

Abstract
Objectives
Excitotoxicity plays a significant role in the pathogenesis of perinatal brain injuries. Among the consequences of excessive activation of the N-methyl-d-aspartate (NMDA)-type glutamate are oxidative stress caused by free radical release from damaged mitochondria, neuronal death and subsequent loss of connectivity. Drugs that could protect nervous tissue and support regeneration are attractive therapeutic options. The hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein I (HIP/PAP) or Reg3α, which is approved for clinical testing for the protection and regeneration of the liver, is upregulated in the central nervous system following injury or disease. Here, we examined the neuroprotective/neuroregenerative potential of HIP/PAP following excitotoxic brain injury.
Methods
We studied the expression of HIP/PAP and two of its putative effectors, cAMP-regulated phosphoprotein 19 (ARPP19) and growth-associated protein 43 (GAP-43), in the neonatal brain, and the protective/regenerative properties of HIP/PAP in three paradigms of perinatal excitotoxicity: intracerebral injection of the NMDA agonist ibotenate in newborn pups, a pediatric model of traumatic brain injury, and cultured primary cortical neurons.
Results
HIP/PAP, ARPP19, and GAP-43 were expressed in the neonatal mouse brain. HIP/PAP prevented the formation of cortical and white matter lesions and reduced neuronal death and glial activation following excitotoxic insults in vivo. In vitro, HIP/PAP promoted neuronal survival, preserved neurite complexity and fasciculation, and protected cell contents from reactive oxygen species (ROS)-induced damage.
Interpretation
HIP/PAP has strong neuroprotective/neuroregenerative potential following excitotoxic injury to the developing brain, and could represent an interesting therapeutic strategy in perinatal brain injury.
Introduction
Excitotoxicity plays a key role both in acute insults to the brain such as perinatal brain damage,1,2 stroke3,4 and traumatic brain injury (TBI),5 and in various neurodegenerative disorders,6,7 and is considered a major target for neuroprotection. During excitotoxicity, the excessive activation of glutamate receptors, particularly N-methyl-d-aspartate (NMDA) receptors, leads to massive calcium influx, damage to cellular membranes and leakage of reactive oxygen species (ROS) from mitochondria, inducing neural cell death, and the loss of neurites and connectivity.8–10 However, due to the important function of NMDA receptors, their indiscriminate targeting induces localized neuronal death and behavioral deficits in adulthood11,12 as well as massive and diffuse neuronal death in the developing brain.13,14 Preventing excitotoxic cell death or promoting neuritogenesis and neuronal connectivity without directly interfering with NMDA receptors is therefore an appealing therapeutic alternative. To this end, several candidate molecules with neuroprotective and neuroregenerative properties have been evaluated.15,16
The hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein I (HIP/PAP), a 16 kDa acute-phase response protein belonging to the regenerating gene (Reg) family of C-type lectins, is expressed in the central and peripheral nervous systems.17 HIP/PAP expression is increased following brain or peripheral-nerve injury in adult rodents and humans.18–20 HIP/PAP protects cerebellar, cortical, and hippocampal neurons against H2O2-induced cell death,19 promotes Schwann cell proliferation during motoneuron regeneration21 and exerts neurotrophic effects on motoneurons themselves.22 Its dual role – neuroprotection against oxidative stress and neurotrophism – suggests that it could be useful in preventing or reversing the effects of excitotoxicity in the developing brain. In addition, HIP/PAP has an advantage over other putative neuroprotective agents in that it has already been approved for clinical testing. A recombinant human HIP/PAP (rcHIP/PAP, ALF5755) was well tolerated in healthy volunteers in a phase 1 clinical trial,23 and is currently being evaluated in a double-blind randomized multicenter phase 2 clinical trial for acute liver failure (NCT01318525).
Even though the regenerative properties of HIP/PAP have been known for years, no specific effector has been identified so far. However, the cAMP-regulated phosphoprotein 19 (ARPP19) has been identified by Copper-IMAC ProteinChip/SELDI-TOF as being upregulated in a HIP/PAP-dependent manner during the course of liver regeneration. ARPP19 is a protein phosphatase inhibitor that specifically inhibits protein phosphatase 2A.24 It is also known to dock on and stabilize the 3′-untranslated region of the mRNA for growth-associated protein 43 (GAP-43), leading to an overall increase in GAP-43 protein.25 In the central nervous system, GAP-43 is essential for axon guidance, synaptic plasticity, and neuroregeneration,26 and we have previously demonstrated its key role in the plasticity induced by brain-derived neurotrophic factor (BDNF) after excitotoxic lesions of the perinatal mouse brain.27
In the present study, we used three experimental systems, (1) excitotoxic perinatal brain injury by ibotenate injection, (2) a model of pediatric TBI that is predominantly mediated through excitotoxicity,28 and (3) primary cortical neuronal cultures exposed to NMDA, to investigate the protective effects of HIP/PAP on excitotoxic cell death and postexcitotoxic neuronal plasticity.
Materials and Methods
Experimental protocols were approved by institutional and local ethics committees, and carried out in accordance with the Guide for the Care and Use of Laboratory Animals (U.S. National Institutes of Health).
Animals and drugs
Male and female Swiss pups (Iffa Credo, L’Abresle, France) were used for in vivo experiments and male and female C57Bl/6 mice for in vitro experiments. All animals were housed under the same temperature (25°C) and photoperiod (12 h:12 h light–dark cycle) conditions, and given free access to food and water.
