

Abstract
In this study, we present a remarkable clonal cell line, 32080, derived from a CD2-Lmo2 transgenic T-cell leukemia with differentiation arrest at the transition from the intermediate single positive (ISP) to double positive (DP) stages of T-cell development. 32080 cells had a striking variegated pattern in CD4 expression. There was cell-to-cell variability with some cells expressing no CD4 and others expressing high CD4. The two populations were isogenic and yet differed in their rates of apoptosis and sensitivity to glucocorticoid. We sorted the 32080 line for CD4 positive or negative cells and observed them in culture. After one week, both sorted populations showed variegated CD4 expression like the parental line, showing that the two populations could interconvert. We determined that cell replication was necessary to transit from CD4+ to CD4- and CD4- to CD4+. Lmo2 knockdown decreased CD4 expression, while inhibition of intracellular Notch1 or HDAC activity, induced CD4 expression. Enforced expression of Runx1 repressed CD4 expression. We analyzed the CD4 locus by H3 chromatin immunoprecipitation and found silencing marks in the CD4- cells, and activating marks in the CD4+ population. The 32080 cell line is a striking model of ISP to DP T-cell plasticity and invokes a novel mechanism for Lmo2's oncogenic functions.
Introduction
The LIM domain Only-2 (LMO2) oncogene is deregulated in the majority of human T-cell acute lymphoblastic leukemias (T-ALL). LMO2 was also the target of frequent integration by replication-defective gene therapy vectors used for treatment of X-linked severe combined immunodeficiency and Wiskott-Aldrich syndrome (1-3). In these cases, the integrations occurred in transduced hematopoietic stem and progenitor cells, but only T-cell progenitors were clonally expanded (2). LMO2 induced T-ALL with cooperativity from oncogenic events such as chromosomal rearrangements or the transgenes themselves (4, 5).
Multiple LMO paralogs have been causally implicated in human cancers (6) but Lmo2 is the best characterized member that has been extensively studied in mouse models where it is a master regulator of hematopoiesis. Lmo2 knockout mice die in utero at E9.5 due to absent erythropoiesis(7) and Lmo2-/- ES cells do not contribute to hematopoietic tissues postnatally in chimeric blastocysts(8). Additionally, Lmo2 is not required for T-cell or B-cell development (9). The Lmo2 protein has two Zinc-coordinating LIM domains that are responsible for protein-protein interactions. These domains are responsible for binding to class II basic helix-loop-helix proteins, Tal1 or Lyl1, and to GATA factors 1-3, and to LIM domain binding 1 (Ldb1) protein. Interestingly, the knockout mice for these factors have remarkably similar phenotypes, affecting primitive and adult hematopoiesis (10-14). Thus, Lmo2 and its associated macromolecular complex are critical for the specification of primitive and adult hematopoietic stem cells.
Importantly, Lmo2's stem cell function may also play a role in the pathogenesis of T-ALL. Recent studies on T-cell progenitors in two independently constructed CD2-Lmo2 transgenic mouse models demonstrated differentiation arrest, increased self-renewal, and an HSC-like transcriptional signature preceding overt leukemia(15, 16). Several groups have shown that enforced expression of Lmo2 induces a specific block in the differentiation of T-cell progenitors. Early T-cell differentiation is divided into 5 stages prior to the expression of CD4 and CD8 co-receptors which occurs at the double positive (DP) stage. Cells newly migrated from the bone marrow to the thymus are called Early T-cell progenitors (ETP) which transit through the thymus from double negative stages, DN2-DN4, with an intermediate single positive (CD8+, ISP) stage prior to the DP (CD4+CD8+) stage (17, 18). Lmo2 is expressed at high levels in hematopoietic stem cells, multipotent progenitor cells, and in ETPs, but is downregulated at the DN2 stage and not expressed in subsequent T-cell progenitor cells or mature T cells (19, 20). Lmo2 overexpression causes a specific block at the DN3 stage which is also the point of beta selection, where T-cell progenitors with productively rearranged T-cell receptors proliferate and are blocked from apoptosis (15, 16). Beta selection is not required for Lmo2 to induce leukemia since Lmo2 overexpression in Rag1-/- mice induces T-ALL with the same penetrance and latency as Rag1+/+(21).
Despite the DN3 differentiation block, T-ALLs caused by Lmo2 overexpression express CD4 and CD8 suggesting that they can originate from various stages of T-cell differentiation (4, 22, 23). Immunophenotypic heterogeneity was observed in primary human T-ALLs engrafted in immunodeficient mice (24, 25). One major question in these studies was whether genetic (clonal) heterogeneity could account for differences in immunophenotype, leukemia initiation, and even drug sensitivity (26). To address this and to explore the relationship between T-cell differentiation and Lmo2 oncogene function, we established clonal cell lines from four independent T-ALLs derived from CD2-Lmo2 transgenic mice representing various stages of T-cell differentiation, DN, DP, and ISP. We found at least 2 clonal lines with a remarkable variegated pattern in CD4 expression. In this study, we present analyses on one of these lines, 32080, which models the ISP to DP transition. Most remarkably, the CD4- and CD4+ populations of this line interconvert and these two populations are biologically disparate in their rates of apoptosis and sensitivity to glucocorticoid. The 32080 cell line is a striking model of ISP to DP T-cell plasticity in T-ALL and invokes a novel mechanism for Lmo2's oncogenic function.
Materials and Methods
Mice
B6.CD2-Lmo2 transgenic mice develop T-ALL with 100% penetrance and have been previously described (16, 27). NSG and nude mice were purchased from Jackson Laboratories (Bar Harbor, Maine) and were bred and maintained at Vanderbilt in SPF facilities in accordance with protocols approved by the Institutional Animal Care and Use Committee.
Cell culture, plasmids and transfection
Single-cell suspensions of mononuclear cells were prepared from T-ALL lymph nodes or thymi harvested from B6.CD2-Lmo2 mice according to standard protocols. Cells were maintained in Iscove's Modified Dulbecco's Medium (Gibco Cat# 12200-028) at 37°C with 5% CO2 at high density until they could be serially passaged without loss of growth. Cell lines were confirmed if they could be passaged serially for 1 year. pcDNA3.1-Runx1 was kindly provided by Dr. Scott Hiebert. CD4 gene reporter plasmids have been previously described and were kindly provided by Drs. Marc Ehlers (University of Lübeck, Lübeck, Germany) and Remy Bosselut (NIH, Bethesda, Maryland) (28). The GIPZ lentiviral shRNAmir-GFP V2HS-62634 (LMO2 shRNA) showed the most consistent knockdown but multiple shRNAs were tested (see Figure S1), all purchased from Open Biosystems (Cat# RHS1764-9099730). Cells were transfected with 6 μg CD4 reporter constructs and then selected in G418-containing medium for 10-14 days followed by single cell cloning. For knockdowns, 6 μg pGIPZ lentiviral shRNAmir-GFP V2HS-62634 (LMO2 shRNA), was transfected into 32080 cells with Lipofectamine 2000 per manufacturer's instructions (Invitrogen). Cells transfected with knockdown constructs were selected in 1 μg/ml puromycin-containing medium for 5 days prior to analysis. Dexamethasone (Sigma) experiments were performed with ethanol as control. Cells with trypan blue exclusion were counted by hemocytometer. Curve fitting and dose-response relationships were generated with GraphPad Prism.
FISH
We probed the murine CD4 gene with BAC 121J20 and the Lmo2 transgene by labeled plasmid pCD2-Lmo2 (27). We labeled these DNA by nick translation using the Vysis kit (Cat # 32-801300). The hybridization procedure was done as previously described(27).
Chromatin Immunoprecipitation
Cells were fixed with formaldehyde for 10 minutes at 37°C before the reaction was stopped with 0.12M glycine and washed with PBS. Cells were resuspended and incubated on ice for 10 minutes in lysis buffer (50mM HEPES (pH 7.9), 140mM NaCl, 1mM EDTA, 1% Triton-X-100, 0.1% Na-deoxycholate) containing protease inhibitors. Lysates were then sonicated to ∼500bp and chromatin pre-cleared, followed by immunoprecipitation with antibodies and 50ul Protein A Agarose. The beads were washed as previsously described (29). DNA was reverse cross-linked and cleaned with the Illustra GFX PCR columns and gel band purification kit (GE Healthcare Cat# 28-9034-71). Samples were used for quantitative PCR on the MyIQ thermocycler (Biorad).
Gene expression analysis
Total RNA was purified by TRIzol (Invitrogen Life sciences) per manufacturer's instructions. First strand cDNA was synthesized using olgo-dT primer and Superscript II transcriptase enzyme (Invitrogen). Primers used for RT-PCR for CD4, CD8, Lmo2, and other genes are listed in the supplemental data file. Cell lines 03027 and 32080 were also analyzed by RNA-seq on the Illumina HiSeq as previously described(16). Quantitative ChIP analysis was performed on Biorad's MyIQ machines with Sybr green with primers listed in the supplemental data file (Biorad). T-cell receptor Jβ2 rearrangements were detected by PCR on genomic DNA using primers and conditions as previously described (29).
