

Abstract
Objective
The mechanism of action of anti-B cell therapy in multiple sclerosis (MS) is not fully understood. Here, we compared the effect of anti-CD20 therapy on microglial activation in two distinct focal rat models of MS.
Methods
The effect of anti-CD20 therapy on lesion formation and extralesional microglial activation was evaluated in the fDTH-EAE (experimental allergic encephalomyelitis) model, which is a focal demyelinating type-IV delayed-type hypersensitivity lesion. For comparison, effects were also assessed in the focal humoral MOG model induced by intracerebral injection of cytokine in myelin oligodendrocyte glycoprotein immunized rats. Microglial activation was assessed in situ and in vivo using the TSPO SPECT ligand [125I]DPA-713, and by immunostaining for MHCII. The effect of treatment on demyelination and lymphocyte recruitment to the brain were evaluated.
Results
Anti-CD20 therapy reduced microglial activation, and lesion formation in the humoral model, but it was most effective in the antibody-independent fDTH-EAE. Immunohistochemistry for MHCII also demonstrated a reduced volume of microglial activation in the brains of anti-CD20-treated fDTH-EAE animals, which was accompanied by a reduction in T-cell recruitment and demyelination. The effect anti-CD20 therapy in the latter model was similarly strong as compared to the T-cell targeting MS compound FTY720.
Interpretation
The suppression of lesion development by anti-CD20 treatment in an antibody-independent model suggests that B-cells play an important role in lesion development, independent of auto-antibody production. Thus, CD20-positive B-cell depletion has the potential to be effective in a wider population of individuals with MS than might have been predicted from our knowledge of the underlying histopathology.
Introduction
Multiple sclerosis (MS) is an inflammatory demyelinating and neurodegenerative disease of the CNS central nervous system (CNS) with both focal and diffuse pathology.1 An aberrant T-cell response has been assumed to be the predominant pathophysiological mechanism2 and current standard therapies such as interferon, glatiramer acetate, natalizumab, and FTY720 (fingolimod) have been developed based on this concept.3 However, numerous histopathological studies have established that focal lesion formation is a heterogeneous process that can be divided into four distinct patterns.4 About half of the lesions are characterized by mechanisms that result in complement deposition, including C1q and terminal complement complex (TCC), which, when coupled to other long-known features (elevated IgG index, presence of oligoclonal bands, and specific autoantibodies in cerebrospinal fluid (CSF)), hint at the involvement of antibody in these lesions.5 The presence of B-cells in lesions,6 the formation of meningeal tertiary follicle-like regions containing B-cells,7 and evidence for their clonal expansion in CSF and brain tissue8 provide additional conceptual foundation for therapeutic targeting of B-cells in MS. Furthermore, in the long-term course of MS, the presence of meningeal B-cells seems to be linked to the degree of cortical microglial activation and neuronal loss, the latter being the morphological substrate for clinical disease progression. The first clinical trials in relapsing-remitting MS (RRMS) using the anti-CD20 antibody rituximab were initiated based on the hypothesis that the elimination of B-cells would indirectly improve the disease course by the eventual reduction of auto-antibody formation.9 However, in hindsight, this is unlikely to be the dominant mode of action: firstly, the suppression of new lesion formation in magnetic resonance imaging occurred rapidly (for instance within 12 weeks after start of therapy), which is too short to be explained by the reduction of auto-antibody levels, given their half-life time in circulation.10 Secondly, the fraction of patients that did not develop new lesions (84%) well exceeded the fraction of patients expected to have T-cell plus humoral pathology. Subsequently, these striking results have been reproduced with two other anti-CD20 compounds (ocrelizumab, ofatumumab).11 In a 24-week phase II trial in 220 RRMS patients, ocrelizumab 600 mg treatment resulted in an 89% reduction in Gd-enhancing lesions and an 80% reduction in annualized relapse rate versus placebo.12 The aims of the present study were threefold: (1) to determine whether anti-CD20 therapy would inhibit lesion formation in a robust cell-mediated antibody-independent model of MS, in comparison with a humoral model (focal myelin oligodendrocyte glycoprotein [fMOG]-induced experimental allergic encephalomyelitis [EAE]), (2) to determine whether anti-CD20 therapy can reduce microglial activation (a marker of sustained neurodegeneration) in lesional and extralesional normal-appearing white matter, using a novel radioligand ([125I]DPA-713) that is targeted toward the 18kD translocator protein (TSPO), and (3) to compare anti-CD20 therapy with fingolimod, an MS therapy compound that exerts its activity mainly via T-cells.
Subjects/Materials and Methods
Animals
Male Lewis rats (Harlan, Bicester, UK) were at least 3-weeks old at the time of experimentation. Ethical committee approval was obtained for all experiments (UK Home Office License 30/2524).
fMOG-EAE and fDTH-EAE lesion
In both cases, lesion induction was established as previously described13 (see Data S1 for more details).
Therapy
5 mg/kg of anti-CD20 monoclonal antibody (Genentech, South San Francisco, USA) or IgG2a isotype-matched control (Genentech, USA) was administered intravenously on days 0 and 7. One group of focal delayed-type hypersensitivity (fDTH)-EAE animals received oral fingolimod 0.1 mg/kg per day from days 5 to 12 (Sigma-Aldrich, Gillingham, UK).
Tissue processing and immunohistochemistry
Animals were killed on Day 12. Tissue was either perfusion-fixed with periodate-lysine-paraformaldehyde (PLP) as previously described14 or used as fresh tissue, which was post-fixed as cryosections. Immunohistochemistry was performed to quantify the number of T-cells (anti-Phycoerythrin, OX22; Sigma-Aldrich), B-cells (anti-WAG-Rjj; AbCam Milton, UK) and activation of microglia (anti-MHCII, OX6; Serotec, Kidlington, UK). We also stained the tissue from the Control, fDTH-EAE, and fMOG-EAE lesioned animals for the presence of C5b-9 TCC. Deposition was only observed in while matter in the MOG-EAE lesioned animals (Fig. S1). (For more detailed methodology see Data S1.)
