

Abstract
Interferon-γ receptor 2 (IFN-γR2) deficiency is a rare primary immunodeficiency characterized by predisposition to infections with weakly virulent mycobacteria, such as environmental mycobacteria and BCG vaccines. We describe here two children with IFN-γR2 deficiency, from unrelated, consanguineous kindreds of Arab and Israeli descent. The first patient was a boy who died at the age of 4.5 years, from recurrent, disseminated disease caused by Mycobacterium simiae. His IFN-γR2 defect was autosomal recessive and complete. The second patient was a girl with multiple disseminated mycobacterial infections, including infection with M. simiae. She died at the age of five years, a short time after the transplantation of umbilical cord blood cells from an unrelated donor. Her IFN-γR2 defect was autosomal recessive and partial. Autosomal recessive IFN-γR2 deficiency is life-threatening, even in its partial form, and genetic diagnosis and familial counseling are therefore particularly important for this condition. These two cases are the first of IFN-γR2 deficiency associated with M simiae infection to be described.
Keywords: MSMD, Mycobacterium simiae, interferon and IFN-γR2 deficiency
Introduction
Mendelian susceptibility to mycobacterial disease (MSMD) is a rare disorder predisposing otherwise healthy individuals to severe clinical disease upon infection with weakly virulent mycobacteria. These mycobacterial species include: Mycobacterium bovis Bacille Calmette-Guerin (BCG) vaccine substrains and non-tuberculous environmental mycobacteria (EM), such as M. fortuitum, M. chelonae, M. abscessus, M. avium complex, M. kansasii, M. marinarum, M. scrofulaceum, M. smegmatis, M. peregrinum, M. tilburgii and, more rarely, M. simiae [1-4]. Affected individuals, usually young children, are also susceptible to Salmonella [4-7] and to the more virulent mycobacterium M. tuberculosis, in endemic areas [1, 4, 7-9]. A high degree of consanguinity was observed in MSMD patients [2, 5]. The genetic dissection of MSMD has revealed germline mutations in nine genes (IFNGR1, IFNGR2, STAT1, IL12B, IL12RB1, IRF8, ISG15, NEMO and CYBB), the products of which are involved in IFN-γ-mediated immunity [5, 6, 10-13]. GATA2 mutations also confer a predisposition to mycobacterial infection, but no patient with a strict MSMD phenotype and such a mutation has yet been reported [14, 15]. Mutations in the gene encoding the second chain of the interferon-γ receptor (IFNGR2) have been identified as one of the genetic causes of MSMD [16-18]. AR complete IFN-γR2 deficiency displays complete penetrance and the most severe phenotype of any IFN-γR2 deficiency, with frequent recurrences and high mortality [18-23]. The partial form is generally less severe but it also displays complete clinical penetrance [18, 23, 24]. A heterozygous mutation in IFNGR2 caused autosomal dominant (AD) IFN-γR2 deficiencies through haploinsufficiency with incomplete penetrance [17]. A dominant-negative effect has been demonstrated in vitro for one mutation in a healthy individual [22]. In total, 12, 5 and 2 cases of AR complete, partial, and AD forms of MSMD, respectively, have been reported to date [17, 19-25]. We report here the cases of two unrelated children suffering from MSMD and M simiae infection, caused by AR complete and partial IFN-γR2 deficiencies due to two previously unknown homozygous mutations, one resulting in a complete absence of detectable receptor expression and completely abolished signaling upon stimulation with IFN-γ, and the other involving residual receptor expression and reduced signaling. As in other cases of AR IFN-γ-R2 deficiency [18, 21, 24], both patients have high level of IFN-γ in plasma.
