS stable at these temperatures was subjected to centrifugation test.
Centrifugation test
Passed SMEDDS were centrifuged at 3500 rpm for 30 min using digital centrifuge (Remi motors Ltd). If SMEDDS did not show any phase separation was taken for freeze-thaw stress test.
Freeze-thaw cycle
Three freeze-thaw cycles between −21°C and +25°C with storage at each temperature for not <48 h was done for SMEDDS.
Conversion of self-microemulsifying drug delivery system into solid-self-microemulsifying drug delivery system
Solid-SMEDDS were prepared by mixing liquid SMEDDS containing telmisartan with microcrystalline cellulose (MCC) in 1:1 proportion. Liquid SMEDDS was added dropwise over MCC and homogenized using glass rod to ensure uniform distribution of formulation in a china dish.
Physiochemical characterization of solid-self-microemulsifying drug delivery system
Micromeritic properties
Prepared solid-SMEDDS was evaluated for micromeritic properties such as angle of repose, bulk and tapped density, compressibility index and Hausner ratio (HR).[21] Globule size, PDI and zeta potential for solid-SMEDDS were determined in the same way as SMEDDS.
Scanning electron microscopy
Scanning electron microscopy (SEM) for telmisartan and prepared solid-SMEDDS was taken using scanning electron microscope (JEOL, Tokyo, Japan) at accelerating voltage at 3-5 kV to study surface topography.[22]
Differential scanning calorimetry
Physical state of telmisartan in solid-SMEDDS was characterized using differential scanning calorimeter. Thermograms of telmisartan and solid-SMEDDS were obtained using differential scanning calorimetry (DSC) (TA Instruments SDT-2960, USA).
X-ray diffraction study
The X-ray diffraction (X-RD) of telmisartan were obtained using X-RD instrument XPERT-PRO with Ni-filtered Cu radiation, at a voltage of 45 kV and current of 40 mA. The scanning speed was 2°/min between 5θ and 50θ.[23]
In vitro release studies
Dissolution study was carried out using USP Type II apparatus (Paddle method) at 50 rpm, and at 37°C ± 0.5°C. The dissolution medium was PBS 7.4 and methanol in the ratio of 9:1. Prepared SMEDDS and solid-SMEDDS with equivalent amount of drug 20 mg were placed in 900 ml of dissolution medium respectively. A sample of 5 ml were withdrawn at regular time interval of 5, 10, 15, 30, 60, and 120 min and filtered using 0.45 μm filter. An equal volume of respective dissolution medium was added to maintain sink conditions. Drug content from sample was analyzed using UV-spectrophotometer at 296 nm.
Release kinetics
To study the release kinetics, data obtained from in vitro dissolution study was fitted in various kinetic models: Zero order as cumulative percent of drug released versus time, first order as log cumulative percentage of drug remaining versus time and Higuchi's model as cumulative percent drug released versus square root of time, Hixon crowel describes the release from systems when there is a change in a surface area and diameter of particles. To determine the mechanism of drug release, the data was fitted into Korsmeyer and Peppas equation as log cumulative percentage of drug released versus log time and the exponent n was calculated from slope of the straight line. For slab matrix, if exponent is 0.5, then diffusion mechanism is Fickian; if 0.5 < n < 1.0, then it is anomalous transport. If n is 1.0, it is case II transport and if n > 1.0, then it is super case II transport.[24]
Selection and comparison of best batch of self-microemulsifying drug delivery system, solid-self-microemulsifying drug delivery system with pure drug
Selection of best batch of SMEDDS and solid-SMEDDS was done on the basis of their globule size, zeta potential, PDI, in vitro drug release studies. Then the optimized formulation of SMEDDS and solid-SMEDDS were compared with pure drug.
Accelerated stability studies of solid-self-microemulsifying drug delivery system
Stability studies for solid-SMEDDS were studied at different temperature conditions according to ICH guidelines at 25°C ± 2°C/60% ± 5% relative humidity (RH) and at 40°C ± 2°C/75% RH ± 5%. The samples were withdrawn at different time intervals as 0, 7, 15, 30, 60, 90 days. Formulation equivalent to 20 mg of the drug was dissolved in methanol, diluted approximately and estimated for the drug content spectrophotometrically at 296 nm using methanol as blank. Effect of storage conditions on drug release was also studied.[25]
Statistical analysis
Graph pad prism 5 (Graph Pad Software Inc., La Jolla, CA, USA) was used for statistical analysis. All studies were done in triplicates unless specified and data represent the mean ± standard deviation. The statistical analysis was performed using Student's t-test. A difference below the probability level was considered as statistically significant (P < 0.05).
RESULT AND DISCUSSION
Screening of components
To develop SMEDDS of telmisartan, it should possess good solubility in the oil, surfactants and co-surfactants of system. The solubility of telmisartan in various oils, surfactants and co-surfactants was investigated. Telmisartan had significantly higher (P < 0.05, t-test) solubility in castor oil (8.17 ± 1.06 mg/ml) than olive oil, cod liver oil, arachis oil, oleic acid. Among surfactants and co-surfactants, tween 20 (107.63 ± 3.81 mg/ml) and propylene glycol (80.72 ± 2.14 mg/ml) respectively showed highest solubilities [Table 1]. Therefore, castor oil was screened as oil phase based on solubility studies.
Table 1.