Cell Cycle Analysis
BrdU incorporation was analyzed per manufacturer's instructions (BD Biosciences). For propidium iodide (PI) incorporation cells were fixed in 70% EtOH, pelleted and resuspended in 50 μg/ml PI, 0.1mg/ml RNase A and 0.05% Triton X-100 and incubated for 40 minutes at 37C. Cells were then washed with 3mL PBS and analyzed by flow cytometry.
Flow cytometry and cell sorting
Antibodies for staining and flow cytometry are listed in the supplemental data file. FACS assays were performed by a BD FACSAria and analyzed with CellQuest (BD Biosciences) and Flojo (Tree Star). Cells were sorted for CD4 expression by flow or by using Dynal magnetic separation beads (Invitrogen) per manufacturer's instructions using unconjugated anti-mouse CD4 (BD Pharmingen 560468 or 553727).
Results
CD4 expression is variegated in Lmo2-induced T-cell leukemia
Transgenic B6.CD2-Lmo2 mice express Lmo2 cDNA from the T-cell specific CD2 promoter/enhancer and develop T-cell acute lymphoblastic leukemias (T-ALL) that originate from T-cell progenitors at diverse developmental stages (16). We established 5 stable cell lines in vitro from transgenic T-ALLs and analyzed them for cell surface expression of CD4 and CD8 antigens (Figure 1). All the lines overexpressed Lmo2 protein and had clonal rearrangement of the T-cell receptor (Tcr) Jβ2 (Figure 1C). We observed cell lines that correlated with normal developmental counterparts such as double negative cells (03027 line, CD4-CD8-, DN), intermediate single positive cells (03007 line, CD8+, ISP), and double positive cells (03020 line, CD4+CD8+, DP). Two cell lines did not fit this pattern and instead showed marked variability in cell surface expression of CD4 despite multiple rounds of singe cell cloning and staining with various anti-CD4 antibodies. The 32080 line was striking in that CD8 expression was uniform, however, CD4 expression ranged continuously from high to negative expression (Figure S4). We screened 9 human LMO2-overexpressing T-ALLs and observed 2 with a pattern of CD4 expression similar to the 32080 line (one shown in Figure 1B). The 32080 line's cell-to-cell variability in CD4 expression was reminiscent of variegated expression resulting from position effects of transgene integrations (30). Thus, we analyzed the 32080 line by FISH using probes against CD2-Lmo2 and the endogenous CD4 gene. As shown in Figure 1D, the probes did not co-localize, ruling out position effect from the CD2-Lmo2 transgene. The 32080 cell line had a clonal rearrangement of T-cell receptor Jβ2, expressed CD24+ (heat stable antigen, HSA) but not CD3- or CD5-. Tcrβ was expressed in low amounts (32% of total) but consistently more in CD4+ than CD4- cells (Figure 1E-F). Thus, the 32080 CD4- cells resembled the ISP stage and the CD4+ population resembled the DP stage of normal T-cell development.
Figure 1. Lmo2 induces T-cell leukemia with diverse patterns of CD4 and CD8 expression and variegation in CD4.
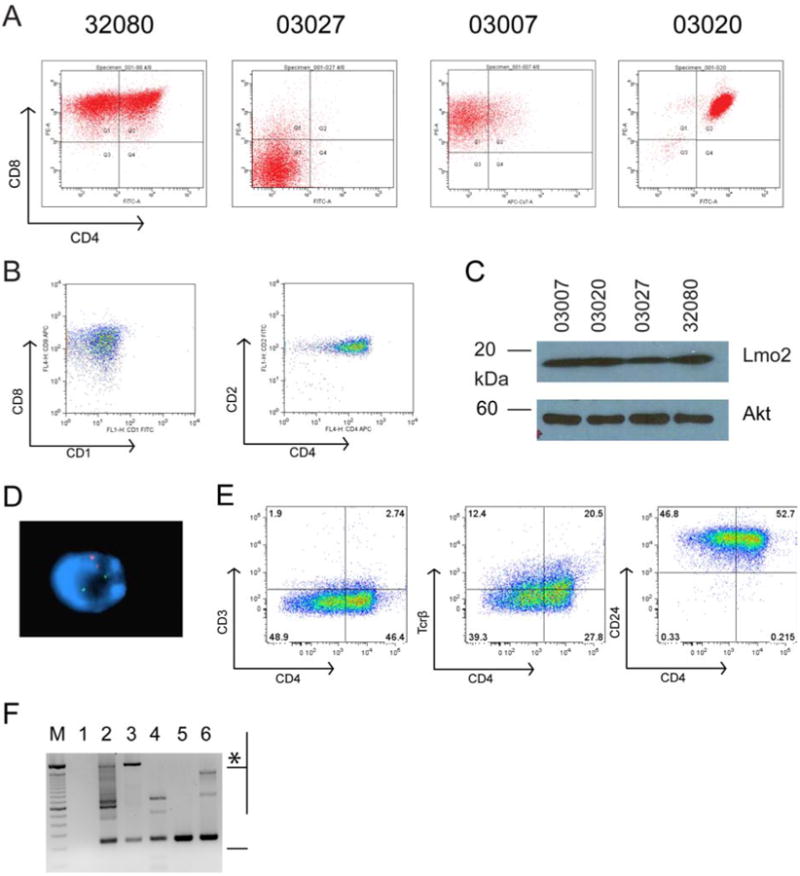
Single cell suspensions of T-ALLs from CD2-Lmo2 transgenic mice were cultured in vitro and single cell cloned by two rounds of limiting dilution. A, shows FACS analysis of CD4 and CD8 antigen expression on cell lines 32080, 03027, 03007, and 03020. B, shows FACS plot of a human T-ALL sample that overexpresses LMO2. Left panel shows analysis of CD8 and CD1 and right panel shows CD2 and CD4. C, shows SDS-PAGE followed by Western blot analysis for Lmo2 protein and Akt protein as a loading control for the murine cell lines shown in A with equivalent protein from whole cell lysates loaded. D, shows a representative 32080 interphase cell analyzed by FISH for the murine CD4 gene (red) and the CD2-Lmo2 transgene (green). E, FACS plots of 32080 cells stained for CD4 and CD3, Tcrβ, and CD24 (HSA). F, agarose gel of PCR products on genomic DNA from the cell lines using primers flanking Dβ and Jβ2. M designates a 100 bp marker; top intense band is 2 kb. Lane 1, H2O; lane 2, thymic genomic DNA from WT B6 mouse; lane 3, tail genomic DNA showing the 2 kb germline band (asterisk) with some rearranged Jβ2 probably from blood contamination; lane 4, line 03007; lane 5, line 03020; and lane 6, line 32080. Bracket designates products with rearranged Jβ2.
CD4+ and CD4- populations differ in apoptosis rates but not replication or leukemia initiation
Murine models of Lmo2-induced T-ALL and human T-ALL have demonstrated immunophenotypic and biological heterogeneity, observations that support the presence of leukemic stem cells in T-ALL (23, 25). We analyzed the 32080 line to see whether CD4- and CD4+ cells were similar in their rate of apoptosis, growth, and leukemia initiation. 32080 cells were stained for Annexin V and propidium iodide (PI) and gated on CD4+ or CD4- cells under normal culture conditions without the addition of any drugs; we also stained for CD8 which stained homogeneously positive. On average, 27% of CD4+ cells were undergoing cell death compared with 6.3% of the CD4- fraction, a statistically significant difference (Figure 2B, P=.0009). The cell death was apoptotic as confirmed by intracellular caspase 3 staining (data not shown). The 32080 cells were treated with various concentrations of dexamethasone and stained for Annexin V, PI, CD4, and CD8. The CD4+ cells had a higher baseline rate of apoptosis compared to CD4- cells (30% v. 8%, Figure 2A) and also showed increased sensitivity to dexamethasone (Figure 2C, IC50=7.2×10-9 M for CD4+ IC50=3.2×10-8 M for CD4-, P<.0001). We flow sorted the CD4+ and CD4- cells into separate cultures and observed similar cycling fractions and Sphase transit by bromodeoxyuridine (BrdU) pulse-chase analysis (data not shown). To test leukemia-initiating activity, sorted CD4+ and CD4- were injected into nude or NSG mice at 105 cells/mouse. Leukemias developed from both sorted CD4+ and CD4- transplanted cells in 2 weeks (Figure S2). Most remarkably, the transplanted leukemias expressed CD4 in the same variegated pattern that was present in the parental 32080 line (compare Figure S2 with Figure 1A) and not the uniform pattern of expression of the CD4+ or CD4- sorted populations that were injected.
Figure 2. CD4- and CD4+ populations of 32080 cells have differential levels of apoptosis and glucocorticoid sensitivity.