Case report
Patient 1 (P1) (born in 2009): This male child, was born after 40 weeks of gestation, by normal spontaneous vaginal delivery. He weighed 3.6 kg at birth and was the 5th child of consanguineous Israeli Arab parents (Figure 1A). The parents and siblings were all healthy. The mother had 13 siblings, eight of whom had died before six weeks of age of unknown cause. The other five siblings remain healthy. All the other members of the extended family are healthy and were vaccinated with BCG without complications. At the age of 13 months, P1 was admitted to the hospital with left lower lobe pneumonia and mild pleural effusion not responding to oral amoxicillin treatment. The patient recovered after treatment with ceftriaxone and azithromycin. At the age of two years, he was hospitalized for right middle and lower lobe pneumonia with pleural effusion. He recovered after drainage of the pleural effusion and treatment with cefuroxime. No pathogen was isolated from the pleural effusion or blood cultures. At the age of 2.5 years, P1 was admitted to the hospital for an episode of fever and abdominal pain. He underwent surgery, which revealed a mass of lymph nodes close to the ileo-cecal valve. These lymph nodes were cultured, yielding non-typhoidal Salmonella (group C) that was susceptible to ampicillin, erythromycin and ceftriaxone and M. simiae displaying a high level of resistance to all drugs other than cycloserine (CYC). Blood culture was also positive for M. simiae. P1 was treated with clarithromycin (CLR) for the Salmonella infection, and with rifabutin (RIF), ethambutol (EMB), CYC and CLR for the mycobacterial infection. His condition gradually improved over a six-month period with negative blood cultures, the resolution of fever, weight gain and improvements in markers of inflammation, such as complete blood count (CBC), C-reactive protein (CRP) concentration and erythrocyte sedimentation rate (ESR). P1, still on the same drug regimen, was re-admitted at the age of 3.5 years, due to right upper and middle lobe pneumonia and mild pleural effusion. An enlarged spleen and liver were noted on physical examination, together with an enlargement of abdominal lymph nodes. Blood and sputum cultures again yielded M. simiae. The treatment was replaced with CYC, CLR, moxifloxacin and high-dose trimethoprim-sulfamethoxazole, with no improvement. Immunological evaluation included testing for immunoglobulins (Ig) and subclasses of IgG, immunophenotyping and assessments of the T-cell response to mitogens, HIV and neutrophil function tests, to rule out chronic granulomatous disease. Negative results were obtained in all these tests. The patient died at home at the age of 4.5 years, and no information is available concerning the immediate cause of death.
Fig. 1. Loss-of-function IFNGR2 alleles associated with MSMD.
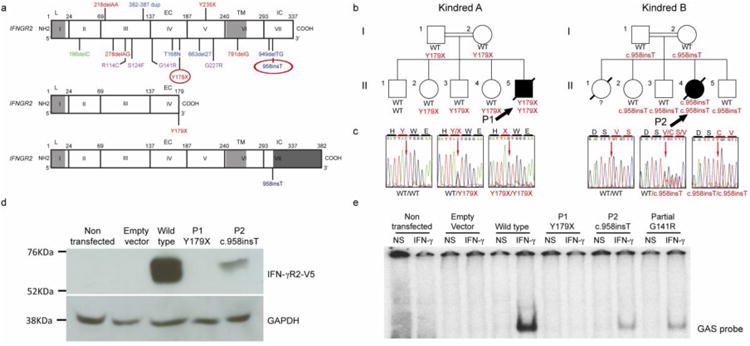
a) Schematic representation of the human IFNGR2 gene and corresponding protein with all previously described MSMD-associated mutations: in blue, mutations causing AR complete IFN-γR2 deficiency with detectable surface expression; in red, AR IFN-γR2 deficiency with no receptor expression; in purple, AR partial IFN-γR2 deficiency, and in green, AD IFN-γR2 deficiency. The different domains of the protein are indicated as L (leader peptide), EC (extracellular domain), TM (transmembrane domain) and IC (intracellular domain). Below this diagram, the consequences of the two mutations described here are shown: in the middle panel, a short IFN-γR2 caused by the creation of a premature stop codon (Y179X) and at the bottom, a longer predicted protein caused by the c.958insT variant. The extra portion of the protein due to the frame shift is indicated at dark gray at the end of the diagram.
b) Familial segregation of the two mutations (Y179X and c.958insT) found in IFNGR2. The index case (P1 and P2) of each pedigree is indicated with solid symbols and an arrow. Each kindred is designated by a capital letter (A-B), and each generation by a Roman numeral (I-II).
c) Electropherograms of a wild-type homozygous individual, a heterozygous carrier and a homozygous carrier are shown in this panel, in which the position of the mutant form is indicated with an arrow.
d) Overexpression of the WT and mutant alleles of IFNGR2 in IFN-γR2-deficient SV40-fibroblasts, revealing a lack of expression of Y179X and the residual expression of 958insT. The results presented are from one experiment representative of the three independent experiments carried out.
e) EMSA was carried out after IFN-γ stimulation of the same transfected cells as in D. A complete absence of DNA binding was observed for the nonsense mutation. The frameshift insertion showed a partial DNA binding, which was comparable to a previously reported partial deficient mutation. The results shown are representative of five independent experiments.