Solubility of telmisartan in oils, surfactants and co-surfactants
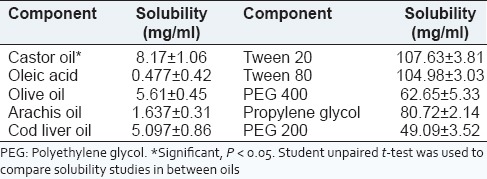
Surfactant and co-surfactant were selected on the basis of percent transmittance.[16] Out of various surfactants and co-surfactants screened, tween 20 revealed 96.34% ± 0.24% transmittance, whereas other surfactants tween 80, span 80 and span 20 showed 80.41 ± 0.66, 50.66 ± 0.69 and 37.09% ± 1.09% transmittance, respectively. As shown by outcomes, tweens are showing significantly higher (P < 0.05, t-test) transmittance values. This can be attributed to the higher hydroplilicity of the tweens as compared to the spans due to the presence of polyoxyethylene chains in the molecule of tweens. Among tweens, tween 20 showed higher transmittance with telmisartan. Hence this surfactant was selected for development of the formulation. This is also compliant with the purpose of SMEDDS which has to form o/w emulsion in situ. In case with co-surfactants, propylene glycol resulted in higher percent transmittance (88.58% ± 0.27%) (P < 0.05, t-test) than PEG 400 (81.21% ± 0.63%), glycerol (80.66% ± 0.58%) and PEG 200 (74.98% ± 0.57%). Therefore, tween 20 and propylene glycol were selected as surfactant and co-surfactant, respectively, for the phase study.
Construction of ternary phase diagram
The aim for constructing ternary phase diagram was to explore the microemulsion region.[26,27,28] Castor oil was used as oil phase. Surfactant co-surfactant mixture was composed of tween 20 as surfactant and propylene glycol as co-surfactant. The phase diagram was constructed in the absence of drug, telmisartan. Initially, 35 formulations were made and diluted with 100 ml of water and on the basis of opaqueness observed visually only seventeen formulations were selected, rest were turbid and rejected. The selected formulations were further carried for zeta sizer and PDI. Formulations F1-F13 without drug was selected for constructing the ternary diagram F14-F17 were rejected as they don’t lied in the size range [Table 2]. Different ratios for these final thirteen formulations were placed in the pro sim ternary diagram software and diagram was plotted. The microemulsion region was demarcated using particle size studies and showed that the formulations lie in this region [Figure 1]. The rest of the region on the phase diagram represents the turbid and conventional emulsions.[19]
Table 2.
Data for the construction of ternary phase diagram without drug
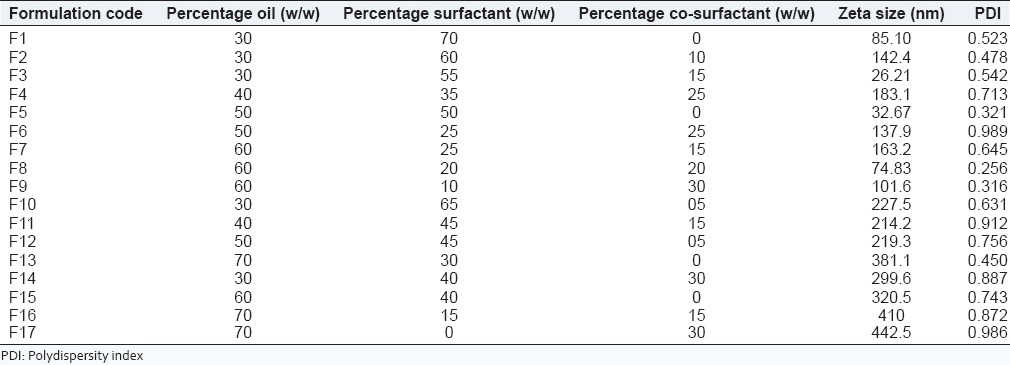
Figure 1.
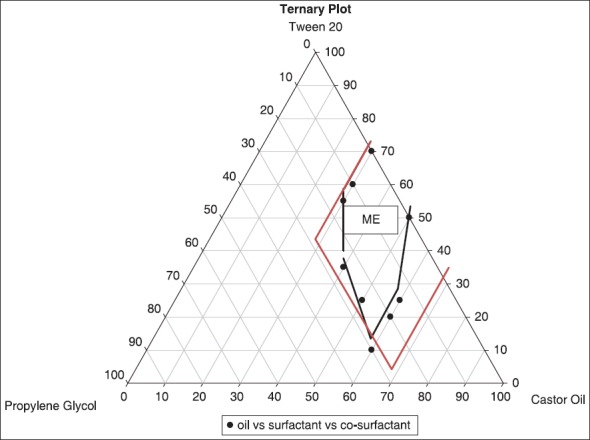
Ternary phase diagram
Formulation of self-microemulsifying drug delivery system
Thirteen formulations (F1-F13) with different concentrations of oil, surfactant and co-surfactant, each containing telmisartan at a final loading of 20 mg of drug were prepared by ultrasonication method and were evaluated.[16]
Physicochemical characterization of self-microemulsifying drug delivery system
Drug content of self-microemulsifying drug delivery system
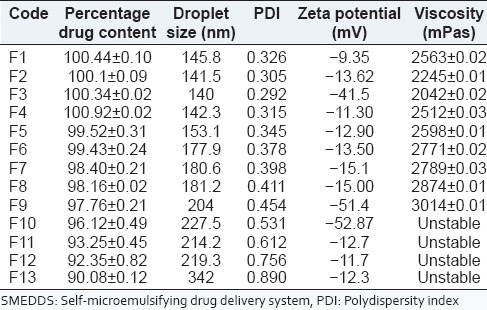
Irrespective of ratios of oil and surfactant used, the drug content in the thirteen formulations (F1-F13) was found in the range of 90.38-100.34% [Table 3], indicating uniform dispersion of drug in formulations.
Table 3.
Physicochemical characterization of SMEDDS
Morphology of self-microemulsifying drug delivery system
Transmission electron microscopy of SMEDDS revealed that spherical microemulsion of uniform size were formed with no signs of coalescence even after 24 h of post dilution [Figure 2]. Furthermore, no signs of drug precipitation were observed inferring the stability of formed microemulsions. Closer analysis of TEM images revealed that F3 globules are surrounded by a thick layer. We can hypothesize that the formed thick layer provides a mechanical barrier to prevent the coalescence of formed microemulsion and precipitation of drug.[29] Results revealed the slight aggregation of globules, which may be due to more concentration of oil (60% w/w).
Figure 2.