Ibotenate and NMDA were purchased from Sigma (St. Louis, MO). Ibotenate was diluted in phosphate buffered saline (PBS) containing 0.01% acetic acid. NMDA was diluted in PBS. The rcHIP/PAP (ALF5755) protein is a recombinant human protein that corresponds to the addition of one amino-terminal methionine to the sequence of the secreted form (i.e., lacking the 26 amino acid signal sequence) of endogenous human HIP/PAP (NP_620355). It was produced in Escherichia Coli, purified to ≥99% and released in batches in compliance with the clinical grade manufacturing process, by PX’Therapeutics Grenoble, France.
Quantitative RT-PCR analysis of HIP/PAP, ARPP19, and GAP-43
Experimental tissues came from the forebrain and hindbrain of embryonic day (E) 14 embryos (n = 6), and the neopallium and cerebellum of P0 (n = 6), P5 (n = 6) and P40 (n = 5) mice. RNA extraction, cDNA synthesis, and RT-PCR were performed and analyzed as previously described,29,30 using the 60S acidic ribosomal protein gene P0 (RPLP0) as a housekeeping gene. Primer sequences are given in Table 1.
Table 1.
Sequences of primers used for real-time PCR
| HIP/PAP | |
| Sense primer | 5′-ATACCCTCCGCACGCATTAGTT-3′OH |
| Antisense primer | 5′-AAGCTCTTGACAAGCTGCCACA-3′OH |
| ARPP19 | |
| Sense primer | 5′-AAGGCAAGGTATCCTCACTTGG-3′OH |
| Antisense primer | 5′-GTCACCAGTGACCTCTGTCTTA-3′OH |
| GAP-43 | |
| Sense primer | 5′-TCAAAGGCGAGAAGAAGGGTGA-3′OH |
| Antisense primer | 5′-AGCAGGCACATCGGCTTGTTTA-3′OH |
| RPLP0 | |
| Sense primer | 5′-AGATGCAGCAGATCCGCAT-3′OH |
| Antisense primer | 5′-GTTCTTGCCCATCAGCACC-3′OH |
Neonatal excitotoxic brain lesions
We induced excitotoxic brain lesions by injecting ibotenate (10 μg) into the developing mouse neopallial parenchyma on postnatal day (P) 5 as previously described.2,31,32 Pups were sacrificed on P10 and following formalin fixation and paraffin embedding of the brain, 15-μm-thick serial coronal brain sections were cut. Every third section was stained with cresyl violet to determine the maximal fronto-occipital extent of the lesion and used as an index of lesion severity.33 Lesion size was determined by an investigator blind to the treatment group.
Pediatric TBI model
P7 mouse pups were deeply anesthetized with isoflurane and subjected to a unilateral closed contusion head trauma as previously described.34,35 In brief, a 10 g weight falls from a height of 10 cm onto a circular footplate (2 mm diameter) resting on the animal’s skull, through a skin incision made to expose the surface of the skull. The footplate is oriented parallel to the left parietal bone with the center positioned 2 mm anterior and 1 mm lateral to lambda. The device was regulated to depress the skull surface by 1 mm. After surgery, animals were kept at 37°C to recover, and returned to their mothers 30 min later. On P8, P12, and P28, whole brains were fixed in 4% formalin for 4–5 days at room temperature, processed for paraffin embedding and coronal sections 16 μm thick were cut and stained with cresyl violet. Brain atrophy induced by the degeneration of neurons after TBI was measured through the dilatation of the ipsilateral ventricle when compared to the contralateral ventricle. Measurements were made (NIH Image JNIH, Bethesda, MD, USA) on 12 sections per subject, at six different levels (two sections per level separated by 320 μm), covering the entire extent of the ventricle. The 12 values were considered as dependent replicates.
Analysis of cell death, astrocytosis, microgliosis, and plasticity
Antibodies used were cleaved caspase-3 (1/500; Cell Signaling Technology, Beverly, MA), apoptosis-inducing factor (AIF, 1/500; Santa Cruz Biotechnology, Santa Cruz, CA), glial fibrillary acidic protein (GFAP, 1/500; Dako, Carpinteria, CA), Iba1 (1/1000; Wako Chemicals, Richmond, VA), and phosphorylated-Ser41 GAP-43 (pGAP-43) (1/100; Novus Biologicals, Littleton, CO),27 and labeling was detected with avidin-biotin horseradish peroxidase kits (Vector Laboratories, Burlingame, CA). Adjacent sections were used for Fluoro-Jade B (Histo-Chem Inc., Jefferson, AR) staining. Five animals were included in each group and for each time point studied. Analyses were carried out by two different investigators independently.
Primary neuronal cultures
On P0 primary neurons were cultured as previously described.36 Cells were plated at a density of 2 × 104 cells/cm2 onto Poly-d-Lysine-coated Labtek chamber slides (Nunc, Roskilde, Denmark). After 24 h of incubation (DIV2) at 37°C in 5% CO2, serum-containing medium was removed. The cells were then cultured in serum-free medium for 12 h after which they were treated with NMDA (300 μmol/L) or H2O2 (30 or 100 μmol/L) in the presence or absence of HIP (0.3–10 μg/mL) for the next 24 h. Following treatment, the cells were rinsed with PBS and then fixed in 4% PFA.