Patient 2 (P2) (born in 2009): This female child was born from consanguineous parents of Palestinian descent. She was the fourth out of five children. The oldest had developed an undiagnosed recurrent infection that led to progressive respiratory failure at the age of three months, leading to his death. The rest of the siblings and parents are healthy and were vaccinated with BCG at the age of one month. At the age of five months, P2 was admitted to the hospital while suffering an episode of severe diarrhea and a hemoglobin (HgB) concentration of 5g per 100ml of blood. Upon physical examination, a significantly enlarged axillary lymph node was observed. Lymph node and bone marrow biopsy were both positive for M. bovis. Combined antimycobacterial treatment was started with Isoniazid (INH), RIF and EMB, and continued for 27 months until P2 presented with high fever, limping, excessive leukocytosis and an increase in acute-phase reactants (ESR and CRP). Extensive testing was carried out and a few lytic bone lesions and hyperechogenic liver lesions were observed. Bone biopsy culture was negative, but liver biopsy culture was positive for M. simiae resistant to INH, RIF and EMB. However, M. simiae was susceptible to CYC, and therefore, was immediately added to the antimycobacterial treatment. At three years of age, she presented with significant axillary lymphadenopathy, whose culture was positive for M. fortuitum. This new episode was treated by adding macrolide to the regimen, which achieved some lymph node improvement. At five years of age, the patient was taken to the local hospital due to focal seizure. Lumbar puncture (LP) did not detect any pleocytosis or increase in protein content. However, the computed tomography (CT) revealed the presence of a few brain abscesses. Biopsy of these abscesses was positive for M. simiae. A massive antimycobacterial treatment was implemented, but the patient's condition worsened and she lost her vision from one eye and developed hemiparesis. In addition, brain imaging showed a progression of the abscesses despite repeated surgical debridement. As a last effort, umbilical cord blood (UCB) transplantation was performed from an unrelated donor, which was shown to be successfully engrafted at day +17. However, the progression of the mycobacterial brain abscess continued, and the patient's neurological status continued to deteriorate until her death on day +61 after transplantation.
The clinical presentation of these two cases led us to hypothesize that both patients suffered from MSMD. We carried out whole-exome sequencing (WES) on genomic DNA (gDNA) from P1 (kindred A, P1)[26]. Using this method and the human gene connectome [27], we identified a homozygous nonsense mutation in IFNGR2. This mutation was located in exon 4 and replaced the C residue in position 537 with a G, leading to the insertion of a premature stop codon (Y179X) (Figures 1B-C). In the case of P2 (kindred B, P2), we performed whole-genome linkage analysis on the family, and IFNGR2 was found to be located within one of the linkage regions. Sanger sequencing revealed the insertion of a single nucleotide, a T residue in position 958, c.958insT, resulting in a frameshift leading to the production of a predicted protein of 382 amino acids, rather than the 337-amino acid wild-type protein (319Sfs382X) (Figures 1B-C). These mutations were not found in any of the public databases searched — dbSNP, the 1000 Genomes database (1092 subjects) or the NHLBI GO exome sequencing project database (6503 subjects) — or in our own exome database containing data for more than 1800 patients with other diseases. The familial segregation pattern (Figures 1B-C) showed that there were no other homozygous carriers of the mutations. We introduced a wild-type or each mutant IFNGR2 cDNA into a pcDNA3.1 (Invitrogen) vector containing a C-terminal V5-tag. Overexpression of these constructs in IFN-γR2-deficient SV40-immortalized fibroblasts (SV40-fibroblasts) showed that the Y179X mutation led to a complete lack of IFN-γR2 expression whereas the c.958insT variant