Transmission electron microscopy images (a) F9 (b) F3 formulation showing globule size
Globule size determination
Globule size ranged from 140 nm to 342 nm [Table 3]. The lowest globule size was observed of F1-F4 formulation due to decreased concentration of co-surfactant. The particle size of F3 formulation was lesser in size in comparison to F1, F2 and F4. FI and F2 formulations cannot be considered among best formulations because of high concentration of surfactant which can cause irritational properties in mucosa. Further in F4, ratio of co-surfactant is high in comparison to F3 due to which its droplet size is slightly increased therefore F3 [Figure 3] was considered to be best among all with optimum ratio of surfactant (tween 20) and co-surfactant (propylene glycol).
Figure 3.
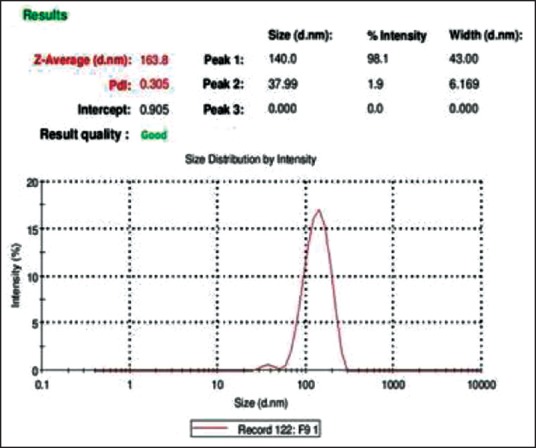
Zeta sizer report of F3 self-microemulsifying drug delivery system
The type of surfactant did not considerably affect the droplet size, while the co-surfactant (propylene glycol) containing microemulsions produced largest droplets as well as highest viscosity.[30] An increase in the ratio of the oil phase (castor oil) also resulted in a proportional increase in particle size. It is well-known that the addition of surfactants to these systems causes the interfacial film to stabilize and condense, while the addition of co-surfactant causes the film to expand; thus, the relative proportion of surfactant to co-surfactant has varied effects on the globule size.[25] Furthermore, it has been reported that the smaller particle size of the emulsion globules may lead to more rapid absorption and improve the bioavailability.[31]
The PDI obtained for all the formulations varied from 0.292 to 0.890. PDI below 0.3 indicates good uniformity in the globule size distribution after dilution with water.[32,33] PDI of F3 (0.292) was found to be lowest than other formulations.
A dividing line between stable and unstable aqueous dispersions is generally taken at either ±30 mV. Particles with zeta potentials more negative than −30 mV are normally considered stable.[32] Zeta potential was found in the range between −9.35 mV and −52.87 mV. Formulations F3 may be considered stable as it was having more negative zeta potential then −30 mV [Figure 4].
Figure 4.
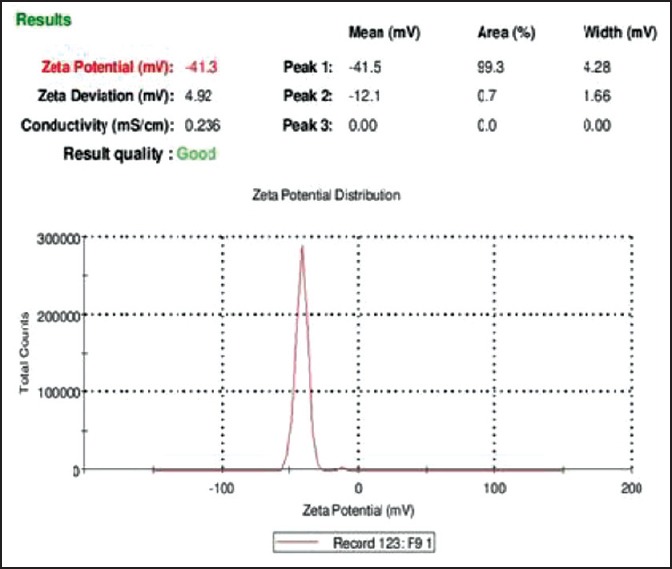
Zeta potential report of F3 self-microemulsifying drug delivery system
Cloud point measurement of self-microemulsifying drug delivery system
Cloud point of prepared SMEDDS formulations F1-F9 was found to be higher than 85°C, which indicates that micro emulsion will be stable at physiological temperature without risk of phase separation. However, F10-F13 formulations showed phase separation after 50°C. It may be due to precipitation of drug. The results [Table 3] showed that F11-F12 had the highest drug encapsulation efficiency after F5, but they cannot be considered among the good formulations due to its phase separation at higher temperature.
Robustness to dilution
After diluting SMEDDS to 50, 100 and 1000 times with water and buffer pH 7.4 and storing for 12 h, it was observed that there was no sign of phase separation or drug precipitation in F1-F9 formulations, but F10-F13 formulations turned hazy after standing for long hours. Hence, formulations F10-F13 were rejected as they were also showing phase separation and became hazy on dilution.
Viscosity determination of self-microemulsifying drug delivery system
The range of the viscosity was found to be 2042-3014 mPas [Table 3]. As F10-F13 were rejected due to precipitation in the formulations viscosity of formulations F1-F9 were only determined. F9 formulation containing 60% oil and 30% co-surfactant was highly viscous due to higher co-surfactant concentration. From viscosity determination, it was observed that as the concentration of co-surfactant (propylene glycol) increased viscosity of the formulation also gets increased.[30]
The concentration of surfactant also increases the viscosity of the formulation.[19] It was expected that FI, F2, and F5 would show the least viscosity due to very low concentration of co-surfactant, but it was not practically obtained as in these formulations surfactant concentration was maximum (70, 60, 50% surfactant respectively). Therefore, F3 (30% oil, 55% surfactant and 15% co-surfactant) due to optimum concentration of surfactant and co-surfactant showed the least viscosity. The sequence of viscosity of prepared SMEDDS batches was shown as: F3 < F4 < F2 < F1 < F5 < F6 < F7 < F8 < F9.
