

Abstract
In an effort to increase the stability and control the platinum reactivity of platinum-texaphyrin conjugates, two Pt(IV) conjugates (4 and 5, Scheme 1) were designed, synthesized, and studied for their ability to form DNA adducts. They were also tested for their anti-proliferative effects using wild-type and platinum-resistant human ovarian cancer cell lines (A2780 and 2780CP, respectively). In comparison to an analogous first generation Pt(II)-texaphyrin chimera (2, Scheme 1), conjugate 4 provided increased stability in aqueous environments.
Using a combination of 1H-NMR spectroscopy and FAAS (flameless atomic-absorption spectrometry), it was found that the Pt(IV) center within conjugate 4 undergoes photo-induced reduction to Pt(II) upon exposure to glass-filtered daylight, resulting in an entity that binds DNA in a controlled manner. Under conditions where the Pt(IV) complex is reduced to the corresponding Pt(II) species, conjugates 4 and 5 demonstrated potent anti-proliferative activity in both test ovarian cancer cell lines.
Keywords: Platinum (IV), Texaphyrin, conjugate, Prodrug, Photo-activation
Introduction
Currently, nearly 50% of all chemotherapeutic regimens given to cancer patients include a platinum drug.[1] Unfortunately, these drugs have a narrow spectrum of activity, which may reflect the diversity of cancerous disease. The three FDA-approved platinum(II) chemotherapeutics, cisplatin, carboplatin, and oxaliplatin, also show a lack of selectivity for neoplastic sites, an effect ascribed inter alia to undesirable interactions with biological nucleophiles.[1] Poor bio-localization is thought to underlie what are often dose-limiting side effects for patients. Another major problem is that tumors can acquire resistance to the FDA-approved Pt(II) drugs.[2] This is particular problematic in the most common regimens that require multiple treatments.
In order to overcome these limitations we recently adopted a strategy that consists of conjugating a platinum(II) entity to a class of tumor localizing agents known as texaphyrins. Certain gadolinium texaphyrins (e.g., motexafin gadolinium (MGd); cf. 1, Scheme 1) are known to enhance MRI signals and act as generators of reactive oxygen species (ROS) in the presence of biological reductants, such as sodium ascorbate or glutathione.[3] These features inspired the synthesis of Pt(II) texaphyrin conjugates. These first generation systems (e.g., 2; cisTEX; Scheme 1) were found to provide increased intracellular uptake and resulted in the formation of more Pt-DNA adducts than did analogous FDA approved Pt agents tested as controls.[4] While effective, it was observed that the Pt moieties present in the first generation system 2 were hydrolytically unstable in aqueous environments, resulting in slow, uncontrolled release of Pt(II).
Scheme 1.

Structures of compounds considered in this study.