resulted in residual expression (Figure 1D). Transfection efficiency was assessed by determining IFNGR2 mRNA levels by RT-qPCR (Supplemental figure 1A). We stimulated these SV40-fibroblasts transfected with each allele and an additional plasmid containing the already described mutant responsible for partial IFN-γR2 deficiency [18] with IFN-γ (104 IU/ml) for 20 minutes and GAF DNA-binding activity was then assessed by electophoretic mobility shift assay (EMSA), as previously described [18]. Cells transfected with the Y179X allele displayed a total abolition of GAF DNA-binding activity, whereas this activity was impaired but not abolished with the c.958insT variant, suggesting complete deficiency for Y179X and partial deficiency for c.958insT (Figure 1E). Overall, cytokine responsiveness via the JAK/STAT pathway was confirmed by stimulation of the cells with Hyper-IL-6, a fusion protein of IL-6 and soluble IL-6R, and subsequent measurement of pSTAT3 [28] (Supplementary figure 1B). The partial deficiency of P2 was confirmed by stimulating with IFN-γ SV40-fibroblasts from this patient and appropriate controls, including the partially IFN-γR2-deficient cells described in a previous study and a complete deficient c.278delAG [18] (Supplemental figure 2A). The cytokine response through the JAK/STAT pathway was assessed by stimulating the cells with Hyper-IL-6 and measuring the phosphorylation of STAT3 by western blot (Supplementary figure 2B). No biological material from P1 was available. As previously described for AR IFN-γR2 deficiency, the patients had high plasma concentrations of IFN-γ (Figure 2) [21, 24]. As both patients died, we were unable to carry out functional assays on primary blood cells of these two patients.
Fig. 2. Plasma IFN-γ concentration.
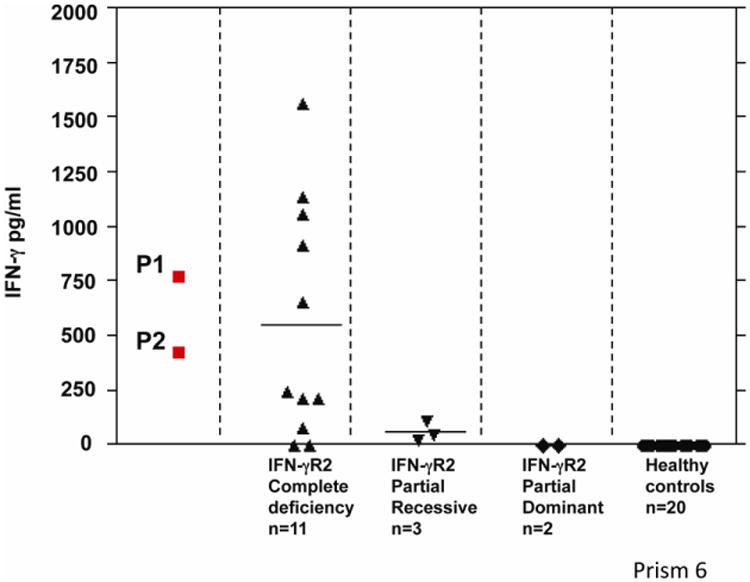
Plasma IFN-γ concentrations of the patients carrying the reported mutations. These concentrations are higher than those in healthy controls. The graph shows IFN-γ concentrations for the different forms of IFN-γR2 deficiency. The values obtained for these two patients (P1, P2) are similar to those reported for other patients with AR IFN-γR2 deficiency.
Discussion
MSMD is a rare, severe condition affecting about 1 in 100,000 individuals [2]. Over the last 20 years, 17 genetic etiologies involving nine different genes have been described, all impairing IFN-γ immunity. In the two unrelated cases described here, the patients presented infection with EM, including M. simiae. Infection with M. simiae is extremely rare and an emerging pathogenic species in the Middle East. Such infections have been reported in only a few patients with various immunodeficiencies, including HIV infection [4, 29-31], in patients with chronic renal failure [31] and, even more exceptionally in immunocompetent individuals [32]. However, to the best of our knowledge, this infection has never before been reported in a context of IFN-γR2 deficiency. The patients reported here carry two different unreported homozygous mutations of the IFNGR2 gene, p.Y179X and c.958insT. These two mutations lead to impaired cellular responses to IFN-γ. These patients have high plasma IFN-γ concentrations, as reported in other cases of AR IFN-γR2 deficiency [16, 19, 22]. The severity of the two forms of AR IFN-γR2 deficiency is further illustrated by the deaths of the two patients described here from infectious complications, despite stem cell transplantation in P2. The development of molecular diagnostic tools potentially allowing physicians to offer preventive treatments to patients and genetic counseling to families suffering from this and other severe infections [33] is of considerable interest. For example, an early diagnosis of IFNGR2 mutation is a contraindication for BCG vaccination and may constitute an indication for hematopoietic stem cell transplantation (for complete defects) or IFN-γ treatment (for partial defects) [15]. The two cases described here provide support for the notion that IFNGR2 mutations or related genetic disorders should be considered in patients with M. simiae disease.