Thermodynamic stability studies
F1-F9 formulations passed all the thermodynamic tests. Thus F1-F9 formulations were used for further studies.[34] Thermodynamic stability study was designed to identify and avoid the unstable systems. SMEDDS are thermodynamically stable systems and are formed at a particular concentration of oil, surfactant and co-surfactant, with no phase separation, creaming or cracking. It is the thermostability, which differentiates nano-or microemulsion from emulsions that have kinetic stability and will eventually phase separate.[35]
Conversion of self-microemulsifying drug delivery system into solid-self-microemulsifying drug delivery system
All SMEDDS were converted to solid-SMEDDS (SF1-SF9) using MCC as an adsorbent in 1:1 w/w ratio with SMEDDS.
Physicochemical characterization of solid-self-microemulsifying drug delivery system
Micromeritic properties
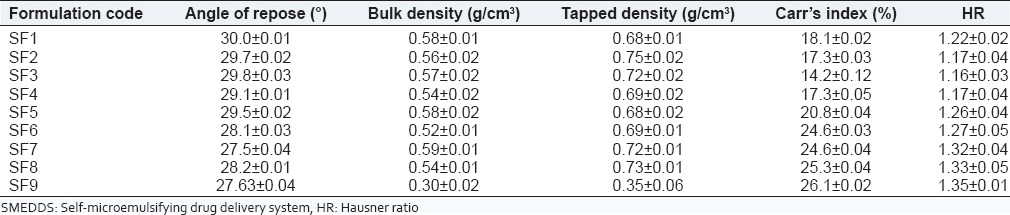
The formulations (SF1-SF9) indicated angle of repose <30 which showed that they had excellent flow properties [Table 4]. Bulk density and tapped density was evaluated to study Carr's index and HR. High concentration of surfactant can also cause loss of flow ability in solid-SMEDDS. Carr's index of SF3 formulation (14.2% ± 0.12%) revealed good flow, that is, 11-15 and formulations SF1, SF2, SF4 had fair flow properties (16-20). Formulations SF5-SF7 bared passable Carr's index (21-25) and SF8, SF9 had poor flow properties (26-31). SF1-SF4 had good flow properties in accordance to HR (1.12-1.18). SF5-SF8 had passable (1.26-1.34) and SF9 formulation was the poorest in flow properties (1.35-1.45).
Table 4.
Micromeritic properties of solid-SMEDDS
Zeta sizer results indicated the size range varied from 163.8 nm to 500 nm [Table 5]. It was observed from the results that globule size range of solid-SMEDDS was higher when compared to SMEDDS. It might be due to the presence of adsorbent (MCC) in solid-SMEDDS, which may lead to increase in globule size during dispersion of formulation. The lowest globule size was observed of SF2-SF4 formulation. The globule size results were found to be correlated with SMEDDS results.
Table 5.
Physicochemical characterization of solid-SMEDDS

The PDI obtained for all the formulations varied from 0.369 to 0.984. PDI of F3 (0.369) again was the lowest and found best in comparison to all other formulations. Zeta potential was found in the range between −8.72 mV and −55.75 mV, which again indicated better stability of formulations (SF3, SF9) [Figure 5].
Figure 5.
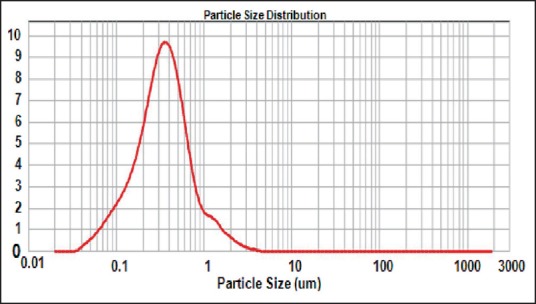
Mean globule size of reconstituted solid-self-microemulsifying drug delivery system
Scanning electron microscope
Solid-SMEDDS appeared as smooth surfaced particles [Figure 6], indicating that the liquid SMEDDS is adsorbed onto the MCC with a lesser amount of aggregation.
Figure 6.

Scanning electron microscopy of solid-self-microemulsifying drug delivery system
Differential scanning calorimetry
The DSC analysis [Figure 7] of pure telmisartan showed a characteristic, sharp endotherm peak at 265.45-268.82°C corresponding to its melting point and indicates the crystalline nature of the drug. The DSC analysis of physical mixture of drug and excipients revealed negligible change in the melting point of telmisartan in the presence excipients, indicating no modification or interaction between the drug and excipients.
Figure 7.
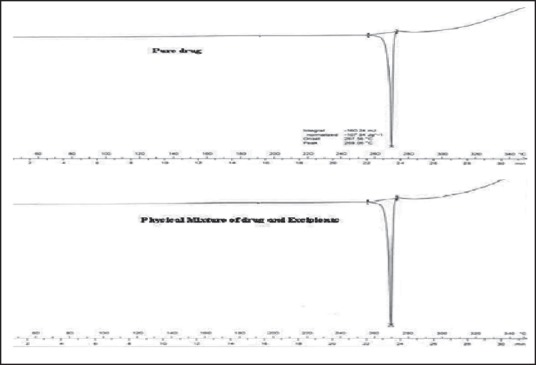
Differential scanning calorimetry (DSC) thermogram of (a) plain TEL, (b) DSC thermogram of pure telmisartan and physical mixture drug and excipients
X-ray diffraction studies
The diffraction pattern of telmisartan revealed several sharp high-intensity peaks at diffraction angles 2θ suggesting that the drug existed as crystalline material. There were few characteristic peaks of telmisartan with a considerable reduction in the peak intensity [Figure 8]. This diminished peak suggests conversion of the drug into an amorphous form. This marked reduction in peak intensities provides may increase dissolution rates of solid-SMEDDS preparation.[36]
Figure 8.