In order to develop a tumor targeting Pt-texaphyrin system capable of controlled Pt release, we have targeted the synthesis of conjugates analogous to 2 but based on Pt(IV). It has been proposed that Pt(IV) is kinetically inert to substitution reactions with biomolecules, such as proteins and DNA, in addition to being relatively stable to hydrolysis.[5] These features militate against premature release in vitro or in vivo. Moreover, the specific rate of release can be tuned at least qualitatively by varying the nature of ligands (i.e., electron-withdrawing vs. electron-releasing).[6] Of particular interest to us is the finding that some Pt(IV) complexes are unstable when exposed to ambient light,[7] since this could provide an alternative means for inducing controlled Pt release. In view of these considerations, we considered it likely that a texaphyrin-Pt(IV) complex analogous to 2 would be inert as prepared but would then act to release a Pt(II) species capable of binding DNA upon photoirradiation or via the reduction of the Pt(IV) center. To test this hypothesis, complexes 4 and 5 (c.f. Scheme 1) were prepared. As detailed below, these conjugates were found to provide a source of active Pt(II) upon treatment with biological reductants or upon exposure to glass-filtered daylight. Evidence for this release came from chemical analyses of the daughter products, the observation of Pt(II)-DNA adducts by FAAS, and demonstrations of anti-proliferative activity in the wild type and cisplatin-resistant human ovarian cancer cell lines A2780 and 2780CP, respectively
Results and Discussion
The asymmetric platinum(IV) complex 3, a derivate of cisplatin originally reported by Lippard,[8] was chosen as the platinum precursor for conjugates 4 and 5. This precursor has the advantage that it contains a difunctional succinic acid moiety that can both serve as the axial ligand for the Pt(IV) center and allow facile conjugation to an amino-functionalized texaphyrin core. Moreover, the presence of an electron-donating hydroxy ligand in the other axial position of complex 3 was expected to make conjugates derived from it more stable than analogues containing carboxylato or halo ligands about the Pt(IV) center, which are known to undergo rapid hydrolysis under biologically relevant conditions.[9]
The synthesis of conjugates 4 and 5 is shown in Scheme 2. Briefly, the mono- and bis-amine derivatives of MGd 1[10] were subjected to carbodiimide coupling conditions in the presence of the Pt(IV)-containing precursor 3; after purification, this gave conjugates 4 and 5 in yields of 40% and 20%, respectively.
Scheme 2.

Synthesis of the platinum(IV)-texaphyrin conjugates 4 and 5. R1 = (OCH2CH2)3OCH3. Conditions: i = 4,4′-dimethoxytriphenylmethylchloride (DMT-Cl), diisopropylethylamine (DIPEA) in dichloromethane, 30%; ii = triphenylphosphine, phthalimide, diisopropylazocaboxylate in dichloromethane, methylamine in methanol/acetonitrile, 25%; iii = N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC•HCl), N-hydroxysuccinimide (NHS) and 3 in water. Yields were 40% (4) and 20% (5).
Conjugate 4 is highly soluble in water. Its stability towards hydrolysis could thus be evaluated using RP-HPLC. This was done by monitoring the decrease in the absorption intensity at 470 nm (Abs470), a peak corresponding to the starting material 4 (Scheme S2). On the basis of these studies, which were carried out in aqueous media at 310 K while minimizing exposure to light, it was concluded that conjugate 4 possesses greater hydrolytic stability (t1/2 > 3 days) than the first generation platinum(II) conjugate, cisTEX (2) (t1/2 = 12 h) when tested under the same conditions. We attribute this greater kinetic stability to the relative inertness of the platinum(IV) complex present in conjugate 4.
Studies were undertaken to investigate the photosensitivity of the Pt(IV) moiety within conjugate 4. Although still under debate,[1,11] it has been proposed that Pt(IV) complexes lose both axial ligands upon reduction. In the case of 4, loss of both axial ligands (i.e., the hydroxyl ligand and the texaphyrin-bearing succinate) would give rise to texaphyrin 6 and cisplatin. After exposure to glass-filtered daylight (see Experimental Section for details), the starting material is transformed (t1/2 = 5 h, ca. 100% after 48 h) to a new compound that on the basis of mass spectrometric and RP-HPLC analyses corresponds to what would be expected for the succinic acid-functionalized texaphyrin 6 (cf. Figure 2 and the Supplementary Information). This daughter compound (6) was independently synthesized, allowing for direct co-injection studies.
Figure 2.

(A) Schematic representation of the photo-activation of 4 followed by formation of a DNA-Pt adduct. Note: The star represents the texaphyrin moiety; (B) stability (RP-HPLC analysis) of 4 in PBS solution in the dark (black circles) or exposed to laboratory light (black squares), The designation 1 indicates the hydrolysis phase (absence of light), while the 2 indicates the phase corresponding to light exposure; (C) quantification by FAAS (Pt) and by nanodrop (DNA) of the number of Pt-DNA adducts formed after reaction of 4 with DNA in the dark (black circles) or exposed to natural light (black squares).
