

SUMMERY
Trypanosomatids represent the causative agents of major diseases in humans, livestock and plants, with inevitable suffering and economic hardship as a result. They are also evolutionarily highly divergent organisms, and the many unique aspects of trypanosome biology provide opportunities in terms of identification of drug targets, the challenge of exploiting these putative targets, and at the same time significant scope for exploration of novel and divergent cell biology. We can estimate from genome sequences that the degree of divergence of trypanosomes from animals and fungi is extreme, with perhaps one third to one half of predicted trypanosome proteins having no known function based on homology or recognizable protein domains/architecture. Two highly important aspects of trypanosome biology are the flagellar pocket and the nuclear envelope, where in silico analysis clearly suggests great potential divergence in the proteome. The flagellar pocket is the sole site of endo- and exocytosis in trypanosomes and plays important roles in immune evasion via variant surface glycoprotein (VSG) trafficking and providing a location for sequestration of various invariant receptors. The trypanosome nuclear envelope has been largely unexplored, but by analogy with higher eukaryotes, roles in the regulation of chromatin and most significantly, in controlling VSG gene expression are expected. Here we discuss recent successful proteomics-based approaches towards characterization of the nuclear envelope and the endocytic apparatus, the identification of conserved and novel trypanosomatid-specific features, and the implications of these findings.
Keywords: Proteomics, nuclear pore complex, flagellar pocket, systems biology, molecular evolution, protein complex, protein interactions
INTRODUCTION
The identification and definition of function remains a highly challenging aspect of modern biology, despite the recent huge technical advances for discovering new genes and their products. One major innovation in the past decade has been the vast improvement in mass spectrometric identification of polypeptides, brought about by a combination of improved instrument sensitivity together with computational resources that allow the assignment of mass spectra based on genome sequences (discussed in Chait, 2011). Coupled with the falling costs of large scale DNA sequencing, many organisms are now amenable to molecular level analysis for the first time as the economic ‘barriers to entry’ have begun to fade. The concept of a ‘proteome’ has emerged through these technological advances, and to a first approximation is equivalent to the protein composition of a defined biological specimen, be it a whole organism lysate, a tissue fluid, an isolated organelle or a specific protein complex. The application of proteomic methods to pathogens has the exciting potential to inform broadly on functions relevant to virulence, host range restriction and immune evasion mechanisms. Examples include the identification of developmental changes through the life cycle, discovery of complexes responsible for signaling, gene expression and intracellular transport and potentially monitoring the responses of a pathogen to a drug or other insult.
The African trypanosomes, Trypanosoma brucei spp, are the causative agents of African sleeping sickness in humans and nagana in cattle (Simarro et al. 2010). These organisms have been, and remain, major causes for concern in terms of public health and agricultural productivity. Unquantified (and likely unquantifiable), but major impacts on the flora and fauna of Africa have resulted from infection of a great many animal species by T. brucei. Related organisms, including Leishmania, Phytomonas and Euglena, most likely also exert huge impacts, both ecologically and economically, on much of the planet (see Camargo, 1999, Duttagupta, et al. 2004 Antinori, et al. 2011, for some examples and discussion). Many of the diseases associated with these organisms have been historically classed as ‘neglected’, in part due to the absence of high quality chemotherapeutic agents or vaccines with which to combat infections, but also due to the absence of a financial insentive as many of the afflicted live in the poorest parts of the world (Wilkinson and Kelly, 2009, Magez and Radwanska, 2009).
TRYPANOSOMES, DIVERGENCE AND PROTEOMIC INSIGHTS
The completion of a genome sequence for one strain of T. brucei yielded many insights into the biology of trypanosomes, and provided the vital framework for going forward with molecular level dissection of trypanosome biology (Berriman, et al. 2005). This has been coupled with the emergence of RNA interference (RNAi) for suppression of gene expression in a conditional manner, RNA sequencing approaches to monitor transcription (Kolev, et al. 2010) and most recently RNAi-based expression knockdown screens (RIT-seq, Alsford, et al. 2010), with the result that our understanding of the cell biology and metabolism of T. brucei has advanced at an accelerated pace during the past five to ten years. However, many of the investigations in this period have been centered around ‘candidate’ based approaches, i.e. mining the genome for gene products with either known functions or at least functions in known processes or pathways or predictions based on similarity of either sequence or domain architectures; transcription, histone modification, intracellular trafficking and the cytoskeleton are all good examples of where this type of approach has been of great value (see Kawahara, et al. 2008, Luz Ambrósio, et al. 2009, Field and Carrington, 2009, Wickstead, et al. 2010). Regardless of how informative, this is still nevertheless an introspective strategy, and thus ignores much of the potential novel biology and therapeutic opportunity within the trypanosome.
It is estimated that up to 50% of the trypanosome protein coding content is ‘divergent’, in the sense that orthology or paralogy with higher eukaryote genes cannot be reliably established, opening up the potential for novel and trypanosome-specific functions. For example the trypanosome kinase families appear highly divergent from higher eukaryotes, with few conserved domain architectures beyond the kinase domains themselves, making functional prediction extremely difficult (Parsons, et al. 2005). It is, however, very likely that this 50% of novel gene products is an overestimate, as many orthologous relationships are simply too divergent to be detected by sequence-based algorithms alone. As we have demonstrated previously, the nuclear pore complex is apparently highly divergent based on just in silico analysis, but is in fact rather well conserved and simply an example of BLAST failing to identify highly diverged sequences (deGrasse, et al. 2009). Thus, targeted proteomic analysis has a major role to play in this regard, as subcellular organelles, macromolecular structures and complexes can be isolated, using evolutionarily conserved handles, i.e. tagged proteins that are sufficiently well conserved for function to be confidently inferred, and potentially less well conserved proteins that are in association can be identified. This has the strategic advantage of finding genuinely trypanosome-specific proteins, but which interact with known proteins. At a minimum, evidence for a likely role in a specific process or pathway is already available for the new protein, facilitating the design of strategies to directly test these hypotheses.