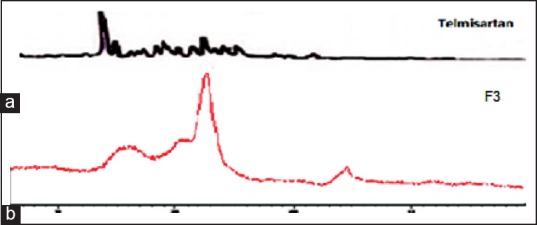
X-ray diffraction of (a) telmisaratan and (b) F3 formulation of solid-self-microemulsifying drug delivery system
In vitro release studies
Self-microemulsifying drug delivery system
The results revealed that formulations F2, F3, and F4 showed 100% drug release within 120 min [Figure 9]. These formulations (F2, F3, and F4) had better release when compared to the other formulations, that is, F1 (93.1% ± 0.05%), F5 (90% ± 0.07%), F6 (89.61% ± 0.11%), F7 (84.93% ± 0.12%), F8 (82.64% ± 0.34%) and F9 (81.32% ± 0.17%) within 120 min.
Figure 9.
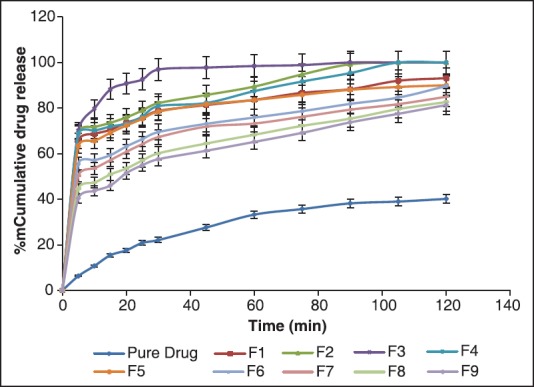
In vitro drug release of pure drug and formulations F1-F9 of self-microemulsifying drug delivery system
The best formulation came out to be was F3 (30% castor oil, 55% tween 20 and 15% co-surfactant), which was showing enhanced release of telmisartan. F2 and F4 were slightly higher in globule size in comparison to F3, thus, their dissolution got slightly reduced; hence, F3 was considered to be the finest formulation among all.
This behavior of drug release was due to their globule size. As we know, globule size is inversely proportional to the surface area that means lesser the globule size more is the surface area and surface area is directly proportional to the dissolution. Thus, least globule size with higher surface area formulation had the highest dissolution. Therefore, F3 was found to release the 100% drug in 120 min from the SMEDDS.
Furthermore, small globule size of resultant microemulsion and solubilized form of drug in lipid and Smix confirms that the solubility of the drug increases several times which may result in higher absorption and improvement in oral bioavailability. Since the drug is present in solubilized form and in the center of lipid core in microemulsion globules, the gastric irritation potential of drug may also get reduced.[37]
Solid-self-microemulsifying drug delivery system
The results of solid-SMEDDS were also in the same pattern as of SMEDDS. The overall dissolution rate was observed to be low in solid-SMEDDS in contrast to SMEDDS because of increased globule size of solid-SMEDDS, and as we know globule size is inversely proportional to the dissolution rate therefore dissolution got reduced. Here still, formulations SF2, SF3 and SF4 showed 100% drug release and there dissolution rate was better among all [Figure 10].
Figure 10.
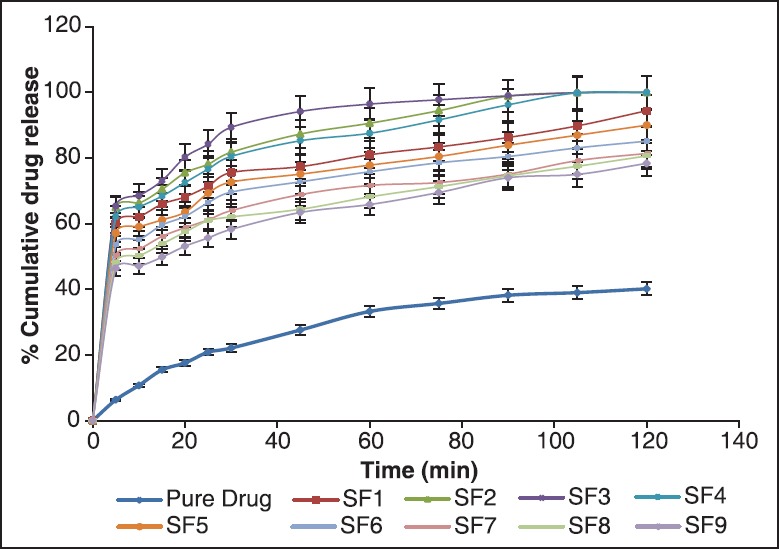
In vitro drug release of pure drug and formulations F1-F9 of solid-self-microemulsifying drug delivery system
This release pattern illustrated here was also due to the globule size. Least the globule size leads to increase in the surface area and more is the dissolution. In consideration with the smallest globule size, SF3 (30% castor oil, 55% tween 20 and 15% co-surfactant; 163.8 nm) formulation showed the ideal and best release out of all nine formulations due to the high concentration of oil and co-surfactant.
Effect of oil on release
The oil ratio in the system has an important role as many physiological parameters depend on on it which eventually affects the dissolution of the drug. Through results, it was revealed that lesser the oil concentration more stable the formulation was as formulations F7-F9 showed least results in all and F13 was rejected even. This was due to the high concentration of oil leads to coalescence or aggregation, which leads to precipitation of the drug. F3 and SF3 (30% castor oil, 55% tween 20 and 15% co-surfactant), respectively showed the best dissolution due to less amount of oil and balanced ration amidst surfactant and co-surfactant.
Effect of co-surfactant on release
Propylene glycol was employed as a co-surfactant in the system which is widely known for increasing solubility along with a surfactant. When the concentration of co-surfactant was increased, the solubility of telmisartan also increased which leads more absorption of the drug in the GI, ultimately enhancing its bioavailability. However, it is also responsible for increasing the size of the globules; hence, the ratio can be increased up to the optimal level only. The results revealed as the propylene glycol in the formulation was increased from 10% w/w to 30% w/w, the dissolution was decreased
Effect of surfactant concentration on the release
In vitro release study in PBS 7.4 shows that the rate of drug release was faster in case of hydrophilic surfactant (tween 20). This is due to the hydrophilic nature of the surfactant. The broadness of the size distribution observed at higher surfactant concentrations could be due to the higher viscosity of the continuous phase, which disperses the stirring energy. Thus, the PDI value increased with increasing surfactant concentrations. Zeta potential increased with increasing concentrations of surfactant. The increase in concentration of surfactant resulted in a slight increase in encapsulation efficiency and loading capacity.