Calculating lesion volume
A camera lucida was used to trace around regions of OX-6 immunopositivity or hypercellularity at the lesion center and at 500, 1000, and 1500 μm either side of the lesion center for the fMOG-EAE lesion or 100 μm intervals between −500 μm and +500 μm for the DTH-EAE lesion. Lesion area was assessed in these sections using NIH ImageJ freeware (available from: http://rsb.info.nih.gov/ij/index.html). Volumes were estimated by integration of lesion areas using the trapezium rule.
In situ hybridization
In vitro autoradiography with [125I]DPA-713 was performed as previously described.15 Briefly, 10 μm-thick brain sections were incubated for 1 h in 1.4 nmol/L [125I]DPA-713 (Genentech, USA). In adjacent sections, 10 μmol/L of racemic cold PK11195 (Sigma-Aldrich, UK) was co-administered with the [125I]DPA-713. Unbound ligand was removed by three washes in phosphate buffered saline (PBS). Slides were air dried and placed on a phosphor analyzer screen. Quantization was carried out with ImageJ software.
Radioligand preparation
[125I]DPA-713 was provided by Molecular Neuroimaging (MNI, New Haven, CT) and synthesized as previously described.15 [123I]DPA-713 was synthesized by a two step reaction from precursor, N,N-diethyl-2-[2-(4-hydroxy-phenyl)-5,7-dimethyl-pyrazolo[1,5-α]pyrimidin-3-yl]-acetamide at the University of Oxford, 37% yield with a radiochemical purity of >99%.
In vivo SPECT/CT imaging
Twelve SPECT/CT measurements were performed with [123I]DPA-713 in fDTH-EAE lesion rat models of MS (n = 6 untreated, n = 6 treated with anti-CD20 therapy). The in vivo scans were performed using a combined nano SPECT/CT (Bioscan, NW Washington, DC, USA). Twenty minutes prior to the imaging, a bolus administration of [123I]DPA-713 was given via the tail vein (19 ± 5 MBq). Static images were then acquired over a time period of 45 min.
In vivo SPECT/CT analysis
The in vivo SPECT images were reconstructed using in vivo scope (Bioscan, USA), utilizing the high noise suppression, and fine iteration settings with a final voxel size of 0.6 mm. All further image processing and analysis was performed in pixalwise modeling software (PMOD v 3.4, Zurich, Switzerland). standardized uptake values (SUV) images were calculated using the following formula:
In order to quantify the binding, identical spherical volumes of interest (14 mm3) were drawn in the injected ipsilateral and contralateral hemispheres for quantification. The binding ratio was then calculated using the following formula:
Statistical analyses
All data were analyzed using a two-tailed Student’s t-test; P < 0.05 was considered significant and carried out in Prism v5 (GraphPad software, La Jolla, CA, USA). Values are expressed as mean ± standard error of the mean (SEM).
Results
Anti-CD20 therapy significantly depletes splenic B-cells
Anti-CD20 therapy significantly reduced the number of B-cells in the spleen by day 12 in rats for both the fMOG-EAE and the fDTH-EAE models compared to controls. B-cells were especially depleted in the germinal center and marginal zone of the splenic follicles (Fig. 1A). Quantitative analysis revealed that there were significantly (P < 0.001) fewer splenic B-cells in anti-CD20-treated rats than in controls in both the fDTH (2074 ± 199 B-cells/mm2 vs. 12198 ± 528 B-cells/mm2) and the fMOG-EAE (2807 per mm2 ± 919 B-cells/mm2 vs. 12,036 ± 704 B-cells/mm2) models (Fig. 1B). Splenic T-cell numbers were then assessed following anti-CD20 therapy and revealed that anti-CD20 therapy had no effect on T-cell numbers in splenic follicles compared with IgG-treated controls (17,456 T-cells/mm2 ± 543 vs. 17,824 T-cells/mm2 ± 555 P = 0.48). Thus, it is likely that the effects of anti-CD20 therapy are consequences of selective B-cell depletion. Administration of FTY720 had no effect on splenic T cell or B cell numbers (Fig. S2).
Figure 1.