The quality of a proteome, and its ultimate utility, rests on sample preparation, and the rather difficult determination or definition of purity (Fig. 1 and legend). There are many strategies for preparing material for mass spectrometric analysis, ranging from whole cell extracts to more specific protein complexes, but in practise the issues remain similar; defining the material being analysed, being aware that there are likely contaminants and validating the proteins that are identified. Given that many complexes are known that participate in multiple interactions, the determination of specificity in itself is a very crucial step.
Figure 1. Considerations for proteomics.
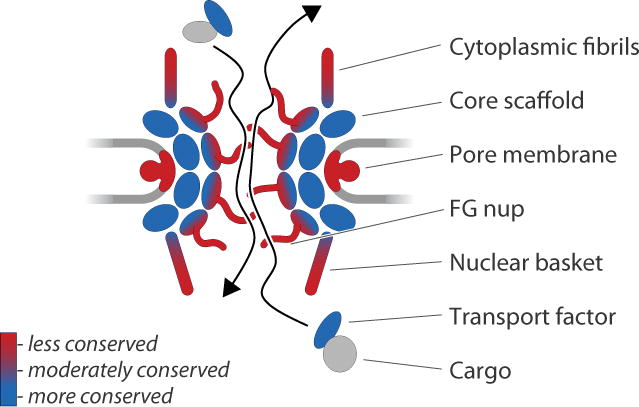
Schematics to illustrate some of the conceptual and technical challenges that need to be addressed or considered when analyzing the protein composition of a cell, organelle or complex. Panel A: Much difficulty can lie in the selection of appropriate gating parameters, with the significant issue that, ab initio, it is almost impossible to know where these parameters should be set. An initial estimate may be gleaned from prior art or scouring the genome sequence for clues, but this can remain a very inaccurate process, and is full of assumptions. In the examples, the gating is used to either stringently eliminate >95% of the experimentally detected proteome (left), to reject >70% as a moderate cutoff (center) or to allow into consideration essentially all of the proteome (right). Depending on the question, the nature of the sample, the quality of the data and the quality of the available predicted proteome database (genome) a gate approximating one (or none) of these possibilities may be appropriate. Obviously, more stringent gating reduces the number of proteins to consider going forward, but will in all probability exclude important information. Panel B: Most proteins participate in a range of interactions, some of which are direct, while others are indirect. Conceptually one can consider a core of tight associations mediating the basic function of a given protein or complex (core, in red). However, this is biologically inaccurate as even tight complexes, organelles or other biological assemblies exist in association with other complexes and systems. These interactions become functionally as well as physically more tenuous, and will eventually come to include proteins that are off target, but which may still retain a genuine affinity for components of the target complex/organelle (light gray and blue). In some cases a given protein may be a bona fide member of several complexes; Sec13 in S. cerevisiae is now known to participate in at least three complexes, COPII, the NPC and the SEA complex. The point at which one considers such interactions to represent contaminants is hard to determine, and to some level is subjective. Panel C: Considerations for the analysis of complex mixtures. Regardless of the appropriateness or not of the gating algorithm used, any initial list of proteins will contain a mixture of genuine components (‘realerons’, red), together with additional moderate or low probability members of the complex/organelle (‘mysterons’, black); probability here equates to additional evidence for membership to a complex. The biological sample will also contain contaminants, which are frequently impossible to exclude, as well as miscalls due to low signals, errors in the predictive algorithms, suppression and database quality problems. All of these criteria are variable (and difficult to quantify, hence ‘mysteron’) in terms of gating and hence any proteome is best considered as ‘a’ proteome for a complex or organelle, rather than definitively ‘the’ proteome. Panel D: Methods and approaches for validation of identified candidates. The top system encompasses in silico methods, primarily designed to discriminate based on known location and function from other studies; again some of these assignments may be very high confidence and others much less so. The lower system includes examples of direct experimental approaches, including validation of the interaction itself by independent methods. The critical point here is that without information from these final validation steps, it is frequently difficult to have confidence in a proteome. Regrettably, this is also the most expensive part of the process in terms of time, resources, and labor.
One approach we have found to be highly successful has been analysis of the trypanosome nuclear envelope. Using as starting material a highly enriched pore complex lamina fraction (Rout and Field, 2001), we were able to identify ~800 distinct polypeptides, amounting to 10% of the predicted T. brucei proteome (DeGrasse, et al. 2009). A recent innovation, pioneered by one of us, is the isolation of protein complexes using a cryogrinding method that was originally developed to facilitate the lysis of Saccharmoyces cerevisiae cells due to the presence of a cell wall that does not dissolve in mild detergents. We have successfully adapted this method to trypanosomes and demonstrated its usefulness using the NPC as a testbed, as well as to elements of the nuclear skeleton/lamina and more recently cytoplasmic complexes. Although still at the development phase, the methodology promises to be generally applicable to trypanosomes (and maybe other protists), with the potental to allow the elucidation of protein-protein interactions in T. brucei in a systematic manner (data not shown).
Here we briefly consider two major cellular systems of trypanosomes, the endocytic apparatus and the nuclear envelope (Fig. 2). Both of these systems are essential, possess numerous diverse functions and likely contribute in unique ways to the biology of trypanosomes. We will also briefly discuss several approaches to the generation of biological samples for proteomics and some advantages and disadvantages of each.
Figure 2. Organelle and systems targets for proteomics in African trypanosomes.