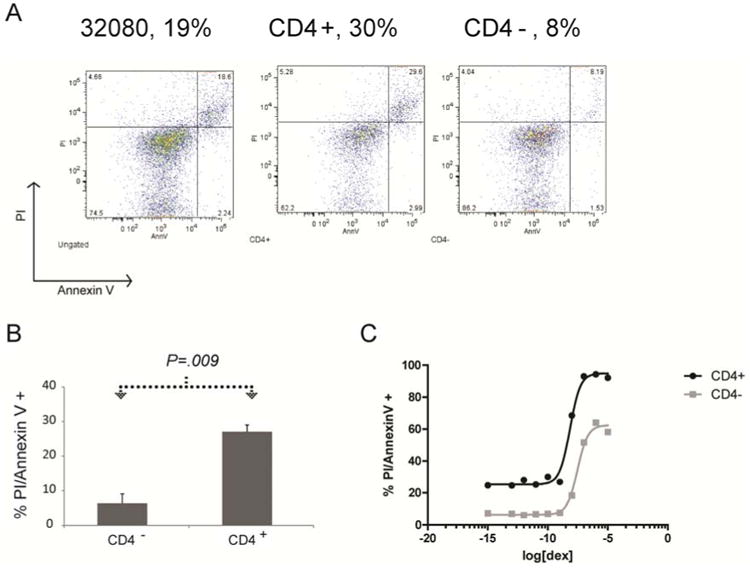
A, 32080 cell line was stained for CD4, Annexin V, and for uptake of propidium iodide (PI). Left panel shows PI and Annexin V staining of the whole population. Middle panel shows gating only on the CD4+ population of 32080 cells; right panel shows gating on the CD4-population. Numbers shown are the percentage of total gated cells positive for both PI and Annexin V. B, graph shows the mean percentage of cells positive for PI and Annexin V at baseline without any drug treatment from 4 independent staining experiments; error bars are the SEM; P value was generated by Student t-test. C, graph shows percentage of cells positive for PI and Annexin V for increasing concentrations of dexamethasone (shown as log values of molarity, M). The black line shows the curve that fit the CD4+ population; and, gray line shows the curve fit for CD4- population. These curves were used to generate IC50 values for CD4+ and CD4- cells explained in the text.
CD4 antigen variegation is due to variegated mRNA abundance and cells show interconversion between CD4+ and CD4- cells
The transplantation experiment on sorted 32080 cells suggested that the CD4+ cells could interconvert with CD4- cells. To test this, we sorted the CD4+ and CD4- cells into separate in vitro cultures. After 7 days, CD4 expression was variegated just like the pre-sort FACS (Figure 3A). Since it was easier to analyze the appearance of CD4 rather than its loss, we next did a time course for CD4 expression by FACS and by RT-PCR for 8 days following CD4- sorting. Flow analysis showed CD4 expression increased daily in the CD4 negative population and returned to pre-sort levels by the final day of analysis (Figure 3B). Similarly, CD4- cells increased in number when starting the in vitro culture with sorted CD4+ cells. RT-PCR analysis showed consistent CD8 expression at these time points, while increases in CD4 mRNA were concordant with CD4 protein expression, confirming mRNA abundance as the source for CD4 protein variability (Figure 3C). These also showed the variegated parental pattern after 8 days in culture. CD4 antigen appearance increased exponentially over time (Figure 3D). This kinetic pattern suggested to us that the appearance of CD4+ cells from CD4- cells (or CD4- from CD4+) was related to replication rather than a stochastic switch. To test this, we sorted CD4- cells from the 32080 line and pulsed them with BrdU. We then gated on cells that acquired CD4 antigen expression over time to analyze whether they were labeled with BrdU. As shown in Figure 3E, 48 hours after the BrdU pulse, 3.23% of cells were CD4+ and BrdU+ compared with 0.456% that were CD4+ and BrdU-. Since the cells were actively cycling and diluting out the BrdU, we performed a 48 hour BrdU pulse and observed 17.2% of total cells were CD4+/BrdU+ whereas only 0.92% were CD4+/BrdU-. Thus, cells that had newly acquired CD4 expression had incorporated BrdU, suggesting that cells with newly expressed CD4 had gone through replication.
Figure 3. CD4- or CD4+ populations of 32080 cells interconvert through replication.
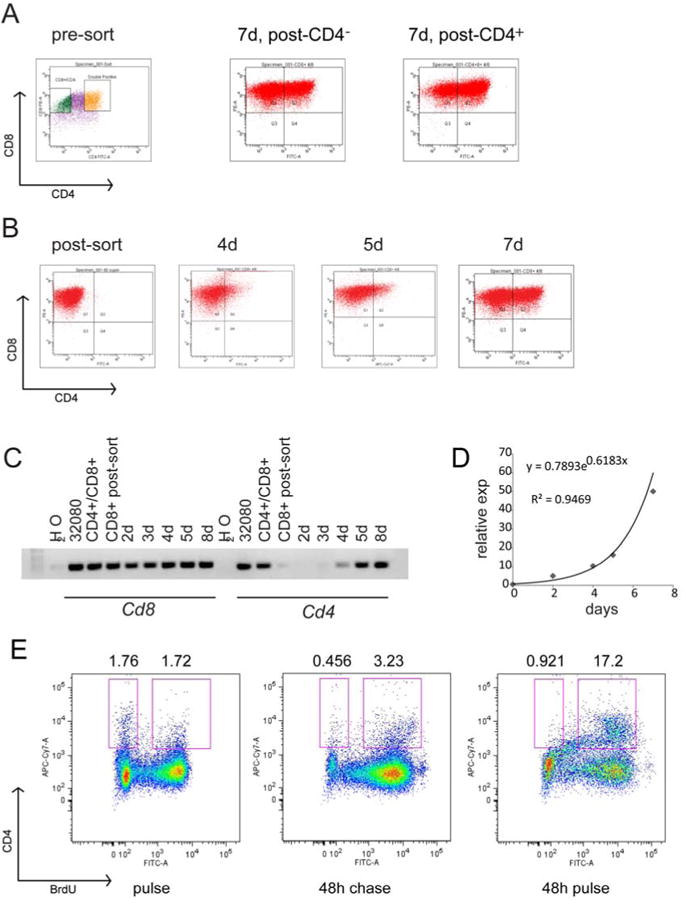
A, shows FACS plots of 32080 cells in the left panel with boxes showing the sorted cells. 32080 cells were sorted into CD4+ (yellow gate) and CD4- (green gate) populations and placed in separate cultures. The FACS plot in the middle shows CD4 and CD8 expression 7 days after sorting for CD4-. The FACS plot on the right shows CD4 and CD8 expression 7 days after sorting for CD4+. B, shows FACS plots of CD4 and CD8 expression of cells from 32080 followed for 4, 5, and 7 days after sorting for CD4-. C, agarose gel shows RT-PCR for CD8 and CD4 mRNAs for the same cells in the experiment shown in B. D, graph shows the expression of CD4 antigen in culture over time for the same experiment shown in B. The exponential curve that fit with its equation and R2 value are shown. E, CD4- cells were sorted from 32080, placed in culture, and pulsed with BrdU for 45 minutes and analyzed immediately (FACS plot on left), or after 48 hours (FACS plot in middle); right panel shows FACS plot of CD4- cells incubated with BrdU for 48 hours. Cells were stained for CD4, CD8, BrdU and 7-AAD. Plots show CD4 at y-axis and BrdU at x-axis. Numbers above the plots are the percentage of cells in the BrdU- and BrdU+ gates (boxes).
CD4 expression depends on Lmo2 and Notch1 oncogenes
The 32080 line was established from a T-ALL in CD2-Lmo2 transgenic mice and overexpressed Lmo2. We analyzed the effect of Lmo2 knockdown in the 32080 line. We transduced the cells with a lentivirus expressing a non-silencing or specific shRNA directed against Lmo2 (after screening several shRNA constructs for efficacy in knockdown, see Figure S1). RT-PCR and Western blot analysis showed effective knockdown of Lmo2 mRNA and protein (Figure 4A). However, the knockdown was transient since after 7 days, the 32080 cells expressed Lmo2 protein at pre-knockdown levels even with stable integration of lentivirus and drug selection. Analysis of 32080 in the time window where Lmo2 protein was knocked down (days 5-7) showed no change in cell cycle entry by BrdU analysis, no change in cell death (data not shown), but decreased transcription of CD4 (Figure 4B). We analyzed these cells for CD4 expression by FACS and observed an increase in the proportion of CD4- cells (Figure 4C). Thus, Lmo2 may directly or indirectly regulate the transcription of the CD4 gene and shifts the 32080 population towards CD4- cells.
Figure 4. Lmo2 knockdown downregulates CD4.