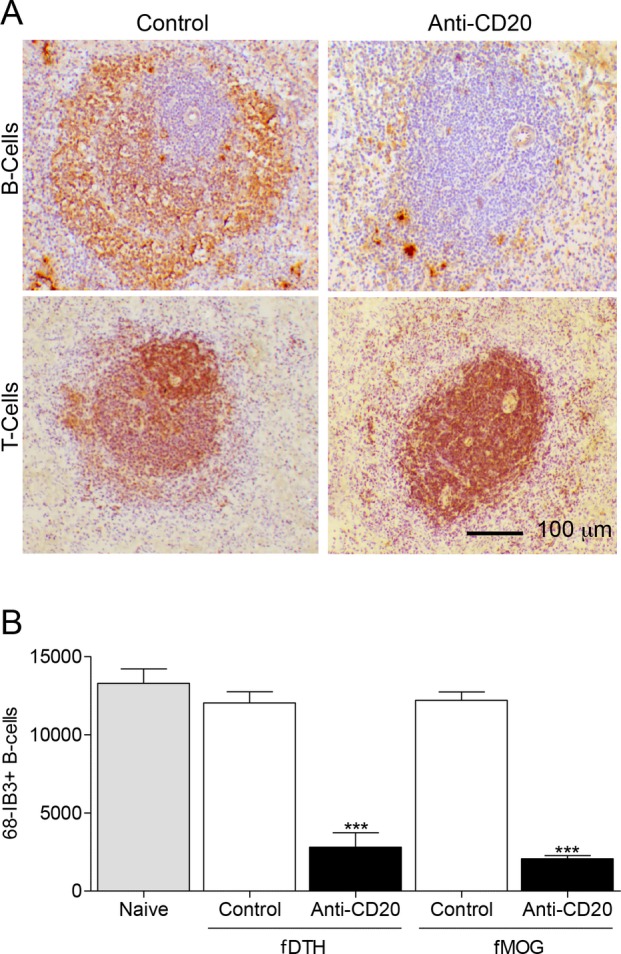
Anti-CD20 therapy significantly reduces splenic B-cells, but not T-cells, in fDTH-EAE and fMOG-EAE model of MS. (A) Immunostaining of 68-IB3-positive B-cells (brown) in rat splenic follicles in an anti-CD20-treated and a control animal. Note the loss of B-cells from the germinal center and marginal zone of the follicle in anti-CD20-treated tissue. Staining for anti-OX22 revealed that anti-CD20 therapy did not alter the number of T-cells present in the spleen. (B) Quantitative analysis of 68-IB3-positive B-cells in splenic follicles in both the DTH-EAE (Naïve, n = 3; Control, n = 4; Anti-CD20, n = 7) and fMOG-EAE (Control, n = 8; Anti-CD20, n = 8) models. ***P < 0.001 versus control in both models. Sections were counterstained with hematoxylin (blue). EAE, experimental allergic encephalomyelitis; MS, multiple sclerosis.
B-cell depletion reduces T-cell infiltration and demyelination in the fDTH-EAE model MS
The number of OX22-positive T-cells present in the lesion was significantly reduced (P = 0.005) by the treatment (1171 ± 80 T-cells/mm2 vs. 1806 ± 178 T-cells/mm2) in the fDTH-EAE animals (Fig. 2A). Demyelination, as determined by Luxol Fast Blue staining, was also reduced by anti-CD20 (0.20 ± 0.03 mm2 vs. 0.33 ± 0.05 mm2) showing that the treatment reduces structural damage (Fig. 2B and C). It should be noted that B-cells were undetectable in fDTH-EAE lesions of controls at this time point (not shown) and hence the observed effect of anti-CD20 therapy results only from its impact on the peripheral immune system. This reduction of microglial activation by anti-CD20 therapy in the fDTH-EAE lesions suggests a role of B-cells in development of antibody-independent MS lesions. Interestingly, the effect size was comparable to the effect of a therapy (fingolimod 0.1 mg kg−1 d−1 per po days 5–12) that directly targets T-cell functions (Fig. 6).
Figure 2.
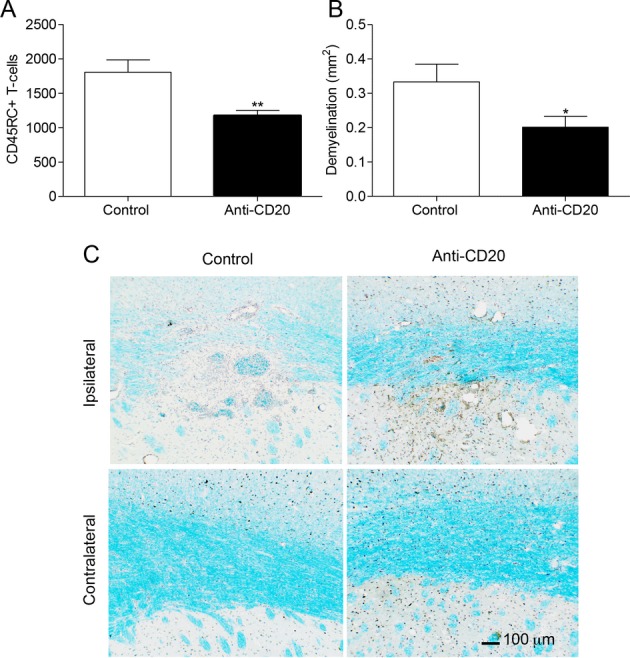
Anti-CD20 treatment reduces T-cell infiltration and demyelination in the fDTH-EAE model of MS. (A) Number of CD45RC+ T-cells in the lesion in the control animals (n = 9) and the anti-CD20-treated animals (n = 9), **P = 0.005. (B) Area of myelin loss within the lesion measured using Luxol fast blue staining in both the control animals (n = 10) and anti-CD20-treated animals (n = 9), *P = 0.049. (C) Photomicrographs demonstrating the greater loss in Luxol fast blue staining (light blue) in the control animals compared to the treated animals at the site of the lesion. A similar loss is not observed in the contralateral hemisphere (not shown). Nuclei were counter stained with Mayer’s haematoxylin (dark blue and brown). EAE, experimental allergic encephalomyelitis; MS, multiple sclerosis.
Figure 6.
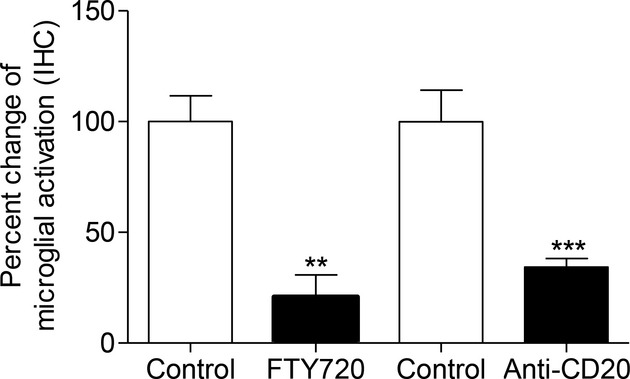
Comparison of the effect of fingolimod and anti-CD20 therapy on microglial activation in the DTH-EAE model. There was a significant reduction when using both fingolimod and anti-CD20 compared to the respective control groups; Key: **P < 0.01, ***P < 0.001. Both treatments were equally effective in reducing microglial activation. EAE, experimental allergic encephalomyelitis.