Top: The flagellar pocket and the endocytic system. At left is a schematic of the organelle, showing the flagellum, pocket collar (red) and transport vesicles. At right are a number of considerations for study of the flagellar pocket and the endocytic system in particular. Lower: Nuclear envelope and associated structures. At left is a schematic of a partial nucleus. In dark gray are filaments that likely participate in maintaining the structure of the nucleus, while heterochromatin is represented as shaded areas. Nuclear pore complexes are shown in red. At right are specific considerations for study of the nuclear envelope. In bold are aspects that are particularly relevant, in terms of either novelty or lack of exploration, to the African trypanosomes.
DEFINING A CLATHRIN INTERACTOME – CLASSICAL ANTIBODY PULLOUT
Based on primary structure-based searches, membrane transport in T. brucei apparently lacks multiple major factors found in other species that are known to play important roles in transport (discussed in Field, et al. 2008). These apparent absences result in an ‘incomplete’, and paradoxically non-functional, predicted protein network with many of the trypanosome endocytic protein orthologues lacking identifiable binding partners (see Field, et al. 2008). As the endocytic system clearly is functional this observation implies either that many critical players are too divergent to be detected by sequence alone, or that truly novel proteins substitute for these factors. To some extent, both of these possibilities have the same consequences and highly divergent proteins are predicted to be operating within the trafficking machinery of trypanosomes.
All endocytosis in trypanosomes proceeds via the flagellar pocket, a membrane invagination surrounding the base of the flagellum and uniquely excluded from the subpellicular microtubule array subtending the bulk of the plasma membrane. The parasite surface is dominated by the GPI-anchored VSG, and endocytosis operates at an extremely rapid rate in the bloodstream stage of the parasite (Pal, et al. 2003, Engstler, et al. 2004). Furthermore, all endocytosis appears to be clathrin-mediated, as RNAi against clathrin completely blocks endocytic activity (Allen, et al. 2003), and many of the proteins that mediate clathrin-independent endocytosis are restricted to metazoan taxa and therefore absent from trypanosomatids and in fact the majority of organisms (Field, et al. 2007). In addition, the AP-2 complex, which mediates recognition of cargo and their concentration in clathrin-coated pits is absent from T. brucei, and the major trans-membrane domain endocytic cargo, the invariant surface glycoproteins 65 and 75, both lack classical AP-based sorting signals and are internalized and degraded via a ubiquitylation-based mechanism (Chung, et al. 2004, Chung, et al. 2008, Leung, et al. 2011). We have also described a putative clathrin-associated sorting protein (CLASP), TbEpsinR, which is clearly required for internalization of surface proteins (Gabernet-Castello, et al. 2009), a second, TbAP180, that is currently under study (MCF and P. Manna, unpublished data) and a conserved protein RME-8 which is also a major player here (L. Koumandou and MCF, unpublished data). Finally, there is good evidence that endocytosis is dynamin-independent in bloodstream forms as RNAi against the single trypanosomal dynamin gene, TbDLP, has no impact on endocytic activity (Morgan, et al. 2004). While evidence for a role in endocytosis in insect stages has been reported (Chanez, et al. 2006), more recently, studies from our group suggest that this endocytic defect could arise indirectly through an impact on the mitochondrion and ATP production, where TbDLP is clearly localised and likely functions (Morgan, et al. 2004, Natesan, et al. 2010).
Overall, while providing some insights into how clathrin operates at the flagellar pocket and potentially elsewhere in the cell, the data above beg the question of if there are novel, or trypanosome-specific factors that operate in the endomembrane system or if trypanosome endocytosis really does function with such a minimal composition? One solution to this question is to interrogate the clathrin interactome ab initio. Global approaches based on immuno-isolation have been reported for mammalian tissue culture cells (Borner, et al. 2006), and have proved insightful, and therefore we asked if such an essentially unbiased approach could be applied to trypanosomes. We chose the bloodstream form parasite as this is the stage where clathrin is more abundant and where potentially relevant proteins may be expressed that are involved in the maintenance of the VSG coat (Morgan, et al 2001, Engstler, et al. 2007, Field and Carrington, 2009).
To isolate clathrin-interacting proteins from the bloodstream trypanosome, co-immunoprecipitation using rabbit anti-clathrin heavy chain antibody (TbCHC, Morgan, et al. 2001) was performed. Subsequently, the isolated complexes were resolved by electrophoresis, silver-stained and the protein bands subjected to MS/MS for identification (Fig. 3). Besides TbCHC, more than twenty distinct proteins were identified in these pullouts, many of which appear to be novel and trypanosome specific (VOA and MCF, unpublished data). While potentially providing insight to the endocytic system, this finding raises concerns that can only be addressed with detailed validation.
Figure 3. Identification of a trypanosome clathrin network.
Panel A: Protein complex immuno-precipitated from BSF and PCF lysates (lane 3 and 4 respectively) using rabbit anti-TbCHC polyclonal antibody (RαTbCHC) was resolved on a 12% SDS-polyacrylamide gel. Beads plus lysate (lane 1) or beads plus antibody alone (lane 2) were used as controls. Silver-staining indicated the presence of the ~175 kDa TbCHC protein in the isolated complexes from BSF and PCF but not in the negative controls. Protein bands migrating above the antibody heavy chain (white arrow) were subjected to mass spectrometry and further investigated. Panel B: Cells expressing C-terminally hemagglutinin (HA) genomic-tagged versions of three candidate clathrin-interacting partners TbHsc70, TbCAP100 and TbCAP161 were generated and prepared for immunofluorescence microscopy. The HA-tagged proteins (green) co-localize with clathrin (red) in the region between the kinetoplast and nucleus, as demonstrated by DAPI (blue) staining for DNA. Panel C: Co-immunoprecipitation of HA-tagged proteins of the candidates was performed and the isolated complexes probed for clathrin by Western blotting. The antibodies used are mouse anti-FLAG (mαFLAG) and mouse anti-HA (mαHA).