Oxidative stress measurement
Untreated and NMDA-treated neurons were assayed for malondialdehyde (MDA), an end product of lipid peroxidation, measured using the thiobarbituric acid (TBA) procedure as previously described.23 Results were normalized using total protein content measured using a Bio-Rad Assay Kit, Marnes-la-Coquette, France.
MTS assay and TUNEL assay
Neuronal viability was quantified using the colorimetric CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI). The TUNEL assay was performed using the In Situ Cell Death Detection Kit (Roche Applied Sciences, Indianapolis, IN). Quantification of TUNEL-positive cells in cultures was performed by counting all TUNEL-positive cells in a total of 10 random fields from two wells each.
Analysis of neurite branching and length
Cultured cells were labeled for beta-III tubulin (1:1000; Promega) at 4°C overnight, incubated with Alexa Fluor 488 anti-mouse secondary antibody (Molecular Probes, Eugene, Oregon, USA) for one hour at room temperature and mounted with Vectashield DAPI (Vector Laboratories). The number of primary, secondary, and tertiary neurites (labeled for beta-III tubulin) as well as the number of branch points were counted in captured images (Fig. S1). Neurite length was measured using Leica IM50 software.
Statistical analysis
All data are expressed as means ± SEM. Data were analyzed using a Mann–Whitney test or a univariate ANOVA (GraphPad Prism version 4.01 for Windows, GraphPad Software, Graphpad Software, San Diego, CA, USA). When ANOVA revealed significant differences between several experimental groups, multiple comparisons between experimental groups were performed using Dunnett’s or Bonferroni’s post hoc test.
Results
HIP/PAP is expressed in the developing brain
Using quantitative RT-PCR, HIP/PAP, GAP-43, and ARPP19 expression levels were measured in the mouse cortex (or forebrain at E14) and cerebellum (or hindbrain at E14) from E14 to P40. HIP/PAP transcripts were barely detected in embryonic samples, but increased 155- and 745-fold (P < 0.01) at P0 in the cortex and cerebellum, respectively (Fig. 1). From P5 to P40, HIP/PAP transcripts decreased progressively to resume their original levels at P40 (Fig. 1). GAP-43 was highly expressed in the cortex and cerebellum at all ages investigated. Its expression levels in the cerebellum were constant, but displayed a 3.6-fold decrease at P40 in the cortex (P < 0.0004) when compared with its original levels (Fig. 1). ARPP19 was expressed at moderate but constant levels in the cortex and cerebellum at all ages (Fig. 1).
Figure 1.
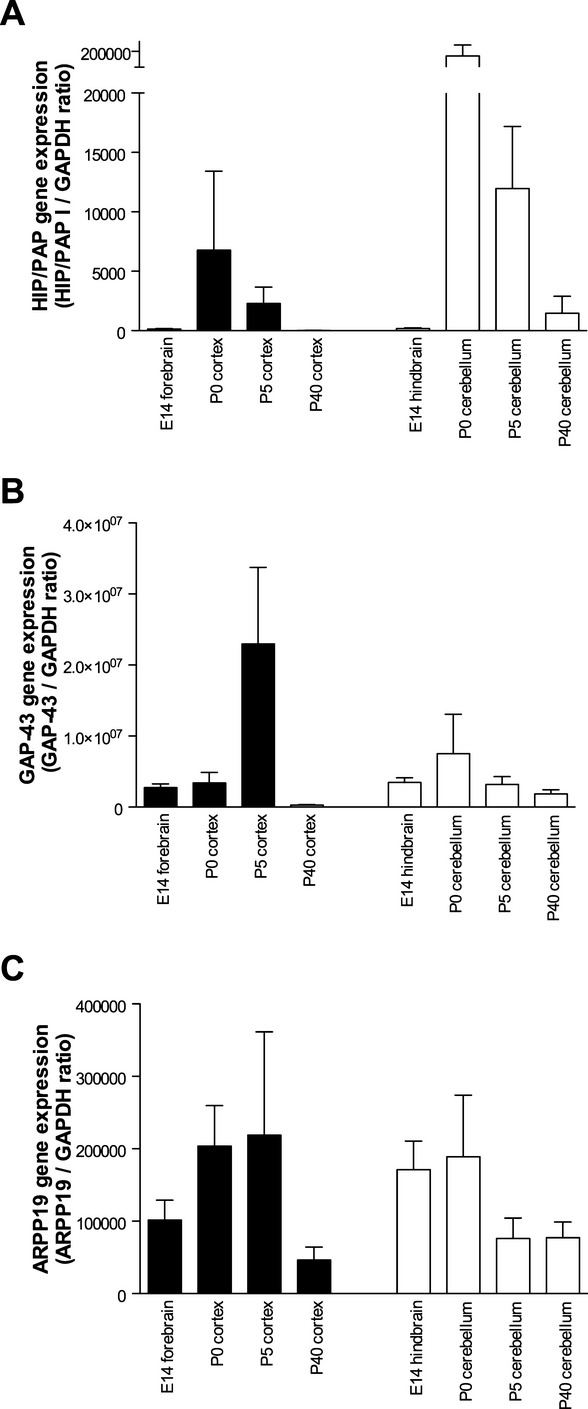
Ontogenic expression of HIP/PAP, GAP-43, and ARPP19 mRNA in murine cerebral cortex and cerebellum, as determined by quantitative RT-PCR. Data are presented as means for the ratio of the gene of interest/RPLP0, ±SEM. HIP/PAP, hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein; GAP, growth-associated protein.
rcHIP/PAP protects the developing neopallium against excitotoxic injury
Overall mortality due to ibotenate excitotoxicity was low in the present study (<3% of the animals injected). No significant difference was observed in a test of contingency (the Fisher exact test) between the different ibotenate-treated groups. Epileptic manifestations including clonic or tonic seizures and apneas were observed in all ibotenate-treated animals. However, treatment with rcHIP/PAP did not induce any detectable difference in the severity or frequency of seizures when compared to controls (data not shown).