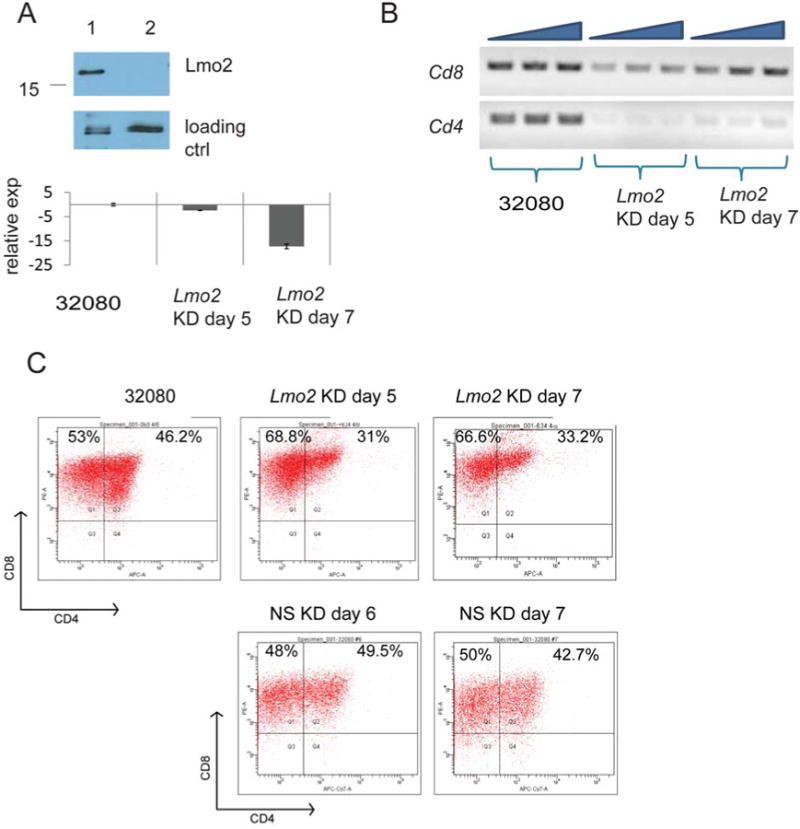
A, SDS-PAGE and Western blot analysis of 32080 cells transduced with an Lmo2 shRNA-expressing lentivirus and selected with 1 μg/ml puromycin for five days. Whole cell lysate is shown. Lower panel shows bar graph with relative expression of Lmo2 mRNA. Whole RNA was prepared from 32080 cells, 5 and 7 days after Lmo2 knockdown for analysis of Lmo2 mRNA by quantitative RT-PCR. Lmo2 expression was 2.5 and 17 times below control (nonsilencing NS shRNA) levels at days 5 and 7, respectively. B, agarose gel of RT-PCR for CD4 and CD8 mRNAs at days 5 and 7 after Lmo2 knockdown. Triangles indicate increasing cDNA concentrations: 100, 200 and 300 ng. C, shows FACS plots of 32080 cells analyzed for CD4 and CD8 expression at days 5 and 7 after Lmo2 knockdown. 32080 cells transduced with non-silencing shRNA were concomitantly selected in puromycin and were analyzed by FACS as shown on days 6 and 7. The plots shown are representative of three independent experiments.
We found activating Notch1 mutations were frequent in CD2-Lmo2 transgenic T-ALLs. We sequenced Notch1 exons 26, 27, and 34 from 32080 genomic DNA. We discovered a hemizygous 68 base pair duplication in exon 34 which added 4 additional amino acid residues and a stop codon after residue 2427 (Figure 5A), which predicted an intracellular Notch1 protein (ICN1) that was smaller than wild type due to truncation of the PEST degradation domain. Western blot analysis of 32080 whole cell lysate with antibody directed against Notch1's NH2-terminal Val1644 showed the constitutive presence of truncated ICN1 (Figure 5B, lane 1). We treated the cells with gamma secretase inhibitor, (DAPT) and probed for ICN1 by Western blot. ICN1 was not detectable after treatment with 1 and 5 μM DAPT (Figure 5B, lanes 5-6). Western blot analysis suggested that Lmo2 protein was increased with increasing concentrations of DAPT. This increase in protein was due to statistically significant upregulation of Lmo2 transcripts as confirmed by qRT-PCR (Figure 5D). At steady state, ICN1 was still detectable at 0.1-0.5 μM DAPT, however, these cells were shifted toward CD4+ (Figure 5C) due to significantly increased CD4 mRNA (Figure 5D). Thus, the downregulation of ICN1 in 32080 cells promoted CD4 expression. 32080 cells were less sensitive to DAPT than other cell lines (i.e. 03020 and 03007) but the loss of ICN1 eventually caused growth arrest and death (data not shown).
Figure 5. 32080 cells have constitutive intracellular Notch1 protein that can be inhibited by gamma secretase inhibition and increase CD4 expression.
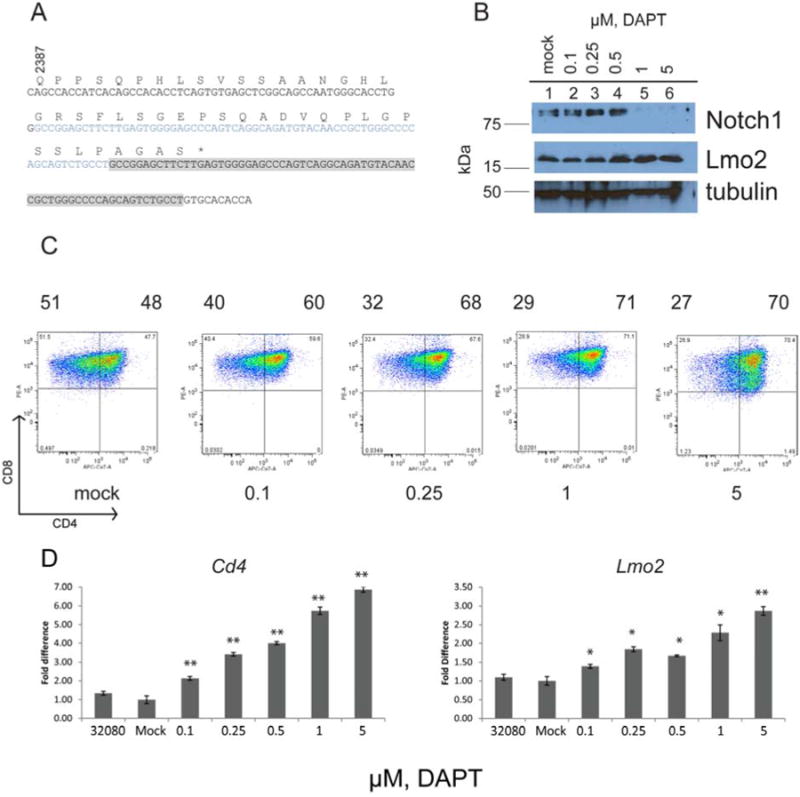
A, exon 34 of Notch1 cDNA was cloned from 32080 cells; wild type sequence is shown in black. Sequence in blue and gray shows a 68 base pair region that was duplicated inducing 4 new residues (AGAS) followed by a premature stop codon (shown at asterisk). B, Western blot analysis of 32080 cells treated with increasing concentrations of DAPT (in μM). Top panel shows blot with anti-Val1744 of Notch1, followed by anti-Lmo2, followed by anti-tubulin as loading control. C, shows FACS plots of CD4 and CD8 expression of 32080 cells treated with increasing concentrations of DAPT. Numbers under the histograms show DAPT concentration in μM. Numbers above the histograms show the percentage of cells in CD4- and CD4+ quadrants. D, graph shows qRT-PCR for CD4 mRNA in the left panel and Lmo2 mRNA in the right panel. Bars represent the mean of triplicate values with SEM. Statistical comparisons were made between the mock treated cells by unpaired t-test. The double asterisks denote P<.001; single asterisks denote P<.006. The graphs represent three independent experiments.
Analysis of cis and trans regulators of CD4 gene expression in 32080 cells
We screened the 32080 cells by RT-PCR for the presence of known epigenetic regulators of the CD4 gene and analyzed their mRNA quantitatively by RNA-seq (see Figure S3). Gene knockouts of some of these regulators have shown variegated CD4 expression in T-cell progenitor cells. Smarce1 (Baf57), Cbx5 (HP1a), Cbfb, Chd4 (Mi-2β), Hes1, and Smarca4 (Brg1) transcripts were present in both CD4+ and CD4- fractions of 32080 cells whereas Zbtb7b (ThPOK) and Runx3, which are active in T-cell development after the DP stage, were not expressed (28, 31). We compared the abundance of these mRNAs between 32080 cells and 03027 cells, which are DN (Figure 1) and have the CD4 gene repressed. RNA-seq analysis showed that the CD4, CD8a, and CD8b1 mRNAs were differentially expressed at a statistically significant levels between 03027 and 32080 cells (Figure S3, 03027 v. 32080, P=1.26×10-8 for CD4, 1.92×10-13 for CD8a, and .0024 for CD8b1) but the other genes were expressed at comparable levels between the cell lines (Figure S1). We got low read counts for Runx1 mRNA transcripts in 32080 cells compared to the other factors (Figure S1). We analyzed the genomic Runx1 coding sequences and found no mutations in 32080 cells. Because of Runx1's central role in CD4 repression, we enforced stable expression of Runx1 in 32080 cells and analyzed the effects on CD4 expression (Figure 6A). 32080 cells overexpressing Runx1 showed decreased expression of CD4 (Figure 6B). Runx1 binds to a silencer element within the first intron of the CD4 gene and deletion of this silencer element induces variegated CD4 expression in vivo (31). The silencer is one of several cis elements that regulate CD4 expression. The proximal promoter and a 5′ enhancer also play a role in CD4 gene regulation. We predicted that these cis elements would recapitulate the regulation of CD4 in 32080 cells. To test this, we used three previously described reporter constructs (Figure 6C): CD4E, where GFP was driven by the CD4 promoter and enhancer; CD4ES, where GFP was regulated by the CD4 promoter, enhancer, and silencer; and CD4ESΔR, which is the same as CD4ES except the silencer's Runx1 binding sites have been deleted (28). We stably transfected these constructs into 03007 cells, which resemble the ISP stage of T-cell development where Runx1 binding to the silencer represses CD4 gene expression. As expected, the CD4E construct expressed a strong GFP signal which was markedly attenuated in the CD4ES construct (Figure 6D). The GFP signal was restored in CD4ESΔR-expressing cells. We compared the GFP expression of multiple independent stable clones of 03007 with CD4E, CD4ES, and CD4ESΔR and found statistically significant decreases in GFP expression in the construct with the silencer element (Figure 6E, P=.026 for CD4ES v. CD4E). These results suggest that the silencer element and its Runx1 binding sites are active in the 03007 line just like they are in ISP cells. CD4E transfected into 32080 cells showed strong GFP expression which was significantly attenuated in stable lines with the CD4ES reporter (Figure 6F, unpaired t test, P<.001, CD4ES v. CD4E). Hence, the silencer markedly decreased expression of GFP but in both CD4- and CD4+ populations (unpaired t test, P=.82, CD4ES in CD4+ v. CD4-). CD4ESΔR did not restore GFP expression as observed in the 03007 line (CD4- cells, unpaired t test P=.35; CD4+ cells, unpaired t test, P=.50, CD4ESΔR v. CD4ES). Thus, although overexpression of Runx1 repressed CD4, the promoter, enhancer and silencer regulatory elements in reporter constructs did not recapitulate the pattern observed in 32080.