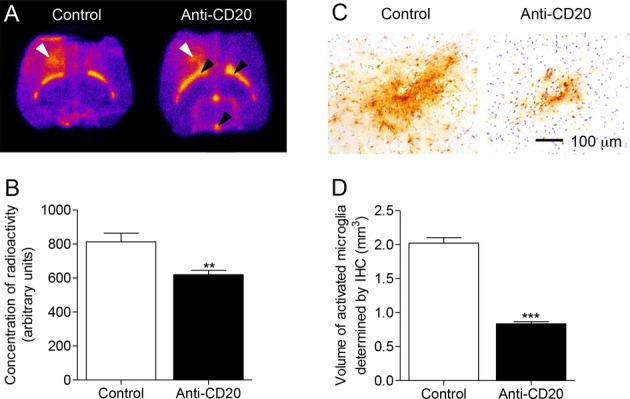
B-cell depletion reduces microglial activation and [125I]DPA-713 binding in the fDTH-EAE model MS
On the basis that B-cells had been successfully depleted in the absence of an effect on T-cells, we sought to discover whether B-cell depletion could also prevent the widespread microglial activation observed in the fDTH-EAE lesions. Ex vivo autoradiography with the TSPO ligand [125I]DPA-713 following the administration of anti-CD20 gave rise to a 53% reduction in [125I]DPA-713 binding (0.28 ± 0.08 au vs. 0.60 ± 0.18 au; P = 0.013, anti-CD20 [n = 6] and control [n = 8], respectively Fig. 3A and B). B-cell depletion reduced the hypercellular MHCII-positive lesion area by 56% at the lesion center in anti-CD20-treated fDTH-EAE rats compared to IgG-treated controls (10.3 ± 0.3 mm3 vs. 23.5 ± 0.5 mm3; P = 0.018, anti-CD20 [n = 9] and control [n = 10], respectively; Fig. 3C) (see Fig. S3 for effect of anti-CD20 on volumetric lesion profile).
Figure 3.
B-cell depletion with anti-CD20 antibody reduces the area of microglial activation in DTH-EAE rat model. (A) In situ hybridization of coronal brain sections with [125I]DPA-713. White arrows indicate the location of the lesion, black arrows demonstrate the binding in the ventricles caused by binding to ependymal cells and the Choroid Plexus. (B) [125I]DPA-713 binding in DTH-EAE lesions (Control, n = 5, anti-CD20 n = 6), *P = 0.012. (C) Immunohistochemistry showing the area of OX-6+ (MHCII, brown staining) staining surrounding a typical lesion in both a control animal and anti-CD20-treated animal. Nuclei visualized using cresyl violet (blue). (D) Volumetry of lesions calculated by measuring the area of OX-6 staining throughout the lesion (Control, n = 9; anti-CD20, n = 10), ***P < 0.001. EAE, experimental allergic encephalomyelitis.
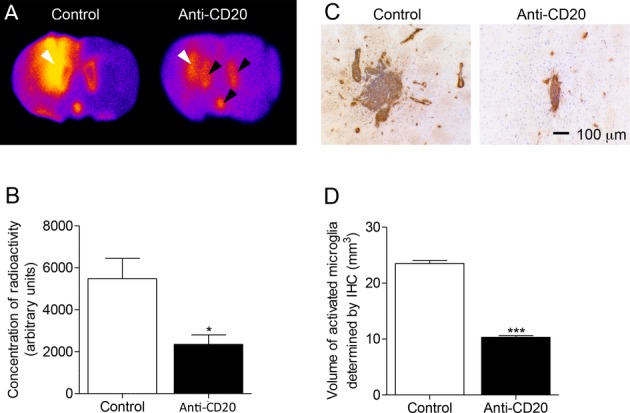
In a new cohort of animals, in vivo imaging of [123I]DPA-713 using SPECT/CT (with 123I, rather than 125I, due to better radiation penetration and shorter half-life of 13 h) showed an increased uptake of radioactivity in the ipsilateral area, as compared with the contralateral side (Fig. 4A and B). The ratio of radioligand uptake in the ipsilateral vs. the contralateral intact areas was significantly reduced in anti-CD20-treated (1.18 ± 0.07) compared with an untreated (1.59 ± 0.05) fDTH-EAE rodent model of MS (P < 0.001; Fig. 4C). These findings were verified in postmortem autoradiographic assessments to confirm the in vivo SPECT imaging data. In untreated animals, binding was significantly higher in the ipsilateral than in the contralateral side (Fig. S4A). The ratio of radioligand uptake in the basal ganglia and overlying cortex in the ipsilateral versus the contralateral hemispheres was significantly reduced in anti-CD20-treated compared with vehicle-treated fDTH-EAE animals (P = 0.047; Fig. S4B).
Figure 4.
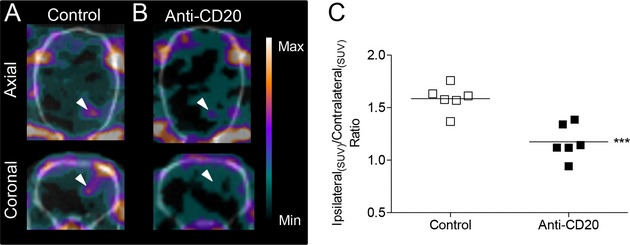
Biplanar in vivo imaging of microglial activation by SPECT. Anti-CD20 therapy reduces activated microglia in a rodent model of MS. (A) Increased activated microglia at the site of the lesion in untreated DTH-EAE model of MS. (B) Decreased activated microglia at the site of lesion in anti-CD20-treated DTH-EAE model of MS. (C) Ratio of radioligand update in the ipsilateral versus contralateral intact areas. ***P < 0.001. EAE, experimental allergic encephalomyelitis; MS, multiple sclerosis.