Mouse pups injected on P5 with intracerebral ibotenate developed typical cortical lesions and periventricular white matter cysts (Fig. 2A). In P5 mice, rcHIP/PAP administered i.c. immediately after ibotenate injection induced significant neuroprotection of the cortical plate and white matter when observed on P10 (Fig. 2A–D). Similarly, rcHIP/PAP administered i.p. immediately after ibotenate injection also induced significant and dose-dependent neuroprotection against ibotenate-induced cortical plate and white matter lesions (Fig. 2C and D), an effect that was still seen at the highest dose of rcHIP/PAP given 3 h after ibotenate injection (Fig. 2C and D). The protective effect of rcHIP/PAP administered i.p. immediately after ibotenate was accompanied by a significant reduction of cell death around the lesion site as measured by cleaved caspase-3, Fluoro-Jade B and AIF labeling 1 day postibotenate injection (Fig. 3), a difference that was still observable at 5 days in the case of cleaved caspase-3. With respect to gliosis, rcHIP/PAP reduced microglial activation as measured by Iba-1 immunolabeling, and both prevented early astrocyte death (1 day postibotenate), delayed astrogliosis (5 days postibotenate) as measured by GFAP immunolabeling (Fig. 4), and increased plasticity (5 days postibotenate) as measured by pGAP-43 immunolabeling (Fig. S2A).
Figure 2.
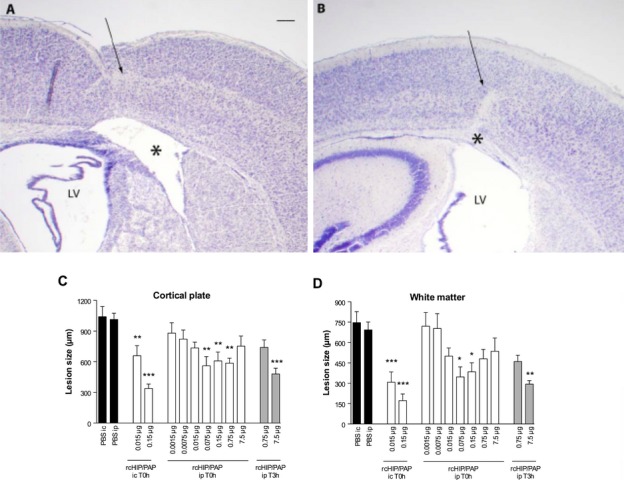
(A and B) Cresyl violet-stained sections showing brain lesions induced by ibotenate injected at P5 and studied at age P10. Brains from pups treated with i.c. ibotenate alone (A) or treated with i.c. ibotenate and rcHIP/PAP (B), showing typical neuronal loss in layers II–VI (arrow) and white matter lesions (*). LV, lateral ventricle. Bar: 40 μm. (C and D) Effect of i.c. and i.p. rcHIP/PAP on ibotenate-induced lesions. Pups were injected on P5 and sacrificed on P10. Drugs indicated on the X axis were injected immediately (T0 h) or 3 h (T3 h) after ibotenate. Values represent the mean length of the lesions ± SEM. Asterisks indicate statistically significant difference from the appropriate controls (black bars); *P < 0.05, **P < 0.01, ***P < 0.001 by ANOVA with Dunnett’s multiple comparison test. HIP/PAP, hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein.
Figure 3.
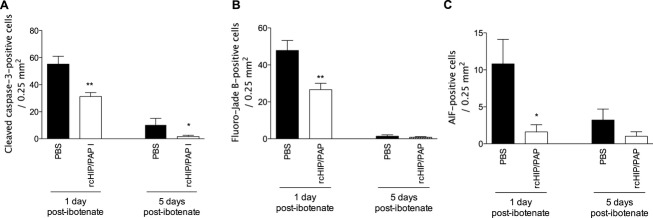
Effect of rcHIP/PAP on ibotenate-induced cell death determined by cleaved caspase-3 immunohistochemistry (A), Fluoro-Jade B staining (B), and AIF immunohistochemistry (C). Pups were injected with ibotenate on P5 and sacrificed 1 or 5 days later. PBS or rcHIP/PAP (0.75 μg) were injected i.p. immediately after ibotenate. Values represent mean density of positive cells ± SEM. Asterisks indicate statistically significant difference from controls (black bars); *P < 0.05, **P < 0.01, ANOVA with Dunnett’s multiple comparison test. HIP/PAP, hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein; AIF, apoptosis-inducing factor.
Figure 4.