Comparison of pure drug, self-microemulsifying drug delivery system and solid-self-microemulsifying drug delivery system
From the drug release studies, it can be clearly seen that pure drug released only 40.14% ± 0.19% up to 120 min and SMEDDS and solid-SMEDDS formulation had shown 100% drug release. This clearly concludes that SMEEDS and solid-SMEDDS formulations enhanced the release of the drug 6-7 folds, which in turn can increase its bioavailability too.
All SMEDDS formulations were in the size range of 140-342 nm and solid-SMEDDS were in the 163.8-500 nm size range which shows that when SMEDDS were converted into solid-SMEDDS their size increased significantly (P < 0.05 t-test) higher, which slightly reduced the dissolution of solid-SMEDDS and PDI, zeta sizer also varied accordingly.
When the in vitro release study of SMEDDS and solid-SMEDDS were compared with each other [Figure 11] it was found that there was an insignificant difference (P > 0.05, t-test) in release behavior of the formulations. In both SMEDDS and solid-SMEDDS, F3 and SF3 (30% castor oil, 55% tween 20 and 15% co-surfactant) formulation respectively was the best release formulation showing 100% release of drug.
Figure 11.
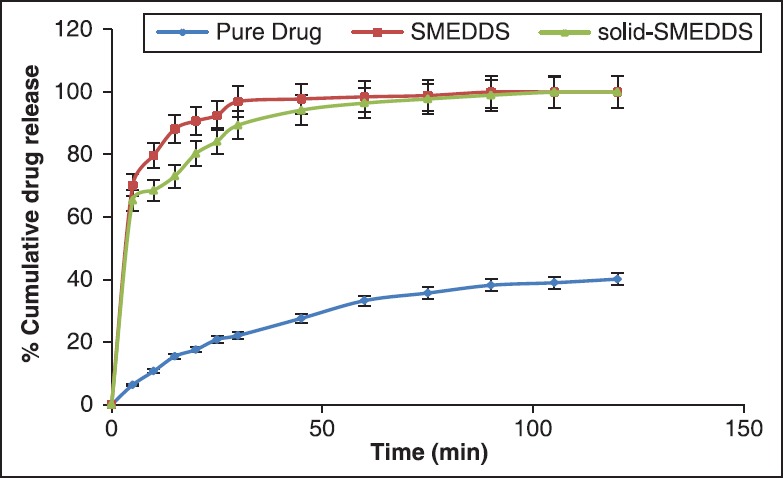
Comparison of in vitro release of pure drug, self-microemulsifying drug delivery system (SMEDDS) (F3) and solid-SMEDDS (SF3)
Release kinetics
To establish a relationship between the release kinetics of the dissolution study of telmisartan in SMEDDS and solid-SMEDDS, data obtained from in vitro dissolution study was fitted into various kinetic models [Tables 6 and 7].
Table 6.
Release kinetics of SMEDDS
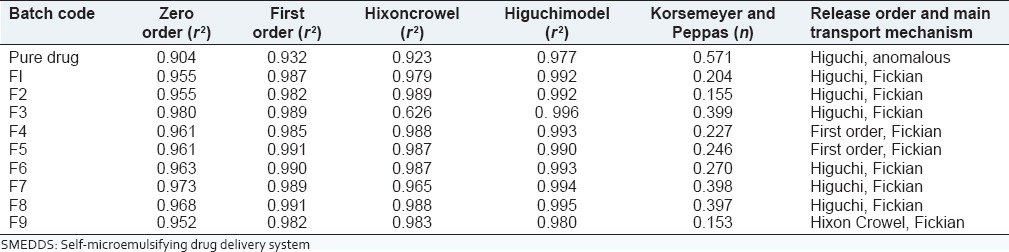
Table 7.
Release kinetics of solid-SMEDDS
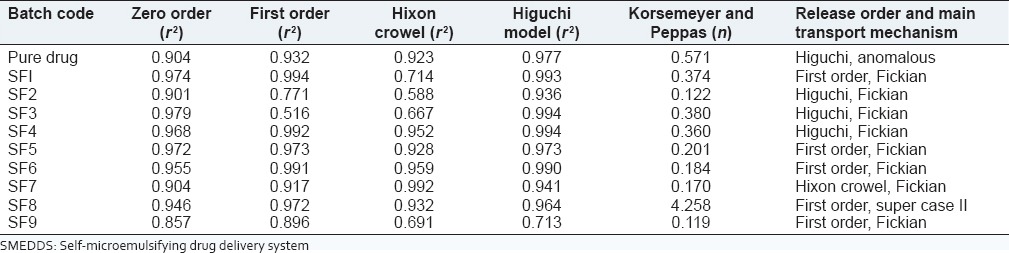
Pure drug showed Higuchi release and the formulations F2-F9 of SMEDDS, SF1-SF4 of solid-SMEDDS were best-fitted Higuchi model, which indicated the drug release by diffusion in slow and sustained way. Formulations F4 and F5 in SMEDDS and SF5, SF6, SF8 and SF9 in solid-SMEDDS followed zero order kinetics and F9 in SMEDDS and SF7 in solid-SMEDDS followed Hixon Crowel, which showed that there was a change in surface area and diameter of particles.
The value of n in all the SMEDDS formulations was close to 0.5 suggesting that the telmisartan was released from the system by Fickian diffusion. Whereas in solid-SMEDDS, except formulation SF8 (n = 4.258, super case II transport) all showed Fickian diffusion. Pure drug showed anomalous transport also known as non Fickian diffusion.