Texaphyrin 6 was also obtained when the Pt(IV) conjugate 4 was subject to reduction using the known biological reductants, sodium ascorbate (NaAsc) and glutathione (GSH). The other reduction product, namely cisplatin, was identified using RP-HPLC after being separated from 6 on a C18 RP HPLC column (Figure S11).
In an effort to understand further the chemistry of Pt(IV) as it bears on conjugates 4 and 5, 1H-NMR spectroscopy was used to probe the effect of light on the texaphyrin-free Pt(IV) complex 3. The choice of 3, rather than 4 or 5, was dictated by the paramagnetic nature of these latter species. The spectrum of 3 was recorded in D2O and is characterized by two triplets at 2.58 and 2.38 ppm corresponding to the four asymmetric methylene protons present in the axially coordinated succinate. In the dark, no change in the spectrum was observed, even after one week. This finding is in line with the comparatively high hydrolytic stability of 4 noted above. In contrast, when precursor 3 was exposed to glass-filtered daylight for 2 days, the two triplets originally present in the spectrum were seen to coalesce into a singlet at 2.39 ppm. This final spectrum corresponds to that of the free succinate anion in water. It was also possible to detect the released succinate anion by ESI-MS analysis (Figure S14). Taken in concert, these findings lead us to suggest that compound 3 is similar to 4 in that exposure to ambient light induces release of the axial succinate ligand. It also provides support for the conclusion that the presence of the texaphyrin localizing group is not necessary to trigger axial ligand release.
An analogous 1H-NMR spectral experiment using 3 in DMSO allowed the peaks corresponding to the coordinated NH3 protons to be monitored. After exposure of 3 to ambient light for 2 days, the initial signals at 5.8 ppm decrease in intensity, while new peaks between 4 and 5.3 ppm appear that are characteristic of a Pt(II) species. These new peaks are similar to those observed when cisplatin is solubilized in DMSO (Figure S15). Analysis of this solution by ESI-MS reveals signals corresponding to [PtII(NH3)2(Cl)(DMSO)]+ (Figure S16). All available data thus support the notion that upon exposure to light, complex 3 is reduced and both axial ligands are released.
To provide further support for the release of a Pt(II) species from 3, and by inference 4, guanosine 5′-monophosphate (5′-GMP) was used as a trapping agent for Pt(II). In the dark, no change was observed in the 1H-NMR spectrum (and RP-HPLC chromatogram) of 3 even after incubating for one-week in the presence of 5′-GMP. Nor were changes in the spectrum of 5′-GMP observed. We take this as evidence that this Pt(IV) complex does not bind to 5′-GMP. In contrast, after exposure to light for 2 days, a new peak is observed at 8.6 ppm (Figure 1, triangle). This peak appears at high frequency, as is typical for the H8 resonance of 5′-GMP in complexes where a diamagnetic cation is coordinated to N7 of 5′-GMP.[12] This new signal was comparable to the signal obtained when sodium ascorbate was added to a mixture of 3 + 5′-GMP or when cisplatin was added to 5′-GMP (Figure 1C).
Figure 1.