The set of candidate clathrin-associating proteins (TbCAPs) includes the heat-shock protein 70 (TbHsc70) and two hypothetical proteins, TbCAP100 and TbCAP161 (Fig. 3). TbHsc70 is a well known clathrin-interacting protein from higher eukaryotes and is responsible for uncoating of clathrin triskelion cages from clathrin-coated vesicles (CCVs) (Rothnie, et al. 2011); identification of this protein thus goes some way towards an indication that the analysis is valid. TbCAP100 is also present in the genomes of a wide range of eukaryotes, and therefore represents an additional component of a potentially conserved core. By contrast TbCAP161 is only found in trypanosomatids. HA genomic-tagged forms of TbHsc70, TbCAP100 and TbCAP161 localize between the kinetoplast and nucleus, the region that contains the vast majority of the endocytic and exocytic organelles (Brickman, et al. 1995, Field, et al. 1998). All three antigens demonstrate significant co-localization with clathrin (Fig. 3 and data not shown). These data suggest that, together with TbHsc70, TbCAP100 and TbCAP161 are potential members of a trypanosome clathrin interactome network. However, as the clathrin interactions likely span endocytosis, endocytic recycling and also post-Golgi complex sorting and transport, the precise functions of TbCAP100 and TbCAP161 cannot be inferred from location alone, and more evidence is needed to clarify the roles of these proteins (Morgan, et al. 2001, Grunfelder, et al. 2003). For further confirmation of clathrin interaction, the HA-tagged proteins were immunoprecipitated from whole cell lysates and these precipitates demonstrated to also contain clathrin by Western analysis (Fig. 3). In summary, bi-directional coimmunoprecipitation and co-localization between TbHsc70, TbCAP100 and TbCAP161 with clathrin provides very strong evidence that TbHsc70, TbCAP100 and TbCAP161 are bona fide clathrin-binding proteins.
The highly significant finding here is that while TbHsc70 and TbCAP100 are clearly well conserved between trypanosomes and higher eukaryotes, TbCAP161 is lineage-specific and cannot be detected by homology searches beyond the kinetoplastida (data not shown). A similar level of conserved and lineage-restricted interaction partners was described recently by us for trypanosome Rab11 (Gabernet-Castello, et al. 2011).
The major advantage of the direct antibody pullout is that it is minimally invasive, with wild-type parasites being the target. The method is also highly accessible – any good polyclonal is a potential starting point, and little in the way of specialised equipment is required. Of course, the approach may fail to capture many associated proteins due to dissociation and the absence of a second affinity step can increase the incidence of off-target hits. In the present example there was a requirement for lysis to provide access to the intracellular antigen, using partial detergent solubilisation, which is potentially disruptive. Finally, there is the significant issue that the antibody resource is finite, and it is (too) easy to consume significant amounts of antibody in such experiments.
THE NUCLEAR ENVELOPE – SUBCELLULAR FRACTIONATION
The nuclear envelope is a defining feature of eukaryotic cells, and serves to separate nuclear activities such as transcription and mRNA processing from translation. Trypanosome nuclei are conventional in that they possess electron dense material at the nuclear periphery, which is most probably heterochromatin, a recognisable nucleolus and a double bilayer nuclear envelope which is contiguous with the endoplasmic reticulum. The limited evidence to hand has suggested that trypanosomes have comparatively conventional systems for RNA and protein import and export, as well as at least one direct study that demonstrated a probable conventional protein import pathway (Marchetti, et al. 2000). However, novel aspects are likely present, for example in mRNA transport, where RNA-recognition motif (RRM) domains appear sufficient to specify nuclear accumulation, likely by binding to a specific RNA (Cassola and Frasch, 2009). Further, ultrastructural analysis of trypanosomes has indicated the presence of nuclear pore complexes (NPCs) that morphologically resemble those in opisthokont taxa (Rout and Field, 2001). The obvious importance of the NPC to mRNA export pathways is enhanced by the assumed absence of promoter control from expression of many protein-coding genes due to polycistronic transcription and the presence of a spliced leader at the 5′ end of mature coding mRNAs.
A similar problem to the identification of the endocytic protein network was initially presented for the analysis of the trypanosome nuclear envelope, and specifically identification of NPC proteins (nucleoporins). Based on BLAST analyses very few nucleoporins could be identified or clear orthologous relationships derived, which raised the possibility that the trypanosome NPC was genuinely highly divergent from metazoan and yeast NPCs (Mans, et al. 2004). The opposing interpretation was that the NPC was in fact conserved, but that the nucleoporins were too divergent to be identified using sequence-based searches. In fact the success in identifying potential NPC orthologues was significantly poorer than for the endocytic network.
A solution has been to pursue a classical subcellular fractionation route, to produce a highly enriched pore complex lamina fraction, essentially the nuclear envelope following mild detergent extraction, and again based on methods originally develped for S. cerevisiae (Rout and Field, 2001). Despite extensive validation of the pore complex lamina fraction by both immunochemistry and electron microscopy which suggested a comparatively simple fraction containing NPCs and some interconnecting fibres, over 800 distinct polypeptides could be identified by a combination of MS/MS and MALDI-MS approaches, with varying degrees of confidence (deGrasse, et al. 2008, deGrasse, et al. 2009). This illustrates rather well an important, and sobering point; despite careful monitoring of the purification, and good evidence that the resulting material was comparatively homogenous, a surprisingly large proteome was uncovered, making the list of proteins identified difficult to discriminate from the cohort that might be identified from a highly impure or heterogenous sample. In part, this is because of the high sensitivity of current mass spectrometric techniques, but in the face of such complexity, some confidence in the starting material, for example a validation of purity, is highly valuable before committing to further analysis with such a dataset. Informatics analysis, based on the observation that secondary structure is better conserved than primary structure, allowed the identification of candidate nucleoporins and finally validation by localization following in situ genomic tagging (Oberholzer, et al. 2006, deGrasse, et al. 2009, Devos and deGrasse, 2010). The important biological outcome of this study was that the trypanosome NPC is substantially more conserved with metazoan and yeast NPCs than sequence searches had indicated, with major implications for the timing of evolution of the nucleus and subnuclear structures. Furthermore, the present data suggests that the NPC is more conserved than the clathrin interactome, despite the poorer return from in silico studies.