Selection of optimized formulation
Both the systems (SMEDDS and solid-SMEDDS), were observed to increase the dissolution of telmisartan and thus, may enhanced the bioavailability. Still solid-SMEDDS would be preferred upon SMEDDS because it has good prospects for development of solid dosage form (tablet, capsules and dry emulsions). Furthermore, solidified dosage forms are more ideal than liquid ones in terms of its stability. In liquid dosage forms (SMEDDS), it is also sometimes necessary to add preservative so as to avoid its oxidation above room temperature, but in case of solid dosage forms (solid-SMEDDS) it is not obligatory. Hence, solid-SMEDDS (SF3) was found to be optimized formulation having in vitro release same as SMEDDS and better dosage form development prospects.
Stability studies
The results of stability studies depicted that the solid-SMEDDS formulation remained clear even after a period of 3 months at temperature 25°C ± 2°C and 40°C ± 0.1°C. There was no phase separation in both the systems at each time. All the formulations were found to be consistent with respect to their drug content, in vitro drug release, phase separation and transparency during the stability study [Figure 12].
Figure 12.

In vitro drug release of solid-self-microemulsifying drug delivery system after stability study
CONCLUSION
Self-microemulsifying drug delivery system is a vital tool in overcoming the formulation difficulties and improving the oral bioavailability of hydrophobic/lipophilic drugs. In this study, SMEDDS and solid-SMEDDS formulations of poorly water-soluble drug, telmisartan were successfully prepared by the ultrasonication method and adsorbent technique respectively for oral administration. Further, they were assessed for in vitro performances. Among various formulations, F3 in SMEDDS and SF3 in solid-SMEDDS showed promising results in the terms of globule size analysis, self-emulsification time, zeta potential, drug loading efficiency and in vitro drug release. It could be summarized that SMEDDS formed from castor oil, tween 20 and propylene glycol as oil, surfactant and co-surfactant is a promising approach to improve the solubility, dissolution rate and hence bioavailability of telmisartan. The optimized formulations showed significantly improved drug release as compared to pure drug. Solid-SMEDDS were preferred over SMEDDS in terms of stable dosage form. It can be concluded that telmisartan solid-SMEDDS offer more predictable and more extensive drug release/absorption than the corresponding conventional formulations. The results from the study showed the utility of solid-SMEDDS to enhance solubility and bioavailability of sparingly soluble compounds like telmisartan, which can be helpful to reduce dose and related side effects of the drug. The present exploratory work successfully illustrates the potential utility of solid-SMEDDS for the delivery of poor water-soluble compounds.
ACKNOWLEDGMENT
The authors are thankful to Central Instrumentation Lab, Panjab University, Chandigarh and NIPER, Mohali, India, for carrying out TEM, X-RD and zeta sizer, SEM, DSC and also thankful to Rayat-Bahra Educational and Research Trust for providing research facilities.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Wolfgang W, Michael E, Jacobus CA, Joachim S, Ulrich B, Thomas E, et al. A Review on telmisartan: A novel, long-acting angiotensin II-receptor antagonist. Cardiovasc Drug Rev. 2000;18:127–54. [Google Scholar]
- 2.Kausalya J, Suresh K, Padmapriya S, Rupenagunta A, Senthilnathan B. Solubility and dissolution enhancement profile of telmisartan using various techniques. Int J Pharm Tech Res. 2011;3:1737–49. [Google Scholar]
- 3.Bhagwat DA, D’souza JI. Development of solid-self micro emulsifying formulation to improve oral bioavailability. Int J Ther Appl. 2012;1:38–41. [Google Scholar]
- 4.Singh A, Chaurasiya A, Warsi Musarrat H, Chaurasiya M, Jain GK, Asati D, et al. Oral pharmacokinetic study of exemestane SMEDDS and suspension in rat plasma by liquid chromatography-mass spectrometric analysis. J Liq Chromatogr Relat Technol. 2012;35:2162–74. [Google Scholar]
- 5.Cole ET, Cadé D, Benameur H. Challenges and opportunities in the encapsulation of liquid and semi-solid formulations into capsules for oral administration. Adv Drug Deliv Rev. 2008;60:747–56. doi: 10.1016/j.addr.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Goyal U, Arora R, Aggarwal G. Formulation design and evaluation of a self-microemulsifying drug delivery system of lovastatin. Acta Pharm. 2012;62:357–70. doi: 10.2478/v10007-012-0022-1. [DOI] [PubMed] [Google Scholar]
- 7.Charman SA, Charman WN, Rogge MC, Wilson TD, Dutko FJ, Pouton CW. Self-emulsifying drug delivery systems: Formulation and biopharmaceutic evaluation of an investigational lipophilic compound. Pharm Res. 1992;9:87–93. doi: 10.1023/a:1018987928936. [DOI] [PubMed] [Google Scholar]
- 8.Spernath A, Aserin A. Microemulsions as carriers for drugs and nutraceuticals. Adv Colloid Interface Sci. 2006;128-130:47–64. doi: 10.1016/j.cis.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 9.Chowdhary KP, Madhav BL. Novel drug delivery technologies for insoluble drugs. Indian Drugs. 2005;42:557–64. [Google Scholar]
- 10.Kim HJ, Yoon KA, Hahn M, Park ES, Chi SC. Preparation and in vitro evaluation of self-microemulsifying drug delivery systems containing idebenone. Drug Dev Ind Pharm. 2000;26:523–9. doi: 10.1081/ddc-100101263. [DOI] [PubMed] [Google Scholar]
- 11.Chow YJ, Choi HK. Enhancement of percutaneous absorption effect of vehicles and adhesive matrix. Int J Pharma. 1998;169:95–104. [Google Scholar]