B-cell depletion reduces microglial activation and [125I]DPA-713 binding in a humoral model of MS
We examined the effect of anti-CD20 therapy on TSPO expression in the humoral fMOG-EAE model in order to compare it with the effect observed in the fDTH-EAE model. [125I]DPA-713 binding was significantly reduced by anti-CD20 therapy in the fMOG-EAE model (Fig. 5A). Interestingly, the reduction of microglial activation seems not to be restricted to lesions but extends to extralesional tissue (although not significant) (Fig. S3).
Figure 5.
DPA-713 binding was reduced by anti-CD20 therapy in rats in the fMOG-EAE model. (A) In situ hybridization with [125I]DPA-713. White arrows mark the area of the lesion, black area demonstrates ventricular binding. (B) Quantification of [125I]DPA-713 binding following anti-CD20 therapy (Control, n = 8, anti-CD20 n = 7), **P = 0.006. (C) Immunohistochemistry showing the area of OX-6+ (MHCII, brown staining) staining surrounding the lesion in both a control animal and a treated animal. Nuclei visualized using cresyl violet staining (blue). (D) Volumetry of lesion determined by the quantification of OX-6 positive area in both the control (n = 7) and treated animals (n = 5), ***P < 0.001. EAE, experimental allergic encephalomyelitis.
In autoradiography control studies, co-incubation of [125I]DPA-713 with nonradioactive PK11195 (a competitive TSPO ligand) led to a significant reduction in 125I signal (Fig. 5B), indicating that [125I]DPA-713 binding to the TSPO receptor is specific. Interestingly, there was prominent ependymal binding of DPA, in both the fMOG-EAE animals and the naïve animals, but to a lesser extent. An in vitro autoradiography assay (mean pixel intensity), demonstrated a 24% reduction in [125I]DPA-713 binding in anti-CD20-treated rats compared with IgG-treated controls (6.2 ± 0.3 au vs. 8.1 ± 0.5; P = 0.006, anti-CD20 (n = 7) and control (n = 8), respectively) (Fig. 5B). OX-6 immunohistochemistry for MHCII revealed a 58% reduction in the volume of microglial activation in anti-CD20-treated fMOG-EAE rats compared with IgG-treated controls (0.84 ± 0.08 mm3 vs. 2.02 ± 0.09 mm3; P = 0.032, for anti-CD20 [n = 5] and control [n = 7], respectively) (Fig. 5A–C). Thus, in these confirmatory experiments, B-cell depletion suppressed the development of CNS disease in the humoral model, and this could be monitored with a novel TSPO ligand. While we could not detect any B cells in the fDTH-EAE or the fMOG-EAE model we were interested to discover whether the antiCD20 treatment might alter the ability of B cells or T cells to directly elicit a TNF response from microglial cells. Co-culture assays with BV2 cells were performed, and the cells isolated from either the fDTH-EAE or the fMOG-EAE anti-CD20-treated animals or control had little effect on TNF production compared to a positive lipopolysaccharide (LPS) control (Fig. S5).
Anti-CD20 therapy reduces microglial activation in the DTH-EAE to a similar degree to fingolimod
We next examined the effect of oral fingolimod 0.1 mg/kg per day from day 7 on microglial activation 12 days after the induction of the fDTH-EAE model in Lewis rats. Fingolimod significantly reduced macrophage/activated microglial staining (79%; P < 0.01) within DTH lesions compared with vehicle-treated controls (Fig. 6). However, there was no significant difference between the reduction observed in the anti-CD20 (65%; P < 0.001) and the fingolimod-treated groups.
Discussion
This study has shown for the first time that anti-CD20 therapy reduces lesion development and extralesional microglial activation in an antibody-independent model of MS and a focal humoral model. These findings help to explain the clinical efficacy of anti-CD20 therapy in most patients in clinical trials and generate a rationale for the use of anti-CD20 therapy in a broad population of MS patients. Interestingly, in our experiments, anti-CD20 therapy is no less effective at reducing lesion formation in a cell-mediated (fDTH-EAE) model than fingolimod, a drug that interferes directly with T-cell trafficking. The reduction of microglial activation by anti-CD20 therapy in the fDTH-EAE lesions also challenges the currently held belief that development and maintenance of cell-mediated MS lesions are B-cell independent. Finally, beyond the immediate suppression of relapse activity, the reduction of microglial activation may have an impact on the long-term disease course of MS, as the novel SPECT radioligand [125I]DPA-713 bound selectively to TSPO demonstrates the effect of therapy on extralesional activity in both models.