The advantages of subcelluar fractination are clear – the target is defined to some degree and selected to be highly biologically relevant, but the study does require a more significant investment in the design and validation of the isolation method than a simple antibody-based immunoisolation. Some disadvantages are perhaps less obvious; surveying a subcellular fraction can be extremely challenging as the composition of the isolated material is likely to be highly complex. Studies of the plasma membrane, flagellum, pore complex lamina fraction and mitochondrial proteomes from trypanosomes have all revealed highly complex proteomes, with the likelihood that much of this complexity is contaminant as well as putative novel components, making discrimination between these two categories vital (Broadhead, et al. 2006, Bridges, et al. 2009, DeGrasse et al. 2009, Panigrahi et al. 2009, Oberholzer et al. 2011). These problems are, of course, all layered upon standard concerns with purity that derive from any subcellular fractionation study.
THE NUCLEAR PORE AND NUCLEOSKELETON – CRYOGRINDING
A fuller understanding of protein function requires identification and characterisation of interaction partners, as all proteins operate in the context of complexes and higher order structures, at least for part of their time. These interactions may be truly stable structures, at least on the seconds to minutes timescale, where examples include cytoskeletal elements and the NPC. More transient complexes are the clathrin triskelion cage and coatomer vesicle coats which assemble and disassemble on the seconds timescale. Examples of rapidly associating and dissociating relationships include small GTPase interactions with their constellation of effector molecules, which exhibit a range of binding affinities that vary from nanomolar to millimolar depending on both the interaction partner and the state of the complex (Will and Gallwitz, 2001, Wu, et al. 2007). Affinities for protein-protein interactions can vary over several orders of magnitude, and koff rates likewise have a broad dynamic range, both of which can confound efforts to isolate intact complexes or reliable portions thereof. Immensely powerful though they are (see, for exampe Panigrahi, et al. 2006), affinity methods including the TAP-tag approach which use a dual affinity tag to attempt to minimise non-specific or off-target associations, suffer greatly from the prolonged period required for the pullout protocol. While facilitating the isolation of stable complexes, this can lead to loss of genuine low affinity interactions, as well as increase non-specific binding. Criticisms of other approaches to map interactions, such as yeast two hybrid, have been well rehearsed in the literature. In vitro or in vivo methods that rely on co-expression of two tagged proteins, either in a homologous system or, for example, in reticulocyte lysates, can suffer from overexpression artefacts as well as frequently non-physiological conditions, and of course, are not directed at the identification of novel factors.
One approach that avoids many of these issues is affinity isolation subsequent to cryogrinding, by virtue of its rapidity, elimination of chaotropes, detergents or other factors from the generation of the initial lysate. This method preserves complexes or dynamic systems at the time of harvest, preventing mixing of components and also reduces problems due to proteases, phosphatases or other potential factors that could degrade the sample as it is captured in liquid nitrogen. Further, inhibitors can be added to resuspension buffers when thawing the cryogrindate, and gentler buffer systems can be used that potentialy preserve complexes that are disrupted under conditions where one biochemically lyses cells with stronger or high concentration detergents or chaotropes.
The method will be published in full elsewhere, but in brief exploits a geological ball mill for the generation of a grindate under essentially native conditions, followed by pullout of the protein handle, normally a genomically-tagged C-terminal GFP fusion protein, using anti-GFP antibodies coupled to magnetic beads, and resolution by 1D SDS-PAGE (Cristea, et al. 2005, Oberholzer, et al. 2006; Oeffinger, et al. 2007; Fig. 4). This method requires substantial quantities of starting material, but can allow the isolation of very high quality complexes. The use of large quantities of starting material also allows exploration of many buffer conditions in parallel. We have successfully used this technique to extensively explore protein-protein interactions within the NPC, as well as to demonstrate interactions between the very large coiled-coil nucleoskeletal protein NUP-1 and the NPC as well as additional coiled-coil proteins that are present within the nucleus (MPR, BTC, MCF and SO, unpublished data). We have also begun to successfully apply the method to several complexes involved in membrane trafficking pathways, including clathrin coats and the exocyst complex.
Figure 4. Principal approach behind cyrogrinding method for determining compositions of complexes.
Trypanosomes are tagged at a single genomic locus with a GFP affinity handle, in frame with the gene of interest (GOI). The localization of the resulting fusion protein is checked by microscopy to ensure faithful targeting, in this case to the nuclear pore complex. The population is then expanded, cells frozen and then ground using a ball mill to produce a cryogrindate. From the grindate the affinity handle can be used to immunoisolate the protein of interest, and associated proteins. This step requires careful assessment of buffer conditions to attain isolation of the complex with minimal background. Mass spectrometric characterization of the resulting proteome may be facilitated using 1D SDS PAGE.