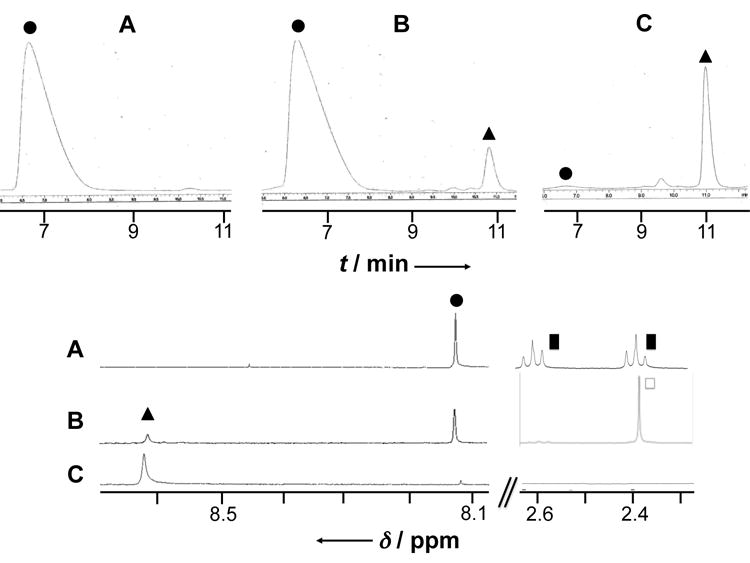
Partial RP-HPLC chromatograms (top) and 1H-NMR spectra (bottom, D2O, pH = 7, 300 K, 400 MHz) of complex 3 recorded in the presence of 5′-GMP (2 equiv.) before (A) and after being exposed to laboratory light for 2 days (B); (C) cisplatin incubated in presence of 5′-GMP (2 equiv.) for 10 h at 37°C; circle = free 5′-GMP; triangle = PtII-coordinated 5′-GMP; black square = PtIV-coordinated succinate anion; square = free succinate dianion.
The solutions used for the NMR spectral studies were also analyzed by RP-HPLC. As can by seen by inspection of the upper part of Figure 1, a new peak appeared after exposing a mixture 3 + 5′-GMP to ambient light for 2 days. This peak has the same retention time (10.9 min) as does the complex formed between cisplatin and 5′-GMP. This leads us to suggest that the same 5′-GMP - Pt(II), complex, namely the known adduct (5′-GMP)2-PtII-(NH3)2,[6,13] is formed upon light-induced reduction of 3.
The influence of light on the interaction between 4 and DNA was also investigated.[14] It was observed that when 4 is exposed to ambient light, 8.5 ± 1.1 times more DNA-Pt adducts are formed (average of 2 independent studies) than when 4 is kept in the dark (see Figure 2C). This result provides further support for the suggestion that the Pt(IV) center present in 4 has to be activated by reduction to Pt(II) prior to binding to DNA. It is thus consistent with the model studies using 3 and 5′-GMP noted above and is in line with recently published studies of Pt(IV) complexes.[15] Nevertheless, even under conditions of photoirradiation with laboratory light, conjugate 4 is much less reactive towards DNA than cisplatin. This finding is thought to reflect the slow light-induced reduction kinetics operative in the case of species such as 3 and 4 and the corresponding release of only a few active platinum(II) complexes at a time.
The anti-proliferative effects of conjugates 4 and 5 were assessed with platinum sensitive human ovarian A2780 cells and the isogenic cisplatin resistant 2780CP cell line. These experiments were carried out in the dark at 37 °C for conjugate 4. Colorimetric cell proliferation assay results (Table 1) lead us to conclude that these new platinum(IV) conjugates are more efficient in inhibiting cancer cell growth in both cell lines than the first generation conjugate cisTEX (2). This enhancement in efficacy is fully consistent with the suggestion that the greater hydrolytic stability of 4 and 5 compared to 2 serves to increase the effective concentration of Pt(II) at locales where it is most effective. The potency of conjugate 5 is roughly equal to that of conjugate 4 on a per platinum basis.
Table 1.
Half maximal inhibitory concentrations (IC50) (micromolar) determined for the Pt(IV)-TEX conjugates 4 and 5, and the texaphyrin-free platinum(IV) precursor complex 3. Also given are data for the platinum(II) complexes cisplatin and cisTEX (2).
| Compounds | IC50 2780CP | IC50 A2780 | Resistance factor[b] |
|---|---|---|---|
| Cisplatin[a] | 7.3 (0.2) | 0.33 (0.02) | 18.25 |
| 2[a] | 17.0 (1.5) | 1.63 (0.20) | 10.42 |
| 3 | 26.88 (2.04) | 6.31 (0.38) | 4.26 |
| 4 | 10.66 (0.63) | 1.28 (0.12) | 8.32 |
| 5 | 4.55 (0.29) | 0.67 (0.06) | 6.79 |
See reference 4b.