CD20 is not found on plasma cells,16 and anti-CD20 therapy does not directly affect antibody production.17 Our results are therefore consistent with an extensive literature regarding the antibody-independent roles of B-cells in MS lesion development.18 Anti-CD20 therapy decreases both B- and T-cell numbers in the CSF19; since we have shown that anti-CD20 therapy depletes only B-cells, it is probable that B-cells have a role in the trafficking of T-cells into the CNS.20 This phenomenon may reflect the capacity of B-cells to activate T-cells, either within the CNS (resulting in T-cell proliferation) or peripherally (resulting in activated T-cells that can more efficiently migrate into the CNS). Interactions between B- and T-cells are considered to be central to MS pathogenesis.18 While a full discussion of this subject is beyond the scope of this manuscript, it has long been known that B-cells can activate autoreactive T-cells by acting as antigen-presenting cells (APCs)21 and there is growing interest in, and evidence for, B-cells acting as APCs in the development, maintenance and progression of MS.22,23 CD80 and HLA-DR (an MHC class II surface receptor) are upregulated in activated B-cells, rendering them more efficient APCs than naïve B-cells.23 B-cells isolated from MS patients and activated in vitro can elicit activation and proliferation of myelin basic protein (MBP)-specific CD4+ T-cells. Interestingly, this activation can be prevented with anti-HLA-DR antibodies, suggesting that antigen presentation by B-cells is necessary for autoreactive T-cell activation.23 Furthermore, CD20+ memory B-cells isolated from a subset (40–50%) of RRMS patients can elicit activation and proliferation of MBP- and MOG-specific CD4+ T-cells without in vitro activation, whereas those isolated from healthy controls cannot.22 It has also been reported that peripheral CD80+ B-cells are enriched in RRMS patients undergoing relapse and absent during remission24 and a high B-cell:monocyte ratio is associated with more rapid disease progression,25 suggesting that B:T-cell interactions are also important in the exacerbation of MS.
Recent evidence suggests that CD20+ memory B-cells may act as potent APCs for myelin auto-antigens including MBP and MOG in 40–50% of RRMS patients, eliciting autoreactive T-cell proliferation and activation, whereas memory B-cells from healthy donors do not elicit these responses.22 This auto-antigen presenting role can be ameliorated by anti-CD20 therapy, which reduces the proliferation and activation of autoreactive CD4+ T-cells26 and IL-17 production27 in the CNS, and reduces clinical severity, in mouse and primate EAE models of MS.27,28 Recently, B-cells from RRMS patients were found to express an exaggerated pro-inflammatory cytokine profile in response to activating stimuli, which may aid T-cell activation. This exaggerated pro-inflammatory response was reduced in rituximab-treated patients, resulting in diminished proliferative and cytokine responses of Th1/Th17 CD4+ and CD8+ T-cells. One may therefore speculate that excessive release of pro-inflammatory cytokines and auto-antigen presentation by B-cells, resulting in aberrant T-cell activation and trafficking, may have a prominent role in the pathophysiology of MS and T-cell-mediated animal models of MS.
Recent studies have shown that the timing of B-cell depletion influences whether it is beneficial or detrimental and several experimental and clinical reports indicate that in certain settings, systemic anti-CD20 may potentially worsen predominantly T-cell-mediated CNS autoimmunity.26,29,30 These effects may refer to the systemic effect of anti-CD20 on other leukocyte populations that do not have a principal role in cell-mediated lesion formation. For example, this may be related to an effect on Breg cells.1,26 However, reductions in new inflammatory disease activity following B-cell depletion in RRMS patients may be sustained in some patients even as the circulating B-cell count returns to normal,31 suggesting a reconstituted population with a higher proportion of anti-inflammatory Breg-cells. B-cells reconstituted in RRMS patients following rituximab treatment produced significantly lower levels of pro-inflammatory cytokines including lymphotoxin and TNFα and higher levels of the immune regulatory cytokine IL-10,31 which has been shown to reduce EAE severity.32 Thus, the balance between pro- and anti-inflammatory B-cells following B-cell depletion will need to be carefully monitored in MS patients receiving anti-CD20 therapy in established MS.
The clinicoradiological paradox in MS refers to the poor association between clinical findings and the detectable radiological extent of lesion load.33 There is growing evidence that widespread, but currently undetectable, pathology is present in the MS brain,33–36 which is thought to underlie clinical progression.36,37 In particular, widespread microglial activation is correlated with axon damage in normal-appearing CNS tissue and increased clinical severity.36 TSPO binding is increased in activated microglia38 and can be visualized with TSPO-binding SPECT radioligands.39 However, pharmacokinetic limitations of PK11195 (an early TSPO-binding radioligand) have prevented its use as a routine imaging agent in MS. Novel TSPO-ligands overcoming these limitations have subsequently been developed, but none has been systematically evaluated in established animal models of MS. Here, we used [125I]DPA-713 to selectively image activated microglia in vitro and showed that binding was significantly reduced by anti-CD20 therapy in both models. In vivo SPECT [123I]DPA-713 images of the DTH-EAE lesions and the in vitro [125I]DPA-713 autoradiograms had similar distributions of activity, and increased radioligand binding could be determined in vivo. While the pattern of binding reflected the increased number of activated microglia in MS-like lesion sites of untreated animals, it is still unclear what proportion of the signal is attributed to microglial or astrocyte binding in this model. However, in the DTH-EAE model, microglial activation is more widespread than astrocyte activation and the spatial extent of the signal suggests that the ligand recognizes both the lesion core and surrounding microglial activation. Evidence from these studies and those in MS with PK11195 suggest that TSPO ligands may provide a more sensitive marker of disease progression through better prediction of the extent of MS pathology. Other potential benefits of TSPO ligands are more sensitive indices of efficacy allowing early identification of nonresponder patients and improved evaluation of efficacy in animal models where determining the level and extent of microglial activation may provide a more relevant outcome measure.
Overall, these findings generate a rationale for the use of anti-CD20 therapy in a broad population of MS patients and show that B-cell depletion is comparable to the efficacy of more traditional T-cell-directed therapy. This is also the first report to show that the novel SPECT radioligand [125I]DPA-713 binds selectively to TSPO and can be used to examine the effect of therapy on extralesional activity in focal models of MS lesions.
Acknowledgments
This study was supported by F. Hoffmann-La Roche Ltd., Basel, Switzerland. Nicholas Seneca and David Leppert are employees of F. Hoffmann-La Roche Ltd.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Supplementary methods.