While requiring an in situ-tagged gene as an affinity handle, this strategy has the advantage of permitting assembly of the handle into the native complex in a minimally invasive manner, as the protein is expressed to normal levels and only one of the two copies in the diploid genome is modified. Remarkably, most proteins appear to tolerate addition of GFP to the C-terminus without major impact on cell viability or function, at least as assessed by proliferation and correct targeting of the tagged protein. Coupled to the near native conditions that can be used for lysis, efficient cryogenic entrapment of complexes and the speed of isolation which aids in recovery of low affinity interactions and minimises non-specific binding, we consider the method to be highly useful, and with great potental. The major downside is the quantity of material curently required, which limits the number of candidate proteins that can be considered. However, the insights that are emerging from this approach auger well for the future exploration of protein complexes in trypanosomes, and the potential identification of novel trypanosome-specific complex members and interactions.
CONCLUSIONS
There are many approaches to the generation of a proteome. The discrimination between contaminant and real component is a fundamental one, and is perhaps even more critical in such a divergent system as Trypanosoma brucei where many of the proteins that are identified may be novel; often this is precisey what the investigator is hoping to find. Frequently there is no a priori basis for a judgment in this context, hence ‘mysteron’ (Fig. 1). Validation is therefore highly critical in this regard, although the lengths to which investigators must go here varies to some extent on the context. For example, two flagellar proteomes have now been published (Broadhead, et al. 2006, Oberholzer, et al. 2011), and the quality of the data here attests to the fact that validation is robust; it is comparatively straightforward to ascertain location as a flagellar protein for example, while this is further assisted by the comparative ease of isolation of a flagellar fraction from these parasites. Internal organelles are substantially less easy to purify, and consequently validation is even more critical as the proteomes described have been extensive and clearly include contamininants as well as valuable novel components; demonstrating the frequency of these latter is important for determining the ultimate usefulness of a proteome (Bridges, et al. 2008, DeGrasse, et al. 2009, Panigrahi, et al. 2009).
New and improved methodologies for the purification of complexes and subcellular organelles to high purity are essential for the exploration of protein-protein interactions. The coupling of genomic-tagging methods, which target a single gene copy in a diploid genome, with cryogrinding has clear advantages over simple antibody-mediated pullouts. Cryogrinding can of course also be applied to a conventional antibody pullout experiment, although the use of a standardised affinity tag does have significant advantages in terms of defining optimal conditions for isolation. There are, however, some obvious issues, not least of which is the amount of material required for an essentially bulk preparation and the skills required to perform the preparations themselves. It is anticipated that the latter can be surmounted relatively quickly with formalization of several of these methods, and that this will offer the community a further tool for unravelling trypanosome biology. With about 50% of the trypanosome proteome assigned as hypothetical, together with unique cellular processes and organelles present in T. brucei, proteomic techniques are key towards realisation of the benefit of the genome sequences. Importantly, and together with other emerging techniques such as RNA interference target sequencing (Alsford, et al. 2010), these approaches will greatly enhance our knowledge of this divergent organism and hasten drug target discovery and validation.
Acknowledgments
Funding for work described in this article has been provided by the Cambridge Commonwealth Scholarships program, the National Institutes of Health (RR00862 and RR022220) and the Wellcome Trust, which is gratefully acknowledged.
References
- Allen CL, Goulding D, Field MC. Clathrin-mediated endocytosis is essential in Trypanosoma brucei. The EMBO J. 2003;22:4991–5002. doi: 10.1093/emboj/cdg481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, Berriman M, Hertz-Fowler C, Horn D. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 2011;21:915–924. doi: 10.1101/gr.115089.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinori S, Schifanella L, Corbellino M. Leishmaniasis: new insights from an old and neglected disease. Eur J Clin Microbiol Infect Dis. 2011 doi: 10.1007/s10096-011-1276-0. in press. [DOI] [PubMed] [Google Scholar]
- Berriman M, et al. The Genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- Borner GH, Harbour M, Hester S, Lilley KS, Robinson MS. Comparative proteomics of clathrin-coated vesicles. J Cell Biol. 2006;175:571–8. doi: 10.1083/jcb.200607164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickman MJ, Cook JM, Balber AE. Low temperature reversibly inhibits transport from tubular endosomes to a perinuclear, acidic compartment in African trypanosomes. J Cell Sci. 1995;108:3611–2361. doi: 10.1242/jcs.108.11.3611. [DOI] [PubMed] [Google Scholar]