Supplementary Material
Acknowledgments
We would like to thank the patients and their families for their collaboration, and both branches of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions and support. We would like to thank the staff of the Pediatric Hemato-oncology and Bone Marrow Transplantation, Hadassah and Pediatric Department, Shaarei Zedek Hospital Jerusalem, where the patients were treated. We would also like to thank Yelena Nemirovskaya, Lahouari Amar, Eric Anderson, Martine Courat and Tiffany Nivare for administrative support. This research was funded in part by the National Institute of Allergy and Infectious Diseases grant number 5R37AI095983, the National Center for Research Resources and the National Center for Advancing Sciences (NCATS) of the National Institutes of Health grant number 8UL1TR000043, The Rockefeller University, Institut National de la Santé et de la Recherche Médicale (INSERM), Paris Descartes University, the St. Giles Foundation and by the French National Research Agency (Agence Nationale de la Recherche ANR13-ISV3-0001-01). RMB was funded by the European Molecular Biology Organization (EMBO) and XFK was supported by Stony Wold-Herbert Fund, Choh-Hao Li Memorial Fund Scholar award and the Shanghai Educational Development Foundation.
Footnotes
Conflict of interest: The authors have no conflict of interest to declare.
References
- 1.Alcaïs A, Fieschi C, Abel L, Casanova JL. Tuberculosis in children and adults: two distinct genetic diseases. J Exp Med. 2005;202:1617–21. doi: 10.1084/jem.20052302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 3.Schepers K, Schandené L, Bustamante J, Vooren JP, Suremain M, Casanova JL, Yombi J, Jacobs F, Mascart F, Goffard JC. IL-12Rβ1 Deficiency and Disseminated Mycobacterium tilburgii Disease. J Clin Immunol. 2013;33:1285–8. doi: 10.1007/s10875-013-9941-y. [DOI] [PubMed] [Google Scholar]
- 4.de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, Al-Muhsen S, Jannière L, Rose Y, de Suremain M, Kong XF, Filipe-Santos O, Chapgier A, Picard C, Fischer A, Dogu F, Ikinciogullari A, Tanir G, Al-Hajjar S, Al-Jumaah S, Frayha HH, AlSum Z, Al-Ajaji S, Alangari A, Al-Ghonaium A, Adimi P, Mansouri D, Ben-Mustapha I, Yancoski J, Garty BZ, Rodriguez-Gallego C, Caragol I, Kutukculer N, Kumararatne DS, Patel S, Doffinger R, Exley A, Jeppsson O, Reichenbach J, Nadal D, Boyko Y, Pietrucha B, Anderson S, Levin M, Schandené L, Schepers K, Efira A, Mascart F, Matsuoka M, Sakai T, Siegrist CA, Frecerova K, Blüetters-Sawatzki R, Bernhöft J, Freihorst J, Baumann U, Richter D, Haerynck F, De Baets F, Novelli V, Lammas D, Vermylen C, Tuerlinckx D, Nieuwhof C, Pac M, Haas WH, Müller-Fleckenstein I, Fleckenstein B, Levy J, Raj R, Cohen AC, Lewis DB, Holland SM, Yang KD, Wang X, Wang X, Jiang L, Yang X, Zhu C, Xie Y, Lee PPW, Chan KW, Chen TX, Castro G, Natera I, Codoceo A, King A, Bezrodnik L, Di Giovani D, Gaillard MI, de Moraes-Vasconcelos D, Grumach AS, da Silva Duarte AJ, Aldana R, Espinosa-Rosales FJ, Bejaoui M, Bousfiha AA, Baghdadi JE, Özbek N, Aksu G, Keser M, Somer A, Hatipoglu N, Aydogmus Ç, Asilsoy S, Camcioglu Y, Gülle S, Ozgur TT, Ozen M, Oleastro M, Bernasconi A, Mamishi S, Parvaneh N, Rosenzweig S, Barbouche R, Pedraza S, Lau YL, Ehlayel MS, Fieschi C, Abel L, Sanal O, Casanova JL. Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Medicine. 2010;89:381–402. doi: 10.1097/MD.0b013e3181fdd832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J, Jouanguy E, Boisson-Dupuis S, Fieschi C, Picard C, Casanova JL. Inborn errors of IL-12/23- and IFN-γ-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. 2006;18:347–61. doi: 10.1016/j.smim.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Haverkamp MH, van de Vosse E, vD JT. Nontuberculous mycobacterial infections in children with inborn errors of the immune system. J Infect. 2014;68:S134–S50. doi: 10.1016/j.jinf.2013.09.024. [DOI] [PubMed] [Google Scholar]