The deposition of complement (C5b-9) on oligodendrocytes in the corpus callosum of the focal MOG model and control animals. (A) No C5b-9 immunoreactivity was present in age-matched animals that had been injected with IFA without MOG peptide. (B) Immunoreactivity in NAWM adjacent to the lesion in animals that had been immunized with IFA/MOG peptide 12 days after the injection of TNF/IFNg into the corpus callosum. (C) Immunoreactivity in the active boarder of the focal MOG lesion at 12 days and (D) immediately adjacent to the lesion. (E) A small number of C5b-9 immunopositive cells were present in the DTH lesion, but (F) no immunopositive cells were present in the NAWM.
Twelve days of FTY720 treatment did not affect the distribution or number splenic B-cells or T-cells. (A) Immunostaining of 68-IB3-positive B-cells (brown) in rat splenic follicles in an FTY720-treated animal. (B) Staining for anti-OX22 revealed that FTY720 therapy did not alter the number of T-cells present in the spleen.
Anti CD20 treatment reduces the area of microglial activation both in the lesion core and the surrounding tissue. This area was calculated by measuring the area of OX6-positive immunohistochemistry staining.
In vivo [123I]-DPA-713 autoradiographic confirmation of decreased activated microglia in anti-CD20-treated DTH model of MS. (A) Increased activated microglia at the site of lesion in untreated DTH model of MS. (B) Decreased activated microglia at the site of lesion in anti-CD20-treated DTH model of MS. (C) Ratio or radioligand uptake in the basal ganglia and overlying cortex in ipsilateral versus contralateral hemispheres, *P = 0.047.
Effect of Anti-CD20 treatment interaction of T cells and B cells with the BV2 microglial cell line. (A) The number of B and T cell recovered from control antibody or anti-CD20-treated animals. (B) Graph of the effect of LPS or T cells from control or anti-CD20-treated animals on TNF release by BV2 cells. (C) Graph of the effect of LPS or T cells from control or anti-CD20-treated animals on TNF release by BV2 cells. Note that LPS elicits markedly increased TNF production compared to the effects of the T or B cells alone. Anti-CD20 therapy had no impact on the outcome.
References
- Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol. 2010;67:452–461. doi: 10.1002/ana.21939. [DOI] [PubMed] [Google Scholar]
- Wiendl H, Gross CC. Modulation of IL-2Ralpha with daclizumab for treatment of multiple sclerosis. Nat Rev Neurol. 2013;9:394–404. doi: 10.1038/nrneurol.2013.95. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Chun J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69:759–777. doi: 10.1002/ana.22426. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C, Bruck W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. 1999;5:170–175. doi: 10.1038/5532. [DOI] [PubMed] [Google Scholar]
- Meinl E, Krumbholz M, Hohlfeld R. B lineage cells in the inflammatory central nervous system environment: migration, maintenance, local antibody production, and therapeutic modulation. Ann Neurol. 2006;59:880–892. doi: 10.1002/ana.20890. [DOI] [PubMed] [Google Scholar]
- Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130(Pt 4):1089–1104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- Obermeier B, Lovato L, Mentele R, et al. Related B cell clones that populate the CSF and CNS of patients with multiple sclerosis produce CSF immunoglobulin. J Neuroimmunol. 2011;233:245–248. doi: 10.1016/j.jneuroim.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Or A, Calabresi PA, Arnold D, et al. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol. 2008;63:395–400. doi: 10.1002/ana.21363. [DOI] [PubMed] [Google Scholar]
- Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548–558. doi: 10.1038/clpt.2008.170. [DOI] [PubMed] [Google Scholar]
- Barun B, Bar-Or A. Treatment of multiple sclerosis with anti-CD20 antibodies. Clin Immunol. 2012;142:31–37. doi: 10.1016/j.clim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378:1779–1787. doi: 10.1016/S0140-6736(11)61649-8. [DOI] [PubMed] [Google Scholar]
- Serres S, Anthony DC, Jiang Y, et al. Comparison of MRI signatures in pattern I and II multiple sclerosis models. NMR Biomed. 2009;22:1014–1024. doi: 10.1002/nbm.1404. [DOI] [PubMed] [Google Scholar]
- McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J Histochem Cytochem. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- Wang H, Pullambhatla M, Guilarte TR, et al. Synthesis of [(125)I]iodoDPA-713: a new probe for imaging inflammation. Biochem Biophys Res Commun. 2009;389:80–83. doi: 10.1016/j.bbrc.2009.08.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stashenko P, Nadler L, Hardy R, Schlossman SF. Characterization of a human B lymphocyte-specific antigen. J Immunol. 1980;125:1678–1685. [PubMed] [Google Scholar]
- Ahuja A, Anderson SM, Khalil A, Shlomchik MJ. Maintenance of the plasma cell pool is independent of memory B cells. Proc Natl Acad Sci USA. 2008;105:4802–4807. doi: 10.1073/pnas.0800555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland S, Monson N. Potential impact of B cells on T cell function in multiple sclerosis. Mult Scler Int. 2011 doi: 10.1155/2011/423971. Article ID 423971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AH, Stark JL, Lauber J, et al. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. 2006;180:63–70. doi: 10.1016/j.jneuroim.2006.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccio L, Naismith RT, Trinkaus K, et al. Changes in B-and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch Neurol. 2010;67:707. doi: 10.1001/archneurol.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera A, Chen C-C, Ron N, et al. Role of B cells as antigen-presenting cells in vivo revisited: antigen-specific B cells are essential for T cell expansion in lymph nodes and for systemic T cell responses to low antigen concentrations. Int Immunol. 2001;13:1583–1593. doi: 10.1093/intimm/13.12.1583. [DOI] [PubMed] [Google Scholar]