Resistance factor = IC50(2780CP)/IC50(A2780)
Conclusion
In conclusion, two new platinum(IV)-texaphyrin conjugates (4 and 5) were synthesized and characterized. The presence of a Pt(IV) center within 4 and 5 provides for increased hydrolytic stability relative to the first generation Pt(II) system 2. On the other hand, the present Pt(IV) conjugates are able to release Pt(II) in a controlled fashion upon exposure to laboratory light or a reducing environment as demonstrated in case of 4 and the control system 3. Both conjugates 4 and 5 demonstrate good anti-proliferative activity in vitro in both wild type and cisplatin-resistant ovarian cancer cell lines. In the case of the conjugate 4 with a single Pt(IV) center, a comparison with the first generation mono-Pt(II) conjugate 2 reveals improvements in the IC50 values for both cell lines, as well as in the resistance factor. In order to check that the gain in stability towards hydrolysis and the ability of conjugate 4 to undergo photoinduced reduction will translate into improved anticancer activity, in vivo studies are necessary. Nevertheless, the present findings lead us to predict that these and other conjugates that combine the targeting features of the texaphyrins with the redox- and photo-release capabilities of appropriately chosen Pt(IV) complexes may have a role to play in treating neoplastic diseases where the utility of current platinum drugs is clinically limited.
Experimental Section
General procedure
Starting materials were purchased from Fisher Scientific or Sigma Aldrich and used without further purification unless otherwise specified. Solvents were purified using a solvent purifier system (Vacuum Atmospheres). Dichloromethane was freshly distilled after being dried over CaH2 under argon. Reaction progress was monitored with Thin Layer Chromatography (TLC) (TLC silica gel 60 F254, Silicycle® UltraPure Silica gels). Texaphyrins and platinum (IV)-texaphyrin conjugates were purified on RP-tC18 SPE (Waters Sep-Pak, waters®) columns containing 10 g of C-18 using increasing gradient of acetonitrile in either 0.1 M ammonium acetate/1% acetic acid aqueous solution or 0.1 M potassium nitrate aqueous solution, depending on which anion (AcO- or NO3-) was desired as ligands on the gadolinium(III) center. HPLC analyses were performed on a Shimadzu Analytical/Preparative HPLC system equipped with PDA detector and a C18 Acclaim™ 3 μm, 120 Å, 2.1 × 150 mm column (Thermo Scientific). An aqueous (0.1% acetic acid)/acetonitrile (0.1% acetic acid) gradient (30-99% acetonitrile over 20 minutes, 0.3 mL/min) was used for the analysis of all texaphyrin-containing compounds (detection at 470 and 740 nm). For analysis of 5′-GMP and PtII(5′-GMP)2(NH3)2, (0.1% acetic acid)/methanol (0.1% acetic acid) gradient (5-55% methanol over 20 minutes, 0.2 mL/min) was used and the detection was done at 254 nm. Mass spectrometric analyses were carried out in the University of Texas at Austin Mass Spectrometry Facility. Low-resolution and high-resolution electrospray mass spectrometric (ESI-MS) analyses were carried out using a Thermo Finnigan LTQ instrument and a Qq-FTICR (7 Telsa) instrument, respectively. Elemental analyses were performed by Atlantic Microlabs Inc. 1H NMR spectra were recorded using a Varian 400 MHz instrument.