- 7.Prando C, Samarina A, Bustamante J, Boisson-Dupuis S, Cobat A, Picard C, AlSum Z, Al-Jumaah S, Al-Hajjar S, Frayha H, Alangari A, Al-Mousa H, Mobaireek K, Ben-Mustapha I, Adimi P, Feinberg J, de Suremain M, Jannière L, Filipe-Santos O, Mansouri N, Stephan J, Nallusamy R, Kumararatne D, Bloorsaz M, Ben-Ali M, Elloumi-Zghal H, Chemli J, Bouguila J, Bejaoui M, Alaki E, AlFawaz T, Al Idrissi E, ElGhazali G, Pollard A, Murugasu B, Wah Lee B, Halwani R, Al-Zahrani M, Al Shehri M, Al-Zahrani M, Bin-Hussain I, Mahdaviani S, Parvaneh N, Abel L, Mansouri D, Barbouche R, Al-Muhsen S, Casanova J. Inherited IL-12p40 deficiency: genetic, immunologic, and clinical features of 49 patients from 30 kindreds. Medicine. 2013;92:109–22. doi: 10.1097/MD.0b013e31828a01f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boisson-Dupuis S, El Baghdadi J, Parvaneh N, Bousfiha A, Bustamante J, Feinberg J, Samarina A, Grant A, Janniere L, El Hafidi N, Hassani A, Nolan D, Najib J, Camcioglu Y, Hatipoglu N, Aydogmus C, Tanir G, Aytekin C, Keser M, Somer A, Aksu G, Kutukculer N, Mansouri D, Mahdaviani A, Mamishi S, Alcais A, Abel L, Casanova J. IL-12Rβ1 deficiency in two of fifty children with severe tuberculosis from Iran, Morocco, and Turkey. PLoS One. 2011;6:e18524. doi: 10.1371/journal.pone.0018524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabarsi P, Marjani M, Mansouri N, Farnia P, Boisson-Dupuis S, Bustamante J, Abel L, Adimi P, Casanova JL, Mansouri D. Lethal tuberculosis in a previously healthy adult with IL-12 receptor deficiency. J Clin Immunol. 2011;31:537–9. doi: 10.1007/s10875-011-9523-9. [DOI] [PubMed] [Google Scholar]
- 10.Casanova JL, Abel L. The genetic theory of infectious diseases: a brief history and selected illustrations. Annu Rev Genomics Hum Genet. 2013;14:215–43. doi: 10.1146/annurev-genom-091212-153448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, Grant AV, Marchal CC, Hubeau M, Chapgier A, de Beaucoudrey L, Puel A, Feinberg J, Valinetz E, Janniere L, Besse C, Boland A, Brisseau JM, Blanche S, Lortholary O, Fieschi C, Emile JF, Boisson-Dupuis S, Al-Muhsen S, Woda B, Newburger PE, Condino-Neto A, Dinauer MC, Abel L, Casanova JL. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. 2011;12:213–21. doi: 10.1038/ni.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, Mansouri N, Okada S, Bryant VL, Kong XF, Kreins A, Velez MM, Boisson B, Khalilzadeh S, Ozcelik U, Darazam IA, Schoggins JW, Rice CM, Al-Muhsen S, Behr M, Vogt G, Puel A, Bustamante J, Gros P, Huibregtse JM, Abel L, Boisson-Dupuis S, Casanova JL. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science. 2012;337:1684–8. doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, Fortin A, Haniffa M, Ceron-Gutierrez L, Bacon CM, Menon G, Trouillet C, McDonald D, Carey P, Ginhoux F, Alsina L, Zumwalt TJ, Kong XF, Kumararatne D, Butler K, Hubeau M, Feinberg J, Al-Muhsen S, Cant A, Abel L, Chaussabel D, Doffinger R, Talesnik E, Grumach A, Duarte A, Abarca K, Moraes-Vasconcelos D, Burk D, Berghuis A, Geissmann F, Collin M, Casanova JL, Gros P. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. 2011;365:127–38. doi: 10.1056/NEJMoa1100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, Arthur DC, Gu W, Gould CM, Brewer CC, Cowen EW, Freeman AF, Olivier KN, Uzel G, Zelazny AM, Daub JR, Spalding CD, Claypool RJ, Giri NK, Alter BP, Mace EM, Orange JS, Cuellar-Rodriguez J, Hickstein DD, Holland SM. GATA2 deficiency: a protein disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123:809–21. doi: 10.1182/blood-2013-07-515528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holland SM, Casanova JL. Inherited disorders of the interleukin-12-interleukin-23/interferon-γ circuit. In: Ochs HD, Smith CIE, JM P, editors. Primary immunodeficiency diseases: a molecular and genetic approach. 3rd. Vol. 1. USA: Oxford university Press; 2014. pp. 450–66. [Google Scholar]
- 16.Dorman S, Picard C, Lammas D, Heyne K, van Dissel J, Baretto R, Rosenzweig S, Newport M, Levin M, Roesler J, Kumararatne D, Casanova J, Holland S. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. 2004;364:2113–21. doi: 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- 17.Kong XF, Vogt G, Itan Y, Macura-Biegun A, Szaflarska A, Kowalczyk D, Chapgier A, Abhyankar A, Furthner D, Djambas Khayat C, Okada S, Bryant VL, Bogunovic D, Kreins A, Moncada-Vélez M, Migaud M, Al-Ajaji S, Al-Muhsen S, Holland SM, Abel L, Picard C, Chaussabel D, Bustamante J, Casanova JL, Boisson-Dupuis S. Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease. Hum Mol Genet. 2013;22:769–81. doi: 10.1093/hmg/dds484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moncada-Vélez M, Martinez-Barricarte R, Bogunovic D, Kong XF, Blancas-Galicia L, Tirpan C, Aksu G, Vincent QB, Boisson B, Itan Y, Ramírez-Alejo N, Okada S, Kreins AY, Bryant VL, Franco JL, Migaud M, Espinosa-Padilla S, Yamazaki-Nakashimada M, Espinosa-Rosales F, Kutukculer N, Abel L, Bustamante J, Vogt G, Casanova JL, Boisson-Dupuis S. Partial IFN-γR2 deficiency is due to protein misfolding and can be rescued by inhibitors of glycosylation. Blood. 2013;122:2390–14. doi: 10.1182/blood-2013-01-480814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogt G, Chapgier A, Yang K, Chuzhanova N, Feinberg J, Fieschi C, Boisson-Dupuis S, Alcais A, Filipe-Santos O, Bustamante J, de Beaucoudrey L, Al-Mohsen I, Al-Hajjar S, Al-Ghonaium A, Adimi P, Mirsaeidi M, Khalilzadeh S, Rosenzweig S, de la Calle Martin O, Bauer T, Puck J, Ochs H, Furthner D, Engelhorn C, Belohradsky B, Mansouri D, Holland S, Schreiber R, Abel L, Cooper D, Soudais C, Casanova J. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet. 2005;37:692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- 20.Vogt G, Bustamante J, Chapgier A, Feinberg J, Boisson Dupuis S, Picard C, Mahlaoui N, Gineau L, Alcaïs A, Lamaze C, Puck JM, de Saint Basile G, Khayat CD, Mikhael R, Casanova JL. Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med. 2008;205:1729–37. doi: 10.1084/jem.20071987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toyoda H, Ido M, Nakanishi K, Nakano T, Kamiya H, Matsumine A, Uchida A, Mizutani H, de Beaucoudrey L, Vogt G, Boisson-Dupuis S, Bustamante J, Casanova JL, Komada Y. Multiple cutaneous squamous cell carcinomas in a patient with interferon γ receptor 2 (IFNγR2) deficiency. J Med Genet. 2010;47:631–4. doi: 10.1136/jmg.2009.072108. [DOI] [PubMed] [Google Scholar]