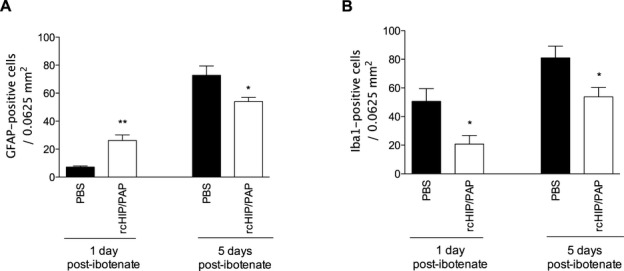
Effect of rcHIP/PAP on ibotenate-induced gliosis determined by immunohistochemistry for GFAP (A) and Iba1 (B). Pups were injected with ibotenate on P5 and sacrificed 1 or 5 days later. PBS or rcHIP/PAP (0.75 μg) was injected i.p. immediately after ibotenate. Values represent mean density of positive cells ± SEM. Asterisks indicate statistically significant difference from controls (black bars); *P < 0.05, **P < 0.01, ANOVA with Dunnett’s multiple comparison test. HIP/PAP, hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein; AIF, apoptosis-inducing factor; PBS, phosphate buffered saline; TBI, traumatic brain injury; GFAP, glial fibrillary acidic protein.
rcHIP/PAP protects the developing brain against TBI
TBI induced a large increase in the cross-sectional area of the ipsilateral ventricle in saline-treated animals, an effect that reached significance 21 days post-TBI, i.e., at P28 (Fig. 5). rcHIP/PAP administered i.p. immediately after TBI largely prevented this TBI-induced ipsilateral ventricular dilatation, but had no detectable effect on the contralateral ventricle (Fig. 5). This protective effect was accompanied by a significant reduction of cell death at 1 day post-TBI as measured by cleaved caspase-3 and Fluoro-Jade B labeling (Fig. 6), reduced astrogliosis at 5 and 21 days post-TBI as measured by GFAP immunolabeling (Fig. 7A–C), reduced microglial activation at 1 day post-TBI as measured by Iba-1 immunolabeling (Fig. 7D–F), and increased plasticity at 5 days post-TBI as measured by pGAP-43 immunolabeling (Fig. S2B).
Figure 5.

(A–C) Cresyl violet-stained sections showing ventricular enlargement (red asterisk) induced by TBI performed on P7 and studied 21 days later. Ipsilateral brain hemispheres from a control animal (no TBI) (A), from an animal with TBI treated with PBS i.p. (B), and from an animal with TBI and treated with rcHIP/PAP (0.75 μg) i.p. (C). Bar: 80 μm. (D–F) Effect of rcHIP/PAP on TBI-induced ventricular enlargement. Pups underwent TBI on P7 and were sacrificed 1 (D), 5 (E) or 21 (F) days later. PBS or rcHIP/PAP was injected i.p. immediately after the TBI. Values represent ventricular area ± SEM. Asterisks indicate statistically significant difference from the contralateral hemisphere of PBS-treated animals (light blue bar); §indicates a statistically significant difference from the ipsilateral hemisphere of PBS-treated animals (dark blue bar); **, §§P < 0.01, ANOVA with Dunnett’s multiple comparison test. HIP/PAP, hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein; TBI, traumatic brain injury; PBS, phosphate buffered saline.
Figure 6.
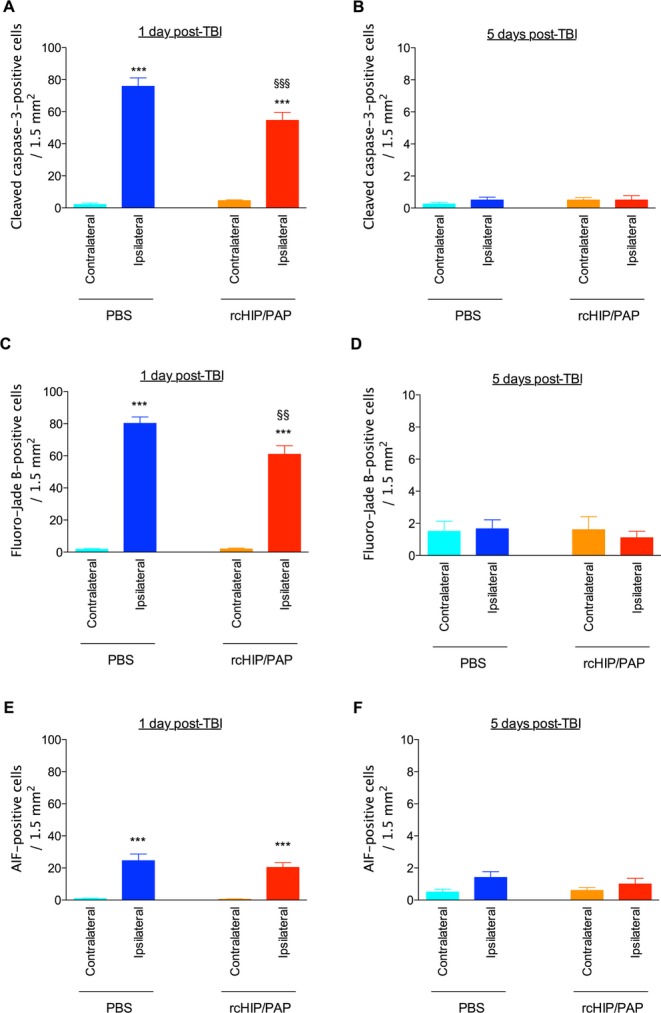
Effect of rcHIP/PAP on TBI-induced cell death determined by cleaved caspase-3 immunohistochemistry (A and B), Fluoro-Jade B staining (C and D), and AIF immunohistochemistry (E and F). Pups underwent TBI on P7 and were sacrificed 1 or 5 days later. PBS or rcHIP/PAP (0.75 μg) was injected i.p. immediately after the TBI. Values represent mean density of positive cells ± SEM. Asterisks indicate statistically significant difference from the contralateral hemisphere of PBS-treated animals (light blue bar); §indicates statistically significant difference from the ipsilateral hemisphere of PBS-treated animals (dark blue bar); **, §§P < 0.01, ***, §§§P < 0.001, ANOVA with Dunnett’s multiple comparison test. AIF, apoptosis-inducing factor; PBS, phosphate buffered saline; TBI, traumatic brain injury; HIP/PAP, hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein.