General procedure for EDC•HCl coupling
EDC•HCl (40 mg, 0.21 mmol) and N-hydroxysuccinimide NHS (24 mg, 0.21 mmol) were dissolved in HPLC submicron filtered grade water (4 mL). Platinum complex 3 (22 mg, 50 μmol) as a suspension in water (2 mL) was added to the mixture (termed “EDC•HCl + NHS”) and left stirring for 30 minutes. Precursor 1ODMTNH2 (60 mg, 42 μmol, RP-HLPC RT = 11.4 min) in CH3CN (5 mL) was added dropwise to the previous solution and the reaction mixture was kept in the dark for 20 h at 40°C. The progress of the reaction was monitored by HPLC (a new peak is formed that is characterized by a RT = 9 min). KNO3 (50 mL of a 0.1 M aqueous solution) was added and the resulting solution was loaded on a C18 column and subject to elution with increasing gradient of acetonitrile in 0.1 M aqueous KNO3. The isolated fraction was loaded on a new C18 column, desalted with water and eluted with pure methanol. The solvent was removed under vacuum to give the product 4 as a dark green powder. (26.4 mg, 40%). For compound 5, the same procedure was followed (see SI for details and characterization data).
Light exposure
For the light-induced release studies, aqueous solutions (of 3 or 4) contained in glass vials were exposed to sunlight in the laboratory behind a window (Viracon®, GL-22), with the following transmittances properties: visible light = 38%, UV light (from 300 to 380 nm) = 12%. A low vapor pressure mercury lamp was also used. In this latter instance, compound 3 is reduced after 15 minutes exposure whereas conjugate 4 required a 15 hour exposure time to be similarly reduced. This could be explained by the fact that the aromatic, paramagnetic texaphyrin chromophore absorbs light well and the resulting excited state undergoes rapid non-radiative decay. These pathways compete effectively with those that would otherwise serve to reduce the Pt(IV) center.
Platinum-DNA binding
Salmon sperm DNA (1.125 mL of 500 μg DNA/mL in Tris-EDTA buffer) was incubated at 37 °C in the dark or exposed to light with platinum complexes (in solution in water; approximately 1 platinum/75 nucleotides as the final ratio). 200 μL aliquots were removed and added immediately to 40 μl of a 10 M aqueous ammonium acetate solution. DNA in samples was precipitated by adding 0.8 mL of absolute ethanol prechilled at -20°C. The samples were left in ice for 1 h and centrifuged at 14000 rpm for 4 minutes. The supernatant was removed carefully and small pellets were dissolved in 50 μl of Tris-EDTA buffer overnight at room temperature. Platinum content was determined by FAAS (model AA300/GTA-96; Varian Instruments, Victoria, Australia) using conditions described previously.[16] Samples were diluted with HCl when the initial Pt concentration was too high. DNA concentrations were determined using a Nanodrop ND-1000 spectrophotometer.
Cell culture
The A2780 line was established from a patient's biopsy prior to initiation of any chemotherapeutic regimen.[17] The resistant cell line, 2780CP, used in this study, was established from A2780 cells, as described previously.[18] The two cell lines have wild-type p53 genotype and/or function. Cells were grown in RPMI containing 10% fetal calf serum and antibiotics (100 μg/mL streptomycin and 100 U/mL penicillin).
Viability tests
The proliferation of exponential phase cultures of A2780 and 2780CP cells was assessed by tetrazolium dye reduction.[19] In brief, tumor cells were seeded in 96-well microliter plates at 700 (A2780) and 1000 (2780CP) cells/well, respectively, and allowed to adhere overnight in RPMI 1640 medium supplemented with 2 mM L-glutamine, 10% heat inactivated fetal bovine serum, antibiotics (200 U/cm3 penicillin and 200 μg/cm3 streptomycin). After 24h, drug was added. After a total incubation time of 6 days at 37°C, 50 μL of a stock solution at 3 mg/ml of the tetrazolium dye, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma Chemical) were added to each well, the plates incubated at 37°C for 4 hours, whereupon the medium was removed, the formazan product dissolved in DMSO (50-100 μL) and absorbance values at 570 nm was measured using a microplate reader (Molecular Devices, Sunnyvale, CA). Absorbance values were corrected for background and then normalized to wells containing untreated cells to allow plate-to-plate comparisons. The growth inhibition data were fitted to a sigmoidal dose-response curve to generate IC50, which is the drug concentration inhibiting cell growth by 50%. The IC50 is presented as mean ± standard deviation.