Figure 6. Runx1 deregulation in 32080 cells.
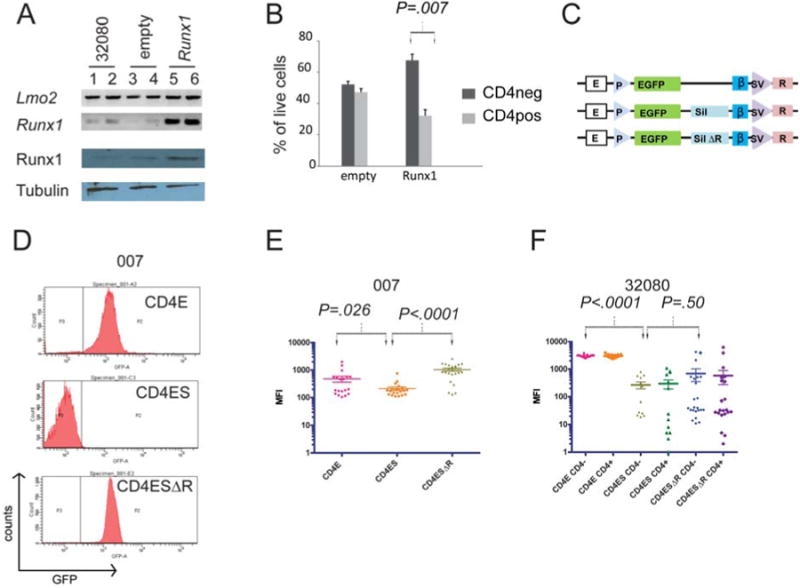
A, shows agarose gels of RT-PCR of 32080 cells (lanes 1, 2) or 32080 cells transfected with empty pCDNA-3.1 (lanes 3, 4) or pcDNA3.1-Runx1 (lanes 4, 5). Lmo2, Runx1 mRNAs were analyzed by RT-PCR using 50 ng (lanes 1, 3, 5) or 100ng (lanes 2, 4, 6) cDNA. Bottom panels show Western blot analysis for Runx1 and tubulin for 32080 and stable lines with empty pcDNA-3.1, or with pcDNA3.1-Runx1. B, stable 32080 cell lines with empty pcDNA-3.1 or with pcDNA-3,1-Runx1 were analyzed by flow cytometry for CD4 and CD8 expression. Bar graph shows 3 independent experiments; y-axis shows the percentage of live cells CD4- or CD4+; dark gray bars are CD4- and light gray bars are CD4+. C, schematic shows 3 reporter constructs stably transfected into 32080 cells. E, CD4 proximal enhancer; P, CD4 promoter; EGFP, reporter gene d2EGFP; sil, CD4 silencer element; silΔR, CD4 silencer element with mutated Runx1 motifs; blue vertical bar, chicken B-globin insulator element; SV, SV40 enhancer/promoter; R, neo resistance gene. D, the 03007 cell line was stably transfected with one of three reporter constructs, CD4-E, CD4-ES, CD4ESΔR, and analyzed for GFP. E, scatter plot of log of mean fluorescence intensity (log [MFI]) of GFP in 03007 cells stably expressing CD4 reporter constructs. Each point represents an independently single cell cloned stable line expressing the GFP reporters. F, 32080 cells were similarly selected in G418 and single cell cloned and analyzed for GFP expression. Clones were stained for CD4 and CD8 and GFP expression was measured by FACS in a minimum of 13 independent clonal lines per reporter construct. Comparisons between the mean expression values were done by Student t-test generating the P values shown.
Chromatin marks at the CD4 gene are dynamic in 32080 cells and reflect on and off states of transcription
We treated 32080 cells with 15 nM of trichostatin A, an inhibitor of class I and II histone deacetylases (HDAC) enzymes, which increased the number of CD4+ cells (Figure 7A). Next, we enforced expression of HP1α, a component of heterochromatin and repressed genes that may be recruited by Runx1 to the CD4 locus (31, 32). HP1α stable expression in 32080 induced a shift toward CD4- cells (Figure 7B). These results suggested that chromatin modification at the CD4 locus may correlate with CD4 transcription. To further test this, we analyzed the CD4 gene promoter (P), 5′enhancer (two separate amplicons, E1 and E2), and silencer (S) for chromatin marks associated with active and repressed transcription (33). We performed multiple independent chromatin immunoprecipitations (ChIP) followed by quantitative PCR for E, P, and S regulatory elements as well as for exons 2 and 10 (ex.2, ex.10). The ChIPs showed multiple marks that were differentially present on CD4- versus CD4+ populations. H3K9 dimethylation (i.e. H3K9me2) was present at higher levels at the promoter in CD4-versus CD4+ cells (P=.04 with one tailed t test and P=.07 with two-tailed t test) consistent with its putative role as a silencing mark. Trimethylation at the same residue (i.e. H3K9me3), a mark associated with heterochromatin, was not significantly different between the CD4- and CD4+ populations. Acetylation of H3 at both K9 and K4 was significantly increased at the promoter (P=.02), 5′enhancer (P=.03 for E1), silencer (P=.05, one tailed), and exon 10 (P=.0013). Trimethylation of H3K27 was increased at the silencer in CD4- cells compared with CD4+ (P=.037, one tail; P=.07, two tailed) cells. Interestingly, RNA PolII occupancy at the promoter was no different between CD4- and CD4+ cells but its occupancy at the silencer was increased in CD4- cells (P=.03, two tailed). This result was strikingly similar to data by Jiang and Peterlin where they observed occupancy of RNA PolII at the promoter in murine cell lines with a CD4 SP phenotype (i.e. 3A9) and with an ISP phenotype (i.e. CD4-1200M) (34). The results here suggest that the CD4 gene undergoes a dynamic shift in histone marks that are known to correlate with active versus repressed transcriptional states. A shift toward H3 acetylation was the major trend in CD4+ versus CD4- cells, which has also been observed in normal DP cells(35).
Figure 7. Chromatin marks at the CD4 locus correlate with CD4 expression or repression.
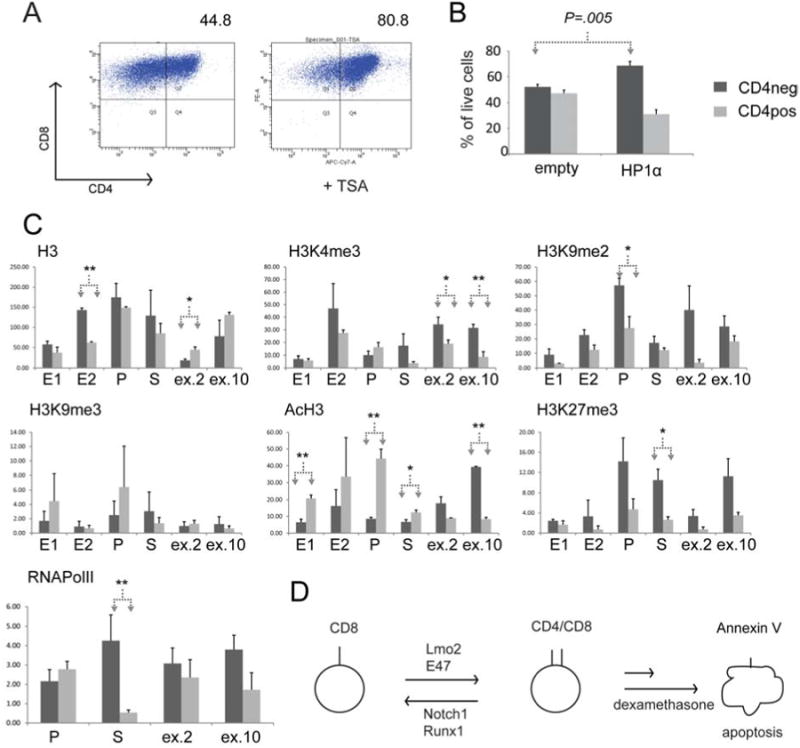
A, shows FACS analysis of 32080 cells treated with 15 nM TSA for 48 hours. Numbers above histograms show percentage of cells in CD4+ quadrant. The plot shown is representative of three independent experiments. B, shows mean values of CD4-expressing cells in percentage in stably lines expressing empty plasmid or HP1α. Dark gray bars show CD4- cells whereas light gray bars show CD4+ cells. C, bar graphs show qPCR analysis of chromatin immunoprecipitations (ChIP) of histone H3 or modified H3 from CD4- (dark gray bars) or CD4+ (light gray bars) cells sorted from 32080 cells. QPCR was done with primers specific for the proximal enhancer (in two amplicons, E1 and E2), promoter (P), silencer (S), exon 2 (ex.2), and exon 10 (ex.10) of the CD4 locus. Arrows show statistical comparisons of mean enrichment using Student t-test. Single asterisk denotes P<.05 for one-tailed test, whereas, double asterisk denotes P<.05 for two-tailed test. D, schematic shows a model of our observations in 32080 cells. This isogenic cell line shuttles between CD4- and CD4+ stages. Lmo2 promotes CD4+ stage whereas Notch1 and Runx1 promote the CD4- stage. The CD4+ stage is prone to apoptosis and is more sensitive to dexamethasone-induced apoptosis as well.