- Bridges DJ, Pitt AR, Hanrahan O, Brennan K, Voorheis HP, Herzykx Herzyk, de Koning HP, Burchmore RJ. Characterisation of the plasma membrane subproteome of bloodstream form Trypanosoma brucei. Proteomics. 2008;8:83–99. doi: 10.1002/pmic.200700607. [DOI] [PubMed] [Google Scholar]
- Broadhead R, Dawe HR, Farr H, Griffiths S, Hart SR, Portman N, Shaw MK, Ginger ML, Gaskell SJ, McKean PG, Gull K. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature. 2006;440:224–7. doi: 10.1038/nature04541. [DOI] [PubMed] [Google Scholar]
- Camargo EP. Phytomonas and other trypanosomatid parasites of plants and fruit. Adv Parasitol. 1999;42:29–112. doi: 10.1016/s0065-308x(08)60148-7. [DOI] [PubMed] [Google Scholar]
- Cassola A, Frasch AC. An RNA recognition motif mediates the nucleocytoplasmic transport of a trypanosome RNA-binding protein. J Biol Chem. 2009;284:35015–28. doi: 10.1074/jbc.M109.031633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chait BT. Mass spectrometry in the postgenomic era. Annu Rev Biochem. 2011;80:239–46. doi: 10.1146/annurev-biochem-110810-095744. [DOI] [PubMed] [Google Scholar]
- Chanez AL, Hehl AB, Engstler M, Schneider A. Ablation of the single dynamin of T. brucei blocks mitochondrial fission and endocytosis and leads to a precise cytokinesis arrest. J Cell Sci. 2006;119:2968–74. doi: 10.1242/jcs.03023. [DOI] [PubMed] [Google Scholar]
- Cristea IM, Williams R, Chait BT, Rout MP. Fluorescent proteins as proteomic probes. Mol Cell Proteomics. 2005;4:1933–41. doi: 10.1074/mcp.M500227-MCP200. [DOI] [PubMed] [Google Scholar]
- DeGrasse JA, Chait BT, Field MC, Rout MP. High-yield isolation and subcellular proteomic characterization of nuclear and subnuclear structures from trypanosomes. Methods Mol Biol. 2008;463:77–92. doi: 10.1007/978-1-59745-406-3_6. [DOI] [PubMed] [Google Scholar]
- DeGrasse JA, Devos D. A functional proteomic study of the Trypanosoma brucei nuclear pore complex: an informatic strategy. Methods Mol Biol. 2010;673:231–8. doi: 10.1007/978-1-60761-842-3_15. [DOI] [PubMed] [Google Scholar]
- DeGrasse JA, DuBois KN, Devos D, Siegel TN, Sali A, Field MC, Rout MP, Chait BT. Evidence for a shared nuclear pore complex architecture that is conserved from the last common eukaryotic ancestor. Mol Cell Proteomics. 2009;8:2119–30. doi: 10.1074/mcp.M900038-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttagupta S, Gupta S, Gupta A. Euglenoid blooms in the floodplain wetlands of Barak Valley, Assam, North eastern India. J Environ Biol. 2004;25:369–73. [PubMed] [Google Scholar]
- Engstler M, Pfohl T, Herminghaus S, Boshart M, Wiegertjes G, Heddergott N, Overath P. Hydrodynamic flow-mediated protein sorting on the cell surface of trypanosomes. Cell. 2007;131:505–15. doi: 10.1016/j.cell.2007.08.046. [DOI] [PubMed] [Google Scholar]
- Engstler M, Thilo L, Weise F, Grünfelder CG, Schwarz H, Boshart M, Overath P. Kinetics of endocytosis and recycling of the GPI-anchored variant surface glycoprotein in Trypanosoma brucei. J Cell Sci. 2004;117:1105–15. doi: 10.1242/jcs.00938. [DOI] [PubMed] [Google Scholar]
- Field MC, Carrington M. The trypanosome flagellar pocket. Nat Rev Microbiol. 2009;7:775–86. doi: 10.1038/nrmicro2221. [DOI] [PubMed] [Google Scholar]
- Field MC, Gabernet-Castello C, Dacks JB. Reconstructing the evolution of the endocytic system: insights from genomics and molecular cell biology. Adv Exp Med Biol. 2007;607:84–96. doi: 10.1007/978-0-387-74021-8_7. [DOI] [PubMed] [Google Scholar]
- Field MC, Lumb JH, Adung’a VO, Jones NG, Engstler M. Macromolecular trafficking and immune evasion in African trypanosomes. Int Rev Cell Mol Biol. 2009;278:1–67. doi: 10.1016/S1937-6448(09)78001-3. [DOI] [PubMed] [Google Scholar]
- Gabernet–Castello C, Dacks JB, Field MC. The single ENTH–domain protein of trypanosomes; endocytic functions and evolutionary relationship with epsin. Traffic. 2009;10:894–911. doi: 10.1111/j.1600-0854.2009.00910.x. [DOI] [PubMed] [Google Scholar]
- Gabernet-Castello C, Dubois KN, Nimmo C, Field MC. Rab11 function in Trypanosoma brucei; identification of conserved and novel interaction partners. Eukaryot Cell. 2011;10:1082–1094. doi: 10.1128/EC.05098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünfelder CG, Engstler M, Weise F, Schwarz H, Stierhof YD, Morgan GW, Field MC, Overath P. Endocytosis of a glycosylphosphatidylinositol-anchored protein via clathrin-coated vesicles, sorting by default in endosomes, and exocytosis via RAB11-positive carriers. Mol Biol Cell. 2003;14:2029–40. doi: 10.1091/mbc.E02-10-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotez PJ, Gurwith M. Europe’s neglected infections of poverty. Int J Infect Dis. 2011 doi: 10.1016/j.ijid.2011.05.006. in press. [DOI] [PubMed] [Google Scholar]
- Kawahara T, Siegel TN, Ingram AK, Alsford S, Cross GA, Horn D. Two essential MYST-family proteins display distinct roles in histone H4K10 acetylation and telomeric silencing in trypanosomes. Mol Microbiol. 2008;69:1054–68. doi: 10.1111/j.1365-2958.2008.06346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev NG, Franklin JB, Carmi S, Shi H, Michaeli S, Tschudi C. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010;6:e1001090. doi: 10.1371/journal.ppat.1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz Ambrósio D, Lee JH, Panigrahi AK, Nguyen TN, Cicarelli RM, Günzl A. Spliceosomal proteomics in Trypanosoma brucei reveal new RNA splicing factors. Eukaryot Cell. 2009;8:990–1000. doi: 10.1128/EC.00075-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magez S, Radwanska M. African trypanosomiasis and antibodies: implications for vaccination, therapy and diagnosis. Future Microbiol. 2009;4:1075–87. doi: 10.2217/fmb.09.65. [DOI] [PubMed] [Google Scholar]