Figure 7.
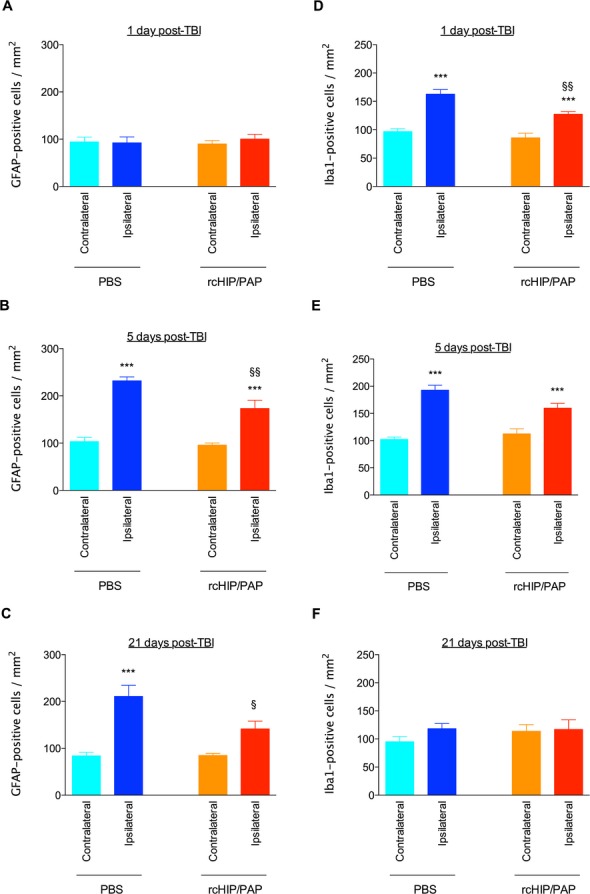
Effect of rcHIP/PAP on TBI-induced gliosis, as determined by immunohistochemistry for GFAP (A–C) and Iba1 (D–F). Pups underwent TBI on P7 and were sacrificed 1, 5, or 27 days later. PBS or rcHIP/PAP (0.75 μg) was injected i.p. immediately after the TBI. Values represent mean density of positive cells ± SEM. Asterisks indicate statistically significant difference from the contralateral hemisphere of PBS-treated animals (light blue bar); §indicates statistically significant difference from the ipsilateral hemisphere of PBS-treated animals (dark blue bar); §P < 0.05, §§P < 0.01, ***P < 0.001, ANOVA with Dunnett’s multiple comparison test. HIP/PAP, hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein; PBS, phosphate buffered saline; TBI, traumatic brain injury; GFAP, glial fibrillary acidic protein.
rcHIP/PAP protects cultured cortical neurons against excitotoxic injury
Exposure of primary cortical neurons to NMDA-induced significant cell death, as determined by MTS and TUNEL assays (Fig. 8A and B). The addition of rcHIP/PAP completely abolished this NMDA-induced cytotoxicity (Fig. 8A and B). As HIP/PAP is a ROS scavenger, its capacity to reduce oxidative damage to NMDA-treated cells was monitored next. The exposure of primary cortical neurons to NMDA induced a significant production of ROS, an effect that was completely blocked by cotreatment with rcHIP/PAP (Fig. 8C). However, rcHIP/PAP under basal conditions did not significantly change the redox status of cells even after 5 h. (Fig. 8C). Further supporting a role for the antioxidant effect of rcHIP/PAP in the protection of neurons against NMDA-induced damage, rcHIP/PAP significantly reduced neuronal cell death induced by H2O2 (Fig. 8D).
Figure 8.