Discussion
In this study, we describe an Lmo2-induced T-cell leukemia line, 32080, that is clonal and yet shows marked immunophenotypic and biological heterogeneity. Most remarkably, the 32080 line has cell-to cell variation in CD4 expression that resembles epigenetic variegation (36). The CD4 gene is known to be epigenetically regulated during normal T-cell ontogeny and the 32080 line provides a remarkable model for the developmental switch between two T-cell progenitor stages, the intermediate CD8 single positive (ISP) stage and the double positive (DP) stage (33). Unlike normal development, however, the 32080 CD4- and CD4+ populations interconvert (Figure 7D). Most strikingly, cells that resemble a more mature stage of development (i.e. DP) can revert to an earlier stage (i.e. ISP) by losing CD4 expression. From sorted pure populations, we show that cells that acquired CD4 antigen expression were also labeled with BrdU suggesting that the transition between on/off transcriptional states requires replication. This makes sense, since replication forks would require chromatin remodeling and erasure of existing marks. In our study, chromatin marks associated with active and repressed transcription were correlated with CD4 mRNA expression. The variation in CD4 antigen expression is most likely a transcriptional effect since the CD4 mRNA varied with perturbations in known transcription factors and co-factors such as Runx1, Lmo2 and Notch1 as well as with trichostatin A; and, the variable pattern in CD4 expression may reflect stochastic silencing of the CD4 gene by spreading heterochromatin. This variegated CD4 expression pattern is observed in the thymocytes of mice with germline deletions of Runx motifs within the CD4 silencer or in thymocytes with conditional deletion of the Runx1 gene itself or other regulators of CD4 gene expression (31, 37-39). If the chromatin marks dictate transcriptional state and if the transcriptional state is heritable then the same marks would need to be re-established in daughter cells. It is likely that, in 32080 cells, the process by which this transcriptional memory is transmitted is deregulated. Thus, the 32080 cells provide an interesting model to study gene silencing and developmental stage transition for T cells.
Runx1 protein appears to be limiting in 32080 cells since its enforced expression represses CD4 gene expression. Runx1 mRNA and the mRNAs of other known CD4 transcriptional regulators were at similar levels in 32080 cells and in 03027 cells, which are DN-like and have repressed CD4, suggesting that the factors are not regulated at the transcriptional level. Runx1 functions by binding to the silencer element within intron 1 of the CD4 gene and recruiting chromatin modifying enzymes to repress transcription (40). Runx1 knockout shows derepression of CD4 expression at DN and ISP stages (31, 41). Interestingly, a reporter with the CD4 promoter, enhancer, and silencer showed strong silencing that did not depend upon Runx1 since it silenced GFP in 32080 cells and deletion of the silencer's Runx1 binding sites did not restore expression. If Runx1 was the only transcriptional regulator of CD4 expression in 32080 cells, then the silencer should not have been functional suggesting that other regulators responsible for binding to the silencer are active. Tfap4 is one such candidate that binds to an E box site within the CD4 silencer and can cooperate with Runx1 to induce repression (42). Even so, the GFP expression of the reporter construct did not track with the expression of CD4 and may be missing other regulatory elements (43).
Lmo2 and Notch1 also induced epigenetic deregulation of CD4. They are both oncogenic drivers of the T-cell leukemia but have opposite effects on CD4 transcription. Intracellular Notch1 protein is constitutively present in 32080 but its levels can be markedly decreased by inhibition of gamma secretase enzyme which induces increased CD4 mRNA. This is consistent with Notch1's proposed function in normal ISP cells, where its downregulation is required for progression to the DP stage (44-46). Notch1 activation may cooperate with Lmo2 to induce transformation of ISP and DP progenitors. For example, in the OP9-DL1 stromal cell assay, we observed a DN block in CD2-Lmo2 transgenic T-cell progenitors which was removed by enforced expression of ICN1(16). Presently, we do not know the mechanisms by which Lmo2 may induce CD4 variegation. Notably, we screened 9 human T-cell leukemias that overexpressed LMO2 and observed similar CD4 variegation in at least one leukemia showing a human T-ALL that resembles 32080. Lmo2 knockdown caused decreased CD4 expression and increased Lmo2 (induced by Notch1 inhibition) increased CD4 expression. These results suggest that Lmo2 promotes CD4 expression which is quite different from observations of Lmo2-induced DN block before leukemia onset (16, 21, 46). Furthermore, we have observed that forced dimerization of an E47/estrogen receptor fusion protein induces CD4 mRNA and protein expression and shifts the 32080 line entirely towards 100% CD4+ cells (see accompanying paper). Hence, Lmo2 knockdown and enforced E47 expression have opposite effects. One hypothesis for Lmo2's oncogenic function proposes that Lmo2 causes a functional deficiency of E47 but the findings in this paper may suggest a novel mechanism for Lmo2's action (6). For example, the results could be explained by Lmo2 directly binding and limiting the function of Runx1 or some other chromatin modifying complex that regulates CD4 gene expression (37, 47). Thus, Runx1's ability to repress CD4 would be compromised by Lmo2, either by direct binding and redirection in the nucleus or indirectly by binding co-repressors required by Runx1 (48-50). RUNX1 ChIP-seq analysis in CCRF-CEM cells did not show specific occupancy at the silencer region which could account for this cell line's expression of CD4 (51). RUNX1 loss of function has been described in Early T-cell Precursor ALLs which do not express CD4 or CD8 (52). However, RUNX1 was required in Jurkat T-ALL cells, which express both CD4 and CD8, and where it was found to directly regulate the transcription of TAL1 and GATA3 (51). Similarly, Runx1 may have multiple transcriptional targets in 32080 cells and the repressive function at the CD4 locus may be compromised if the protein was redirected to other locations within the genome. Thus far, we have not been able to co-immunoprecipitate endogenous Lmo2 and Runx1 proteins in 32080 cells but the proteins have been co-purified from erythroid cells where they appear to be part of a larger multi-subunit complex and they may be binding partners in leukemia as well (53, 54).
Our ChIP experiments showed that the most substantial change was at H3K9 at the promoter, which was dimethylated in CD4- cells but acetylated in CD4+ cells. Acetylation at other elements in the CD4 locus was also increased in CD4+ cells. Similar increased acetylation was observed in normal DP thymocytes in comparison to DN progenitors. Most strikingly, RNA Pol II occupancy at the promoter was the same in CD4- and CD4+ cells but was increased at the silencer only in CD4- cells. Our findings suggest that RNA Pol II is paused at the promoter which has also been described in 1200M murine T-cell leukemia cells that do not express CD4 and resemble ISPs. Runx1 has been proposed to regulate CD4 silencing by potentiating transcriptional elongation (34, 55). Our results would be consistent with this model. In fact, Lmo2's involvement in transcriptional elongation is also suggested by the co-purification of multiple Lmo2 binding partners with TIF1γ, Cdk9 and components of the FACT complex (54, 56). An unbiased purification of the Lmo2-associated complex in 32080 is ongoing and may reveal similar protein partners.
In addition to mechanistic insight into Lmo2's function, our results also have implications for the treatment T-ALL. The interconversion between more differentiated DP-like cells into ISP-like cells argues against targeting one specific immunophenotype within leukemia. However, we may be able to exploit unique biological features of one population subset. For example, in 32080, the CD4- and CD4+ populations differ in their rates of apoptosis and in their sensitivity to dexamethasone. Presently, we do not understand the basis for this differential sensitivity but in normal physiology DP progenitors are more sensitive than DN/ISP cells to glucocorticoid-induced apoptosis (57, 58). Further gene expression analysis and protein signaling data on these two populations is ongoing to probe apoptosis and other mechanisms. Lastly, our results could provide rationale for the use of differentiation agents such as histone deacetylase inhibitors in combination therapy that could shift leukemias toward drug-sensitive states.