- Mans BJ, Anantharaman V, Aravind L, Koonin EV. Comparative genomics, evolution and origins of the nuclear envelope and nuclear pore complex. Cell Cycle. 2004;3:1612–37. doi: 10.4161/cc.3.12.1316. [DOI] [PubMed] [Google Scholar]
- Marchetti MA, Tschudi C, Kwon H, Wolin SL, Ullu E. Import of proteins into the trypanosome nucleus and their distribution at karyokinesis. J Cell Sci. 2000;113:899–906. doi: 10.1242/jcs.113.5.899. [DOI] [PubMed] [Google Scholar]
- Morgan GW, Allen CL, Jeffries TR, Hollinshead M, Field MC. Developmental and morphological regulation of clathrin-mediated endocytosis in Trypanosoma brucei. J Cell Sci. 2001;114:2605–15. doi: 10.1242/jcs.114.14.2605. [DOI] [PubMed] [Google Scholar]
- Morgan GW, Allen CL, Jeffries TR, Hollinshead M, Field MC. Developmental and morphological regulation of clathrin-mediated endocytosis in Trypanosoma brucei. J Cell Sci. 2001;114:2605–26015. doi: 10.1242/jcs.114.14.2605. [DOI] [PubMed] [Google Scholar]
- Morgan GW, Goulding D, Field MC. The single dynamin-like protein of Trypanosoma brucei regulates mitochondrial division and is not required for endocytosis. J Biol Chem. 2004;279:10692–701. doi: 10.1074/jbc.M312178200. [DOI] [PubMed] [Google Scholar]
- Natesan SK, Peacock L, Leung KF, Gibson W, Field MC. Evidence that low endocytic activity is not directly responsible for human serum resistance in the insect form of African trypanosomes. BMC Res Notes. 2010;(3):63. doi: 10.1186/1756-0500-3-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberholzer M, Langousis G, Nguyen HT, Saada EA, Shimogawa MM, Jonsson ZO, Nguyen SM, Wohlschlelgel JA, Hill KL. Independent analysis of the flagellum surface and matrix proteomes provides insight into flagellum signaling in mammalian-infectious Trypanosoma brucei. Mol Cell Proteomics. 2011 doi: 10.1074/mcp.M111.010538. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberholzer M, Morand S, Kunz S, Seebeck T. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol Biochem Parasitol. 2006;145:117–20. doi: 10.1016/j.molbiopara.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Oberholzer M, Langousis G, Nguyen HT, Saada EA, Shimogawa MM, Jonsson ZO, Nguyen SM, Wohlschlelgel JA, Hill KL. Independent analysis of the flagellum surface and matrix proteomes provides insight into flagellum signaling in mammalian-infectious Trypanosoma brucei. Mol Cell Proteomics. 2011 doi: 10.1074/mcp.M111.010538. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeffinger M, Wei KE, Rogers R, DeGrasse JA, Chait BT, Aitchison JD, Rout MP. Comprehensive analysis of diverse ribonucleoprotein complexes. Nat Methods. 2007 Nov;4(11):951–6. doi: 10.1038/nmeth1101. [DOI] [PubMed] [Google Scholar]
- Pal A, Hall BS, Jeffries TR, Field MC. Rab5 and Rab11 mediate transferrin and anti-variant surface glycoprotein antibody recycling in Trypanosoma brucei. Biochem J. 2003;374:443–51. doi: 10.1042/BJ20030469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Ernst NL, Domingo GJ, Fleck M, Salavati R, Stuart KD. Compositionally and functionally distinct editosomes in Trypanosoma brucei. RNA. 2006;12:1038–49. doi: 10.1261/rna.45506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Ogata Y, Zíková A, Anupama A, Dalley RA, Acestor N, Myler PJ, Stuart KD. A comprehensive analysis of Trypanosoma brucei mitochondrial proteome. Proteomics. 2009;9:434–50. doi: 10.1002/pmic.200800477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M, Worthey EA, Ward PN, Mottram JC. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics. 2005;(6):127. doi: 10.1186/1471-2164-6-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothnie A, Clarke AR, Kuzmic P, Cameron A, Smith CJ. A sequential mechanism for clathrin cage disassembly by 70-kDa heat-shock cognate protein (Hsc70) and auxilin. Proc Natl Acad Sci U S A. 2011;108:6927–32. doi: 10.1073/pnas.1018845108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, Field MC. Isolation and characterization of subnuclear compartments from Trypanosoma brucei. Identification of a major repetitive nuclear lamina component. J Biol Chem. 2001;276:38261–71. doi: 10.1074/jbc.M104024200. [DOI] [PubMed] [Google Scholar]
- Simarro PP, Cecchi G, Paone M, Franco JR, Diarra A, Ruiz JA, Fèvre EM, Courtin F, Mattioli RC, Jannin JG. The Atlas of human African trypanosomiasis: a contribution to global mapping of neglected tropical diseases. Int J Health Geogr. 2010;(9):57. doi: 10.1186/1476-072X-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstead B, Carrington JT, Gluenz E, Gull K. The expanded Kinesin-13 repertoire of trypanosomes contains only one mitotic Kinesin indicating multiple extra-nuclear roles. PLoS One. 2010;5:e15020. doi: 10.1371/journal.pone.0015020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson SR, Kelly JM. Trypanocidal drugs: mechanisms, resistance and new targets. Expert Rev Mol Med. 2009;11:e31. doi: 10.1017/S1462399409001252. [DOI] [PubMed] [Google Scholar]
- Will E, Gallwitz D. Biochemical characterization of Gyp6p, a Ypt/Rab-specific GTPase-activating protein from yeast. J Biol Chem. 2001;276:12135–9. doi: 10.1074/jbc.M011451200. [DOI] [PubMed] [Google Scholar]
- Wu YW, Tan KT, Waldmann H, Goody RS, Alexandrov K. Interaction analysis of prenylated Rab GTPase with Rab escort protein and GDP dissociation inhibitor explains the need for both regulators. Proc Natl Acad Sci U S A. 2007;104:12294–9. doi: 10.1073/pnas.0701817104. [DOI] [PMC free article] [PubMed] [Google Scholar]