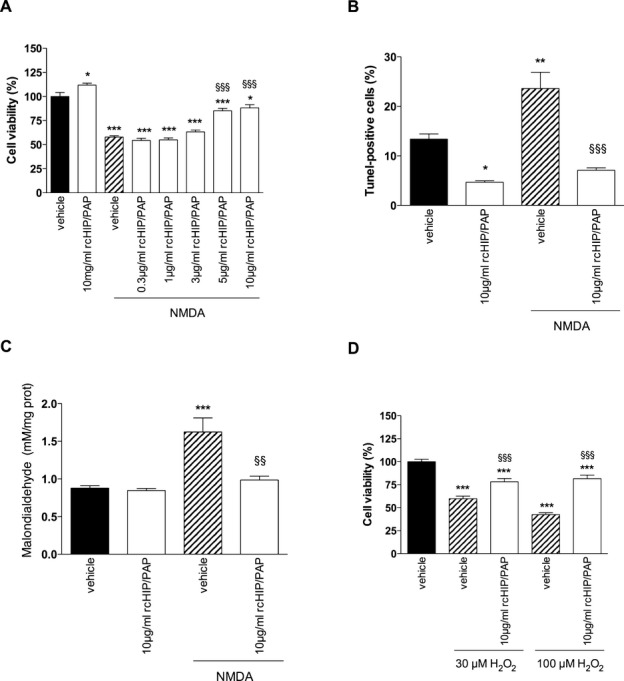
(A and B) Effect of rcHIP/PAP on NMDA-induced cell death of cultured primary cortical neurons, as determined by MTS (A) and TUNEL (B) techniques. (C) Effect of rcHIP/PAP on NMDA-induced oxidative stress in primary cortical neurons, as determined by quantification of lipid peroxidation (malondialdehyde or MDA assays). (D) Effects of rcHIP/PAP on H202-induced cell death of primary cortical neurons determined by the MTS technique. Values represent means ± SEM. Asterisks indicate statistically significant difference from the black bar; §indicates statistically significant difference from the hatched bar; *P < 0.05, **, §§P < 0.01, ***, §§§P < 0.001, ANOVA with Dunnett’s multiple comparison test. HIP/PAP, hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein; MDA, malondialdehyde; NMDA, N-methyl-d-aspartate.
rcHIP/PAP promotes postexcitotoxic plasticity of cultured cortical neurons
Exposure of primary cortical neurons to NMDA induced a significant reduction in the number of primary neurites per cell (Figs. 9 and 10A), the proportion of cells with tertiary neurites (Figs. 9 and 10B), the number of neurite branching points per cell (Figs. 9 and 10C), and mean neurite length (Figs. 9 and 10D). These deleterious effects of NMDA were largely counteracted by cotreatment with rcHIP/PAP (Figs. 9 and 10).
Figure 9.
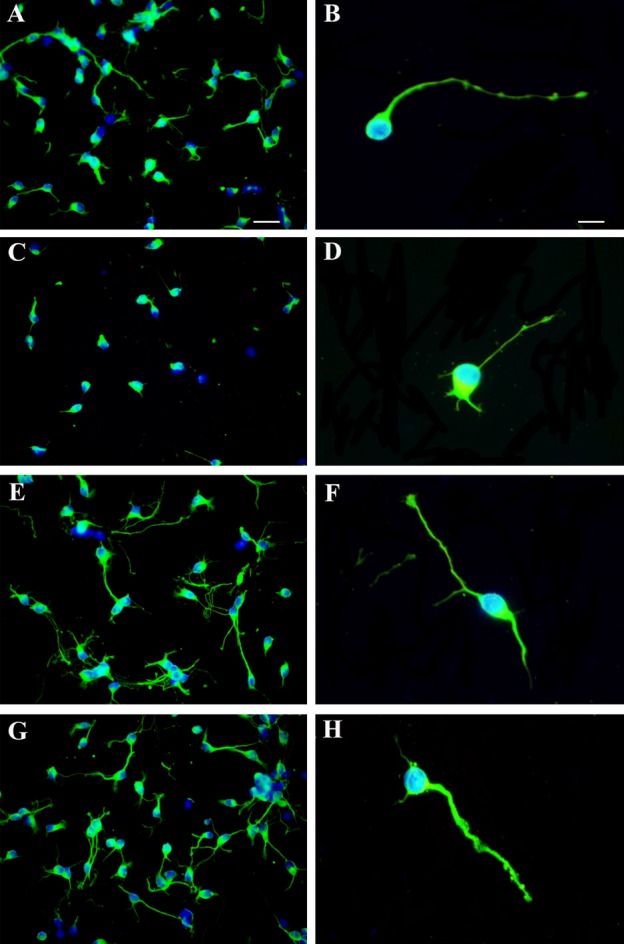
Effects of rcHIP/PAP on neurite extension and branching. (A and B) Cultured primary cortical neurons treated with vehicle. (C and D) Primary cortical neurons treated with NMDA + vehicle. (E and F) Primary cortical neurons treated with rcHIP/PAP. (G and H) Primary cortical neurons treated with NMDA + rcHIP/PAP. Bar: 16 μm (A, C, E, and G) or 8 μm (B, D, F, and H). HIP/PAP, hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein; NMDA, N-methyl-d-aspartate.
Figure 10.
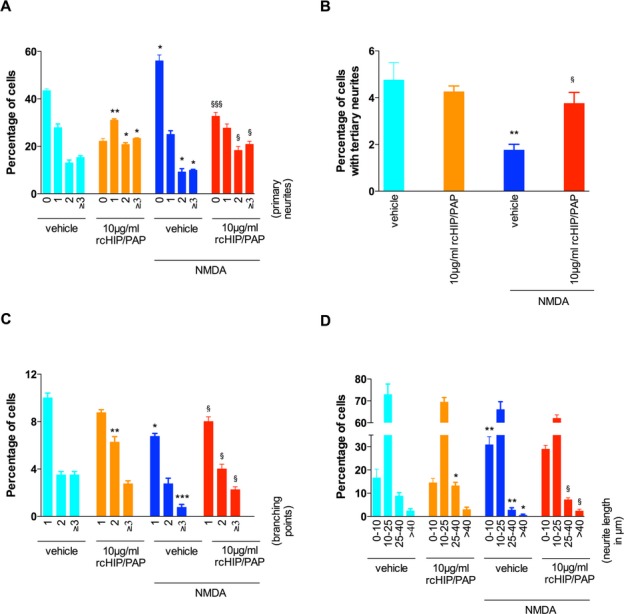
Effects of rcHIP/PAP on the number of primary neurites (A), the percentage of cells with tertiary neurites (B), the number of branching points (C) and neurite length (D) in primary cortical neurons. Values represent means ± SEM. Asterisks indicate statistically significant difference from light blue bar; §indicates statistically significant difference from dark blue bar; *, §P < 0.05, **P < 0.01, ***, §§§P < 0.001, ANOVA with Dunnett’s multiple comparison test. HIP/PAP, hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein.