Supplementary Material
Acknowledgments
This work was supported by the U.S. National Institutes of Health (grants CA160687 and CA 68687 to ZHS and JLS). GT also acknowledges Dr. Cheulhee Jung and Prof. Andrew Ellington for nanodrop experiments.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
Contributor Information
Prof. Zahid H. Siddik, Email: zsiddik@mdanderson.org.
Prof. Jonathan L. Sessler, Email: sessler@mail.utexas.edu.
References
- 1.a) Wexselblatt E, Gibson D. J Inorg Biochem. 2012;117:220–229. doi: 10.1016/j.jinorgbio.2012.06.013. [DOI] [PubMed] [Google Scholar]; b) Wexselblatt E, Yavin E, Gibson D. Inorg Chim Acta. 2012;393:75–83. [Google Scholar]
- 2.a) Kelland L. Nat Rev cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]; b) Rabik CA, Dolan ME. Cancer Treat Rev. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Miller RA, Woodburn K, Fan Q, Renschler M, Sessler JL, Koutcher JA. Int J Radiat Oncol Biol Phys. 1999;45:981–989. doi: 10.1016/s0360-3016(99)00274-6. [DOI] [PubMed] [Google Scholar]; b) Magda D, Gerasimchuk N, Lecane P, Miller RA, Biaglow JE, Sessler JL. Chem Commun. 2002:2730–2731. doi: 10.1039/b208760j. [DOI] [PubMed] [Google Scholar]
- 4.a) Arambula JF, Sessler JL, Siddik ZH. Bioorg Med Chem Lett. 2011;21:1701–1705. doi: 10.1016/j.bmcl.2011.01.092. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Arambula JF, Sessler JL, Siddik ZH. Med Chem Commun. 2012;3:1275–1281. doi: 10.1039/C2MD20206A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hall MD, Hambley TW. Coord Chem Rev. 2002;232:49–67. [Google Scholar]
- 6.Choi S, Filotto C, Bisanzo M, Delaney S, Lagasee D, Whitworth JL, Jusko A, Li C, Wood NA, Willingham J, Schwenker A, Spaulding K. Inorg Chem. 1998;37:2500–2504. [Google Scholar]
- 7.a) Kratochwil NA, Parkinson JA, Bednarski PJ, Sadler PJ. Angew Chem Int Ed. 1999;38:1460–1463. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1460::AID-ANIE1460>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]; b) Farrer NicolaJ, Woods JulieA, Salassa Luca, Zhao Yao, Robinson KimS, Clarkson Guy, Mackay FionaS, Sadler PeterJ. Angew Chem Int Ed. 2010;49:8905–8908. doi: 10.1002/anie.201003399. [DOI] [PubMed] [Google Scholar]; c) Zhao Y, Woods JA, Farrer NJ, Robinson KS, Pracharova J, Kasparkova J, Novakova O, Li H, Salassa L, Pizarro AM, Clarkson GJ, Song L, Brabec V, Sadler PJ. Chem Eur J. 2013;19:9578–9591. doi: 10.1002/chem.201300374. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Petruzzella E, Margiotta N, Ravera MM, Natile G. Inorg Chem. 2013;52:2393–2403. doi: 10.1021/ic302100x. [DOI] [PubMed] [Google Scholar]