- 22.Rosenzweig SD, Dorman SE, Uzel G, Shaw S, Scurlock A, Brown MR, Buckley RH, Holland SM. A novel mutation in IFN-γ receptor 2 with dominant negative activity: biological consequences of homozygous and heterozygous states. J Immunol. 2004;173:4000–8. doi: 10.4049/jimmunol.173.6.4000. [DOI] [PubMed] [Google Scholar]
- 23.Döfflnger R, Jouanguy E, Dupuis S, Fondanèche MC, Stephan JL, Emile JF, Lamhamedi-Cherradi S, Altare F, Pallier A, Barcenas-Morales G, Meinl E, Krause C, Pestka S, Schreiber RD, Novelli F, Casanova JL. Partial interferon-γ receptor signaling chain deficiency in a patient with Bacille Calmette-Guérin and Mycobacterium abscessus Infection. J Infect Dis. 2000;181:379–84. doi: 10.1086/315197. [DOI] [PubMed] [Google Scholar]
- 24.Kilic SS, van Wengen A, de Paus RA, Celebi S, Meziane B, Hafizoglu D, van Dissel JT, van de Vosse E. Severe disseminated mycobacterial infection in a boy with a novel mutation leading to IFN-γR2 deficiency. J Infect. 2012;65:568–72. doi: 10.1016/j.jinf.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Dorman S, Holland S. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101:2364–9. doi: 10.1172/JCI2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Byun M, Abhyankar A, Lelarge V, Plancoulaine S, Palanduz A, Telhan L, Boisson B, Picard C, Dewell S, Zhao C, Jouanguy E, Feske S, Abel L, Casanova JL. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med. 2010;207:2307–12. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itan Y, Zhang SY, Vogt G, Abhyankar A, Herman M, Nitschke P, Fried D, Quintana-Murci L, Abel L, Casanova JL. The human gene connectome as a map of short cuts for morbid allele discovery. Proc Natl Acad Sci USA. 2013;110:5558–63. doi: 10.1073/pnas.1218167110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer M, Goldschmitt J, Peschel C, Brakenhoff JP, Kallen KJ, Wollmer A, Grötzinger J, R-J S. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat Biotechnol. 1997;15:142–5. doi: 10.1038/nbt0297-142. [DOI] [PubMed] [Google Scholar]
- 29.Vitoria M, González-Domínguez M, Salvo S, Crusells M, Letona S, Samper S, Sanjoaquín I. Mycobacterium simiae pulmonary infection unmasked during immune reconstitution in an HIV patient. Diagn Microbiol Infect Dis. 2013;75:101–3. doi: 10.1016/j.diagmicrobio.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Narang R, Narang P, Jain AP, Mendiratta DK, Joshi R, Lavania M, Das R, Katoch VM. Disseminated disease caused by Mycobacterium simiae in AIDS patients: a report of three cases. Clin Microbiol Infect. 2010;16:912–4. doi: 10.1111/j.1469-0691.2009.03021.x. [DOI] [PubMed] [Google Scholar]
- 31.Cortés-Torres N, González-Y-Merchand J, González-Bonilla C, García-Elorriaga G. Molecular analysis of mycobacteria isolated in Mexican patients with different immunodeficiencies in a tertiary care hospital. Arch Med Res. 2013;44:562–9. doi: 10.1016/j.arcmed.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Balkis M, Kattar M, Araj G, Kanj S. Fatal disseminated Mycobacterium simiae infection in a non-HIV patient. Int J Infect Dis. 2009;13:e286–e7. doi: 10.1016/j.ijid.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 33.Alcaïs A, Quintana-Murci L, Thaler DS, Schurr E, Abel L, Casanova JL. Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity? Ann N Y Acad Sci. 2010;1214:18–33. doi: 10.1111/j.1749-6632.2010.05834.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.