Supplementary Material
Figure S1. Screen of multiple shRNAs against Lmo2. Western blot of 32080 cells transfected with lentiviral plasmids expressing shRNAs directed against Lmo2: 634 (V2LHS-62634), 635 (V2LHS - 62635), 822 (V2LMM-20822, and 823 (V2LMM-20823) expressing shRNAs directed against the coding regions of Lmo2. The cells were selected for 5 days with puromycin and whole lysate prepared for SDS-PAGE separation. The gels were transferred to nitrocellulose and probed with antibodies against Lmo2 and tubulin. The molecular weight markers are shown to the left of the blots. shRNAs 634 and 635 induced consistent knockdown of Lmo2 and were used for further studies.
Figure S2. Sorted 32080 cells produce leukemia in NSG mice. 32080 cells were sorted into CD4+ and CD4- fractions. 100K sorted cells were injected into the tail vein of NSG mice resulting in morbidity due to florid leukemia in 2 weeks. Necropsy showed abdominal masses, splenomegaly, and bone marrow infiltration. Single cell suspensions were prepared and analyzed by staining for CD4 and CD8. FACS plots are shown for the masses (top panel) and the bone marrow (bottom panel). Orange cells are those designated CD4+ and blue designate those that are CD4-.
Figure S3. 32080 cells have mRNA expression of CD4 gene regulators at similar levels as a double negative cell line, 03027. Whole RNA was isolated from 32080 and 03027 cells and prepped and labeled for Illumina next generation sequencing (RNA-seq). The 03027 cell line does not express CD4 or CD8 by FACS analysis. Bar graph on left shows comparison of transcript counts between 03027 (light gray) and 32080 (dark gray) for CD4, CD8a, and CD8b1 mRNAs. The y-axis shows RPKM, reads per kilobase of gene per million reads. The right panel shows log of RPKM for transcripts of known CD4 gene regulators in 03027 and 32080 cells. There were no statistically significant differences in transcript levels for these genes between the two cell lines.
Figure S4. Gating scheme for FACS analysis of 32080 cells. Healthy sized cells were gated upon in forward and side scatter and then refined. Typically, anti-CD4 was labeled with APC-Cy7 and anti-CD8 labeled with PE. There was no GFP emission at baseline.
Acknowledgments
We thank Drs. Steve Brandt, Scott Hiebert, Sandy Zinkel, Justin Layer, and Mark Koury for helpful discussions. This work was supported by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, the American Society of Hematology, National Institutes of Health K08HL089403, the Leukemia & Lymphoma Society, and the Vanderbilt Ingram Cancer Center (P30 CA68485) (U.P.D.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NCI or the NIH. Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404).
Footnotes
Conflict of Interest Statement: The authors of this manuscript have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deichmann A, Hacein-Bey-Abina S, Schmidt M, et al. Vector integration is nonrandom and clustered and influences the fate of lymphopoiesis in SCID-X1 gene therapy. J Clin Invest. 2007;117:2225–2232. doi: 10.1172/JCI31659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hacein-Bey-Abina S, C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science (New York, NY) 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 3.Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. The Journal of clinical investigation. 2008 doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dave UP, Akagi K, Tripathi R, et al. Murine leukemias with retroviral insertions at Lmo2 are predictive of the leukemias induced in SCID-X1 patients following retroviral gene therapy. PLoS Genet. 2009;5:e1000491. doi: 10.1371/journal.pgen.1000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dave UP, Jenkins NA, Copeland NG. Gene therapy insertional mutagenesis insights. Science. 2004;303:333. doi: 10.1126/science.1091667. [DOI] [PubMed] [Google Scholar]
- 6.Matthews JM, Lester K, Joseph S, et al. LIM-domain-only proteins in cancer. Nat Rev Cancer. 2013;13:111–122. doi: 10.1038/nrc3418. [DOI] [PubMed] [Google Scholar]
- 7.Warren AJ, Colledge WH, Carlton MB, et al. The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell. 1994;78:45–57. doi: 10.1016/0092-8674(94)90571-1. [DOI] [PubMed] [Google Scholar]
- 8.Yamada Y, Warren AJ, Dobson C, et al. The T cell leukemia LIM protein Lmo2 is necessary for adult mouse hematopoiesis. Proc Natl Acad Sci U S A. 1998;95:3890–3895. doi: 10.1073/pnas.95.7.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCormack MP, Forster A, Drynan L, et al. The LMO2 T-cell oncogene is activated via chromosomal translocations or retroviral insertion during gene therapy but has no mandatory role in normal T-cell development. Mol Cell Biol. 2003;23:9003–9013. doi: 10.1128/MCB.23.24.9003-9013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai FY, Keller G, Kuo FC, et al. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 1994;371:221–226. doi: 10.1038/371221a0. [DOI] [PubMed] [Google Scholar]
- 11.Shivdasani RA, Mayer EL, Orkin SH. Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCK. Nature. 1995;373:432–434. doi: 10.1038/373432a0. [DOI] [PubMed] [Google Scholar]
- 12.Schlaeger T, Mikkola H, Gekas C, et al. Tie2Cre-mediated gene ablation defines the stem-cell leukemia gene (SCL/tal1)-dependent window during hematopoietic stem-cell development. Blood. 2005;105 doi: 10.1182/blood-2004-11-4467. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Freudenberg J, Cui K, et al. Ldb1-nucleated transcription complexes function as primary mediators of global erythroid gene activation. Blood. 2013;121:4575–4585. doi: 10.1182/blood-2013-01-479451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Lee JY, Gross J, et al. A requirement for Lim domain binding protein 1 in erythropoiesis. J Exp Med. 2010;207:2543–2550. doi: 10.1084/jem.20100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCormack MP, Young LF, Vasudevan S, et al. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science (New York, NY) 327:879–883. doi: 10.1126/science.1182378. [DOI] [PubMed] [Google Scholar]
- 16.Cleveland SM, Smith S, Tripathi R, et al. Lmo2 induces hematopoietic stem cell-like features in T-cell progenitor cells prior to leukemia. Stem Cells. 2013;31:882–894. doi: 10.1002/stem.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ceredig R, Rolink T. A positive look at double-negative thymocytes. Nat Rev Immunol. 2002;2:888–897. doi: 10.1038/nri937. [DOI] [PubMed] [Google Scholar]
- 18.Bhandoola A, Sambandam A. From stem cell to T cell: one route or many? Nat Rev Immunol. 2006;6:117–126. doi: 10.1038/nri1778. [DOI] [PubMed] [Google Scholar]
- 19.Herblot S, Steff AM, Hugo P, et al. SCL and LMO1 alter thymocyte differentiation: inhibition of E2A-HEB function and pre-T[alpha] chain expression. Nat Immunol. 2000;1:138–144. doi: 10.1038/77819. [DOI] [PubMed] [Google Scholar]
- 20.Mingueneau M, Kreslavsky T, Gray D, et al. The transcriptional landscape of alphabeta T cell differentiation. Nat Immunol. 2013;14:619–632. doi: 10.1038/ni.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drynan LF, Hamilton TL, Rabbitts TH. T cell tumorigenesis in Lmo2 transgenic mice is independent of V-D-J recombinase activity. Oncogene. 2001;20:4412–4415. doi: 10.1038/sj.onc.1204538. [DOI] [PubMed] [Google Scholar]
- 22.Treanor LM, Volanakis EJ, Zhou S, et al. Functional interactions between Lmo2, the Arf tumor suppressor, and Notch1 in murine T-cell malignancies. Blood. 2011;117:5453–5462. doi: 10.1182/blood-2010-09-309831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tatarek J, Cullion K, Ashworth T, et al. Notch1 inhibition targets the leukemia-initiating cells in a Tal1/Lmo2 mouse model of T-ALL. Blood. 2011;118:1579–1590. doi: 10.1182/blood-2010-08-300343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clappier E, Gerby B, Sigaux F, et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med. 2011;208:653–661. doi: 10.1084/jem.20110105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiu PP, Jiang H, Dick JE. Leukemia-initiating cells in human T-lymphoblastic leukemia exhibit glucocorticoid resistance. Blood. 2010;116:5268–5279. doi: 10.1182/blood-2010-06-292300. [DOI] [PubMed] [Google Scholar]
- 26.Shackleton M, Quintana E, Fearon ER, et al. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 27.Smith SB, Tripathi R, Goodings C, et al. LIM domain Only-2 (LMO2) induces T-cell leukemia by two distinct pathways. PLoS ONE. 2013 doi: 10.1371/journal.pone.0085883. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wildt KF, Sun G, Grueter B, et al. The transcription factor Zbtb7b promotes CD4 expression by antagonizing Runx-mediated activation of the CD4 silencer. J Immunol. 2007;179:4405–4414. doi: 10.4049/jimmunol.179.7.4405. [DOI] [PubMed] [Google Scholar]