- Harp CT, Ireland S, Davis LS, et al. Memory B cells from a subset of treatment-naïve relapsing-remitting multiple sclerosis patients elicit CD4+ T-cell proliferation and IFN-γ production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur J Immunol. 2010;40:2942–2956. doi: 10.1002/eji.201040516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harp CT, Lovett-Racke AE, Racke MK, et al. Impact of myelin-specific antigen presenting B cells on T cell activation in multiple sclerosis. Clin Immunol. 2008;128:382–391. doi: 10.1016/j.clim.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Genc K, Dona DL, Reder AT. Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J Clin Invest. 1997;99:2664. doi: 10.1172/JCI119455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepok S, Jacobsen M, Schock S, et al. Patterns of cerebrospinal fluid pathology correlate with disease progression in multiple sclerosis. Brain. 2001;124:2169–2176. doi: 10.1093/brain/124.11.2169. [DOI] [PubMed] [Google Scholar]
- Matsushita T, Yanaba K, Bouaziz J-D, et al. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monson NL, Cravens P, Hussain R, et al. Rituximab therapy reduces organ-specific T cell responses and ameliorates experimental autoimmune encephalomyelitis. PLoS One. 2011;6:e17103. doi: 10.1371/journal.pone.0017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kap YS, Laman JD, A’t Hart B. Experimental autoimmune encephalomyelitis in the common marmoset, a bridge between rodent EAE and multiple sclerosis for immunotherapy development. J Neuroimmune Pharmacol. 2010;5:220–230. doi: 10.1007/s11481-009-9178-y. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn K, Schleich E, Hertzenberg D, et al. Anti-CD20 B-cell depletion enhances monocyte reactivity in neuroimmunological disorders. J Neuroinflammation. 2011;8:146. doi: 10.1186/1742-2094-8-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MS, Prod’homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 2010;68:369–383. doi: 10.1002/ana.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duddy M, Niino M, Adatia F, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol. 2007;178:6092–6099. doi: 10.4049/jimmunol.178.10.6092. [DOI] [PubMed] [Google Scholar]
- Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58:840–846. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- Barkhof F. The clinico-radiological paradox in multiple sclerosis revisited. Curr Opin Neurol. 2002;15:239–245. doi: 10.1097/00019052-200206000-00003. [DOI] [PubMed] [Google Scholar]
- Serres S, Anthony D, Jiang Y, et al. Systemic inflammatory response reactivates immune-mediated lesions in rat brain. J Neurosci. 2009;29:4820–4828. doi: 10.1523/JNEUROSCI.0406-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JW, Bo L, Mork S, et al. VCAM-1-positive microglia target oligodendrocytes at the border of multiple sclerosis lesions. J Neuropathol Exp Neurol. 2002;61:539–546. doi: 10.1093/jnen/61.6.539. [DOI] [PubMed] [Google Scholar]
- Howell OW, Rundle JL, Garg A, et al. Activated microglia mediate axoglial disruption that contributes to axonal injury in multiple sclerosis. J Neuropathol Exp Neurol. 2010;69:1017–1033. doi: 10.1097/NEN.0b013e3181f3a5b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds R, Roncaroli F, Nicholas R, et al. The neuropathological basis of clinical progression in multiple sclerosis. Acta Neuropathol. 2011;122:155–170. doi: 10.1007/s00401-011-0840-0. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Baraldi M, Guilarte TR, et al. Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Banati RB. Visualising microglial activation in vivo. Glia. 2002;40:206–217. doi: 10.1002/glia.10144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary methods.
The deposition of complement (C5b-9) on oligodendrocytes in the corpus callosum of the focal MOG model and control animals. (A) No C5b-9 immunoreactivity was present in age-matched animals that had been injected with IFA without MOG peptide. (B) Immunoreactivity in NAWM adjacent to the lesion in animals that had been immunized with IFA/MOG peptide 12 days after the injection of TNF/IFNg into the corpus callosum. (C) Immunoreactivity in the active boarder of the focal MOG lesion at 12 days and (D) immediately adjacent to the lesion. (E) A small number of C5b-9 immunopositive cells were present in the DTH lesion, but (F) no immunopositive cells were present in the NAWM.
Twelve days of FTY720 treatment did not affect the distribution or number splenic B-cells or T-cells. (A) Immunostaining of 68-IB3-positive B-cells (brown) in rat splenic follicles in an FTY720-treated animal. (B) Staining for anti-OX22 revealed that FTY720 therapy did not alter the number of T-cells present in the spleen.
Anti CD20 treatment reduces the area of microglial activation both in the lesion core and the surrounding tissue. This area was calculated by measuring the area of OX6-positive immunohistochemistry staining.
In vivo [123I]-DPA-713 autoradiographic confirmation of decreased activated microglia in anti-CD20-treated DTH model of MS. (A) Increased activated microglia at the site of lesion in untreated DTH model of MS. (B) Decreased activated microglia at the site of lesion in anti-CD20-treated DTH model of MS. (C) Ratio or radioligand uptake in the basal ganglia and overlying cortex in ipsilateral versus contralateral hemispheres, *P = 0.047.
Effect of Anti-CD20 treatment interaction of T cells and B cells with the BV2 microglial cell line. (A) The number of B and T cell recovered from control antibody or anti-CD20-treated animals. (B) Graph of the effect of LPS or T cells from control or anti-CD20-treated animals on TNF release by BV2 cells. (C) Graph of the effect of LPS or T cells from control or anti-CD20-treated animals on TNF release by BV2 cells. Note that LPS elicits markedly increased TNF production compared to the effects of the T or B cells alone. Anti-CD20 therapy had no impact on the outcome.