- 12.Cho W, Kim MS, Kim JS, Park J, Park HJ, Cha KH, et al. Optimized formulation of solid self-microemulsifying sirolimus delivery systems. Int J Nanomedicine. 2013;8:1673–82. doi: 10.2147/IJN.S43299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui J, Yu B, Zhao Y, Zhu W, Li H, Lou H, et al. Enhancement of oral absorption of curcumin by self-microemulsifying drug delivery systems. Int J Pharm. 2009;371:148–55. doi: 10.1016/j.ijpharm.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Kommuru TR, Gurley B, Khan MA, Reddy IK. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm. 2001;212:233–46. doi: 10.1016/s0378-5173(00)00614-1. [DOI] [PubMed] [Google Scholar]
- 15.Shen H, Zhong M. Preparation and evaluation of self-microemulsifying drug delivery systems (SMEDDS) containing atorvastatin. J Pharm Pharmacol. 2006;58:1183–91. doi: 10.1211/jpp.58.9.0004. [DOI] [PubMed] [Google Scholar]
- 16.Patel A. Preparation and in vivo evaluation of SMEDDS (Self-Microemulsifying Drug Delivery system) containing finofibrate. AAPS J. 2007;9:344–52. doi: 10.1208/aapsj0903041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khoo SM, Humberstone AJ, Porter CJ, Edwards GA, Charman WN. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int J Pharm. 1998;167:155–624. [Google Scholar]
- 18.Patil P, Paradkar A. Porous polystyrene beads as carriers for self-emulsifying system containing loratadine. AAPS Pharm SciTech. 2006;7:199–205. doi: 10.1208/pt070128. [DOI] [PubMed] [Google Scholar]
- 19.Baboota S, Shakeel F, Ahuja A, Ali J, Shafiq S. Design, development and evaluation of novel nanoemulsion formulations for transdermal potential of celecoxib. Acta Pharm. 2007;57:315–32. doi: 10.2478/v10007-007-0025-5. [DOI] [PubMed] [Google Scholar]
- 20.More HN, Hazare AA. 1st ed. Kolhapur: Manas Prakashan; 2004. Practical Pharmaceutics (Physical pharmacy) pp. 86–105. [Google Scholar]
- 21.Swamy NGN, Rupa V, Abbas Z, Dasankoppa FS. Formulation and evaluation of Nanosuspensions for enhancing the dissolution of poorly soluble Mebendazole. Indian Drugs. 2010;47:47–54. [Google Scholar]
- 22.Surjyanarayan M, Hemangini R, Bhavdip J, Mikesh P, Rajesh KS. Release kinetic modeling of atorvastatin calcium loaded self microemulsifying drug delvery system. Elixir Pharm. 2012;53:11725–9. [Google Scholar]
- 23.Mahajan HD, Shaikh T, Baviskar D, Wagh RD. Design and development of solid self-micro-emulsifying drug delivery system (SMEDDS) of fenofibrate. Int J Pharm Res Sci. 2011;3:163–6. [Google Scholar]
- 24.Alany RG, Tucker IG, Davies NM, Rades T. Characterizing colloidal structures of pseudoternary phase diagrams formed by oil/water/amphiphile systems. Drug Dev Ind Pharm. 2001;27:31–8. doi: 10.1081/ddc-100000125. [DOI] [PubMed] [Google Scholar]
- 25.Chen H, Chang X, Weng T, Zhao X, Gao Z, Yang Y, et al. A study of microemulsion systems for transdermal delivery of triptolide. J Control Release. 2004;98:427–36. doi: 10.1016/j.jconrel.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Liu H, Zhou LL, Wei LL, Hong-Guo , Nie SF, Yang XG, et al. Preparation of budesonide-poly (ethylene oxide) solid dispersions using supercritical fluid technology. Drug Dev Ind Pharm. 2007;33:959–66. doi: 10.1080/03639040601134181. [DOI] [PubMed] [Google Scholar]
- 27.Singh A, Chaurasiya A, Warsi MH, Chaurasiya M, Jain GK, Asati D, et al. Oral pharmacokinetic study of exemestane SMEDDS and suspension in rat plasma by liquid chromatography-mass spectrometric analysis. J Liq Chromatogr Relat Technol. 2012;35:2162–74. [Google Scholar]
- 28.Idrees M, Rahman N, Ahmad S, Ali M, Ahmad I. Enhance transdermal delivery of flurbiprofen via microemulsions: Effects of different types of surfactants and cosurfactants. Daru. 2011;19:433–9. [PMC free article] [PubMed] [Google Scholar]
- 29.Shukla JB, Akshay RK, Ketan MR, Rajesh KP. Self micro emulsifying drug delivery system. Pharm Sci Monit. 2010;1:19–33. [Google Scholar]
- 30.Pouton CW. Effects of the inclusion of a model drug on the performance of self-emulsifying formulations. J Pharm Pharmacol. 1985;37:IP. [Google Scholar]
- 31.Gershanik T, Benita S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur J Pharm Biopharm. 2000;50:179–88. doi: 10.1016/s0939-6411(00)00089-8. [DOI] [PubMed] [Google Scholar]
- 32.Raval M, Patel J, Patel A, Sheth N. Formulation and development of a self-nanoemulsifying drug delivery system of irbesartan. AAPS J. 2011;2:9–16. doi: 10.4103/2231-4040.79799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Charman SA, Charman WN, Rogge MC, Wilson TD, Dutko FJ, Pouton CW. Self-emulsifying drug delivery systems: Formulation and biopharmaceutic evaluation of an investigational lipophilic compound. Pharm Res. 1992;9:87–93. doi: 10.1023/a:1018987928936. [DOI] [PubMed] [Google Scholar]
- 34.Lawrence MJ, Rees GD. Microemulsion-based media as novel drug delivery systems. Adv Drug Deliv Rev. 2000;45:89–121. doi: 10.1016/s0169-409x(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 35.Park S, Baker JO, Himmel ME, Parilla PA, Johnson DK. Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels. 2010;3:10. doi: 10.1186/1754-6834-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deshmukh A, Kulkarni S. Solid self-microemulsifying drug delivery system of ritonavir. Drug Dev Ind Pharm. 2014;40:477–87. doi: 10.3109/03639045.2013.768632. [DOI] [PubMed] [Google Scholar]
- 37.Singh AK, Chaurasiya A, Awasthi A, Mishra G, Asati D, Khar RK, et al. Oral bioavailability enhancement of exemestane from self-microemulsifying drug delivery system (SMEDDS) AAPS PharmSciTech. 2009;10:906–16. doi: 10.1208/s12249-009-9281-7. [DOI] [PMC free article] [PubMed] [Google Scholar]