- 8.a) Feazell RP, Nakayama-Ratchford N, Dai H, Lippard SJ. J Am Chem Soc. 2007;129:8438–8439. doi: 10.1021/ja073231f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiao H, Qi R, Liu S, Hu X, Duan T, Zheng Y, Huang Y, Jing X. Biomaterials. 2011;32:7732–7739. doi: 10.1016/j.biomaterials.2011.06.072. [DOI] [PubMed] [Google Scholar]; c) Xiao H, Yan L, Zhang Y, Qi R, Li W, Wang R, Liu S, Huang Y, Li Y, Jing X. Chem Commun. 2012;48:10730–10732. doi: 10.1039/c2cc34297a. [DOI] [PubMed] [Google Scholar]; d) Dhar S, Daniel WL, Giljohann DA, Mirkin CA, Lippard SJ. J Am Chem Soc. 2009;131:14652–14653. doi: 10.1021/ja9071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wexselblatt E, Yavin E, Gibson D. Angew Chem Int Ed. 2013;52:6059–6062. doi: 10.1002/anie.201300640. [DOI] [PubMed] [Google Scholar]
- 10.Wei WH, Fountain M, Magda D, Wang Z, Lecane P, Mesfin M, Miles D, Sessler JL. Org Biomol Chem. 2005;3:3290–3296. doi: 10.1039/b503664j. [DOI] [PubMed] [Google Scholar]
- 11.a) Sinisi M, Intini FP, Natile G. Inorg Chem. 2012;51:9694–9704. doi: 10.1021/ic300957v. [DOI] [PubMed] [Google Scholar]; b) Nemirovski A, Vinograd I, Takrouri K, Mijovilovich A, Rompel A, Gibson D. Chem Commun. 2010;46:1842–1844. doi: 10.1039/b925721g. [DOI] [PubMed] [Google Scholar]
- 12.a) Berners-Price SJ, Ranford JD, Sadler PJ. Inorg Chem. 1994;33:5842–5846. [Google Scholar]; b) Berners-Price SJ, Frey U, Ranford JD, Sadler PJ. J Am Chem Soc. 1993;115:8649–8659. [Google Scholar]
- 13.a) Van der Veer JL, Peters AR, Reedijk J. J Inorg Biochem. 1986;26 doi: 10.1016/0162-0134(86)80006-x. [DOI] [PubMed] [Google Scholar]; b) Roat RM, Reedijk J. J Inorg Bioch. 1993;52:263–274. [Google Scholar]; c) Choi S, Mahalingaiah S, Delaney S, Neale NR, Masood S. Inorg Chem. 1999;38:1800–1805. doi: 10.1021/ic9809815. [DOI] [PubMed] [Google Scholar]; d) Zöllner P, Zenker A, Galanski M, Keppler BK, Lindner W. J Mass Spectrom. 2001;36:742–753. doi: 10.1002/jms.178. [DOI] [PubMed] [Google Scholar]
- 14.Shi Y, Liu S, Kerwood DJ, Goodisman J, Dabrowiak JC. J Inorg Biochem. 2012;107:6–14. doi: 10.1016/j.jinorgbio.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 15.Montagner D, Yap SQ, Ang WH. Angew Chem Int Ed. 2013;52:11785–11789. doi: 10.1002/anie.201305734. [DOI] [PubMed] [Google Scholar]
- 16.Siddik ZH, Boxall FE, Harrap KR. Anal Biochem. 1987;163:21–26. doi: 10.1016/0003-2697(87)90087-x. [DOI] [PubMed] [Google Scholar]; Siddik ZH, Newman RA. Anal Biochem. 1988;172:190–196. doi: 10.1016/0003-2697(88)90431-9. [DOI] [PubMed] [Google Scholar]
- 17.Godwin AK, Meister A, O'Dwyer PJ, Huang CS, Hamilton TC, Anderson ME. Proc Natl Acad Sci USA. 1992;89:3070–3074. doi: 10.1073/pnas.89.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siddik ZH, Mims B, Lozano G, Thai G. Cancer Res. 1998;58:698–703. [PubMed] [Google Scholar]
- 19.Mosmann T. J Immunol Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.