- 29.Smith S, Tripathi R, Goodings C, et al. LIM Domain Only-2 (LMO2) Induces T-Cell Leukemia by Two Distinct Pathways. PLoS ONE. 2014;9:e85883. doi: 10.1371/journal.pone.0085883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grigliatti T. Position-effect variegation--an assay for nonhistone chromosomal proteins and chromatin assembly and modifying factors. Methods Cell Biol. 1991;35:587–627. [PubMed] [Google Scholar]
- 31.Taniuchi I, Osato M, Egawa T, et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–633. doi: 10.1016/s0092-8674(02)01111-x. [DOI] [PubMed] [Google Scholar]
- 32.Taniuchi I, Ellmeier W, Littman DR. The CD4/CD8 lineage choice: new insights into epigenetic regulation during T cell development. Advances in immunology. 2004;83:55–89. doi: 10.1016/S0065-2776(04)83002-5. [DOI] [PubMed] [Google Scholar]
- 33.Gialitakis M, Sellars M, Littman DR. The epigenetic landscape of lineage choice: lessons from the heritability of CD4 and CD8 expression. Curr Top Microbiol Immunol. 2012;356:165–188. doi: 10.1007/82_2011_175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang H, Peterlin BM. Differential chromatin looping regulates CD4 expression in immature thymocytes. Molecular and cellular biology. 2008;28:907–907. doi: 10.1128/MCB.00909-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu M, Wan M, Zhang J, et al. Nucleoprotein structure of the CD4 locus: Implications for the mechanisms underlying CD4 regulation during T cell development. Proceedings of the National Academy of Sciences. 2008;105:3873–3878. doi: 10.1073/pnas.0800810105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Girton JR, Johansen KM. Chromatin structure and the regulation of gene expression: the lessons of PEV in Drosophila. Adv Genet. 2008;61:1–43. doi: 10.1016/S0065-2660(07)00001-6. [DOI] [PubMed] [Google Scholar]
- 37.Chi TH, Wan M, Zhao K, et al. Reciprocal regulation of CD4/CD8 expression by SWI/SNF-like BAF complexes. Nature. 2002;418:195–199. doi: 10.1038/nature00876. [DOI] [PubMed] [Google Scholar]
- 38.Taniuchi I, Sunshine MJ, Festenstein R, et al. Evidence for distinct CD4 silencer functions at different stages of thymocyte differentiation. Mol Cell. 2002;10:1083–1096. doi: 10.1016/s1097-2765(02)00735-9. [DOI] [PubMed] [Google Scholar]
- 39.Williams CJ, Naito T, Arco PG, et al. The chromatin remodeler Mi-2beta is required for CD4 expression and T cell development. Immunity. 2004;20:719–733. doi: 10.1016/j.immuni.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 40.Reed-Inderbitzin E, Moreno-Miralles I, Vanden-Eynden SK, et al. RUNX1 associates with histone deacetylases and SUV39H1 to repress transcription. Oncogene. 2006;25:5777–5786. doi: 10.1038/sj.onc.1209591. [DOI] [PubMed] [Google Scholar]
- 41.Collins A, Littman DR, Taniuchi I. RUNX proteins in transcription factor networks that regulate T-cell lineage choice. Nat Rev Immunol. 2009;9:106–115. doi: 10.1038/nri2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egawa T, Littman DR. Transcription factor AP4 modulates reversible and epigenetic silencing of the Cd4 gene. Proc Natl Acad Sci U S A. 2011;108:14873–14878. doi: 10.1073/pnas.1112293108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chong MMW, Simpson N, Ciofani M, et al. Epigenetic propagation of CD4 expression is established by the Cd4 proximal enhancer in helper T cells. Genes & Development. 24:659–669. doi: 10.1101/gad.1901610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiong J, Armato MA, Yankee TM. Immature single-positive CD8+ thymocytes represent the transition from Notch-dependent to Notch-independent T-cell development. Int Immunol. 2011;23:55–64. doi: 10.1093/intimm/dxq457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fiorini E, Merck E, Wilson A, et al. Dynamic regulation of notch 1 and notch 2 surface expression during T cell development and activation revealed by novel monoclonal antibodies. J Immunol. 2009;183:7212–7222. doi: 10.4049/jimmunol.0902432. [DOI] [PubMed] [Google Scholar]
- 46.Huang EY, Gallegos AM, Richards SM, et al. Surface expression of Notch1 on thymocytes: correlation with the double-negative to double-positive transition. J Immunol. 2003;171:2296–2304. doi: 10.4049/jimmunol.171.5.2296. [DOI] [PubMed] [Google Scholar]
- 47.Naito T, Pablo, Williams CJ, et al. Antagonistic interactions between Ikaros and the chromatin remodeler Mi-2beta determine silencer activity and Cd4 gene expression. Immunity. 2007;27:723–734. doi: 10.1016/j.immuni.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 48.Lutterbach B, Westendorf JJ, Linggi B, et al. A mechanism of repression by acute myeloid leukemia-1, the target of multiple chromosomal translocations in acute leukemia. J Biol Chem. 2000;275:651–656. doi: 10.1074/jbc.275.1.651. [DOI] [PubMed] [Google Scholar]
- 49.Aronson BD, Fisher AL, Blechman K, et al. Groucho-dependent and -independent repression activities of Runt domain proteins. Mol Cell Biol. 1997;17:5581–5587. doi: 10.1128/mcb.17.9.5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Telfer JC, Hedblom EE, Anderson MK, et al. Localization of the domains in Runx transcription factors required for the repression of CD4 in thymocytes. J Immunol. 2004;172:4359–4370. doi: 10.4049/jimmunol.172.7.4359. [DOI] [PubMed] [Google Scholar]
- 51.Sanda T, Lawton LN, Barrasa MI, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22:209–221. doi: 10.1016/j.ccr.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Riel B, Pakozdi T, Brouwer R, et al. A novel complex, RUNX1-MYEF2, represses hematopoietic genes in erythroid cells. Mol Cell Biol. 2012;32:3814–3822. doi: 10.1128/MCB.05938-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meier N, Krpic S, Rodriguez P, et al. Novel binding partners of Ldb1 are required for haematopoietic development. Development. 2006;133:4913–4923. doi: 10.1242/dev.02656. [DOI] [PubMed] [Google Scholar]
- 55.Jiang H, Zhang F, Kurosu T, et al. Runx1 binds positive transcription elongation factor b and represses transcriptional elongation by RNA polymerase II: possible mechanism of CD4 silencing. Mol Cell Biol. 2005;25:10675–10683. doi: 10.1128/MCB.25.24.10675-10683.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bai X, Kim J, Yang Z, et al. TIF1gamma Controls Erythroid Cell Fate by Regulating Transcription Elongation. Cell. 142:133–143. doi: 10.1016/j.cell.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kong FK, Chen CL, Cooper MD. Reversible disruption of thymic function by steroid treatment. J Immunol. 2002;168:6500–6505. doi: 10.4049/jimmunol.168.12.6500. [DOI] [PubMed] [Google Scholar]
- 58.Screpanti I, Morrone S, Meco D, et al. Steroid sensitivity of thymocyte subpopulations during intrathymic differentiation. Effects of 17 beta-estradiol and dexamethasone on subsets expressing T cell antigen receptor or IL-2 receptor. J Immunol. 1989;142:3378–3383. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Screen of multiple shRNAs against Lmo2. Western blot of 32080 cells transfected with lentiviral plasmids expressing shRNAs directed against Lmo2: 634 (V2LHS-62634), 635 (V2LHS - 62635), 822 (V2LMM-20822, and 823 (V2LMM-20823) expressing shRNAs directed against the coding regions of Lmo2. The cells were selected for 5 days with puromycin and whole lysate prepared for SDS-PAGE separation. The gels were transferred to nitrocellulose and probed with antibodies against Lmo2 and tubulin. The molecular weight markers are shown to the left of the blots. shRNAs 634 and 635 induced consistent knockdown of Lmo2 and were used for further studies.
Figure S2. Sorted 32080 cells produce leukemia in NSG mice. 32080 cells were sorted into CD4+ and CD4- fractions. 100K sorted cells were injected into the tail vein of NSG mice resulting in morbidity due to florid leukemia in 2 weeks. Necropsy showed abdominal masses, splenomegaly, and bone marrow infiltration. Single cell suspensions were prepared and analyzed by staining for CD4 and CD8. FACS plots are shown for the masses (top panel) and the bone marrow (bottom panel). Orange cells are those designated CD4+ and blue designate those that are CD4-.
Figure S3. 32080 cells have mRNA expression of CD4 gene regulators at similar levels as a double negative cell line, 03027. Whole RNA was isolated from 32080 and 03027 cells and prepped and labeled for Illumina next generation sequencing (RNA-seq). The 03027 cell line does not express CD4 or CD8 by FACS analysis. Bar graph on left shows comparison of transcript counts between 03027 (light gray) and 32080 (dark gray) for CD4, CD8a, and CD8b1 mRNAs. The y-axis shows RPKM, reads per kilobase of gene per million reads. The right panel shows log of RPKM for transcripts of known CD4 gene regulators in 03027 and 32080 cells. There were no statistically significant differences in transcript levels for these genes between the two cell lines.
Figure S4. Gating scheme for FACS analysis of 32080 cells. Healthy sized cells were gated upon in forward and side scatter and then refined. Typically, anti-CD4 was labeled with APC-Cy7 and anti-CD8 labeled with PE. There was no GFP emission at baseline.