In vitro, control primary cortical neurons have a tendency to form aggregates, with neurites emerging from these aggregates to form fascicles (Fig. 11). NMDA exposure induced a significant reduction in the number of neurons per aggregate and in the accompanying fasciculation of neurites (Fig. 11). In contrast, rcHIP/PAP reversed this NMDA-induced disaggregation of neurons and increased the accompanying fasciculation of neurites emerging from these aggregates (Fig. 11). Also, rcHIP/PAP alone increased neuronal aggregate formation and neurite fasciculation (Fig. 11).
Figure 11.
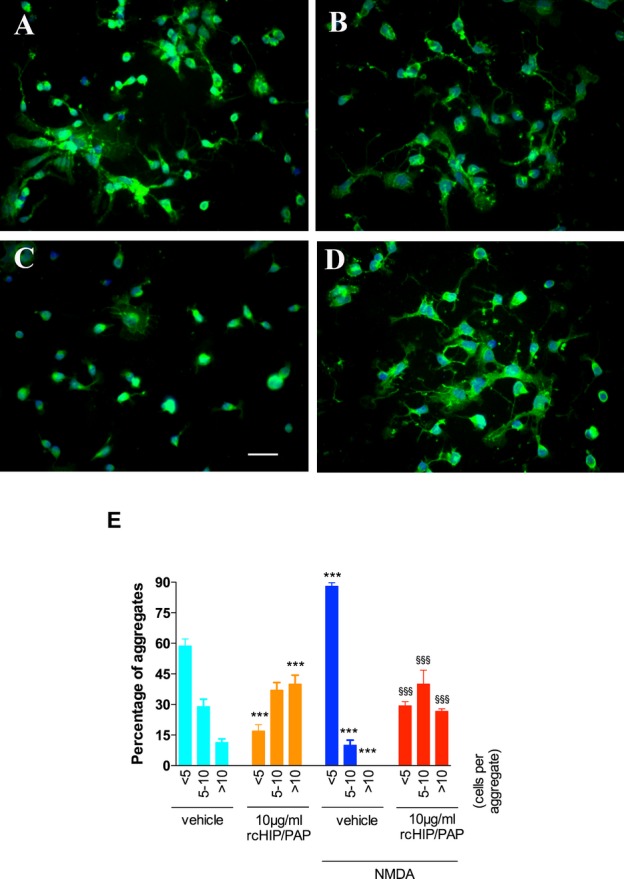
Effects of rcHIP/PAP on neuronal aggregation. (A) Primary cortical neurons treated with vehicle. (B) Primary cortical neurons treated with rcHIP/PAP. (C) Primary cortical neurons treated with NMDA + vehicle. (D) Primary cortical neurons treated with NMDA + rcHIP/PAP. Bar: 16 μm (A–D). (E) Values represent mean percentage of aggregates containing a given number of neurons, ±SEM. Asterisks indicate statistically significant difference from light blue bar; § indicate statistically significant difference from dark blue bar; ***, §§§P < 0.001 in ANOVA with Dunnet’s multiple comparison test. NMDA, N-methyl-d-aspartate; HIP/PAP, hepatocarcinoma intestine pancreas protein/pancreatitis-associated protein.
Discussion
In the current work, we showed that HIP/PAP as well as two of its potential downstream effectors, GAP-43 and ARPP19, are expressed in the mouse cortex and cerebellum during the perinatal and early postnatal period. A single injection of recombinant HIP/PAP protein protects the developing brain and its components against excitotoxic injury. In three different models of perinatal brain injury – ibotenate-induced lesions of the cortical plate and white matter, TBI of the cortex, and NMDA-mediated insult of cultured primary cortical neurons – HIP/PAP had significant neuroprotective effects, reducing cell death and gliosis in vivo, and improving survival, redox status and neuronal morphology in vitro.
The strength of the neuroprotective effects of HIP/PAP lies in its combined antiexcitotoxic and pro-regenerative capabilities. Work from our laboratory with the neurotherapeutic melatonin has shown similar properties.37 However, HIP/PAP is the only molecule tested so far to protect both the gray and white matter from excitotoxic lesions and do so with a delayed therapeutic window. These differences are of particular interest in the context of perinatal or pediatric TBI, for which few treatment options to protect the grey and white matter, especially those that could still be effective when given after a delay of several hours, are available. However, further studies are necessary to confirm that delayed administration of HIP/PAP is protective in the TBI model as TBI has more to its underlying pathology than just excitotoxicity,
HIP/PAP displays a complex dose–response relationship dependent on the time of administration following injury. A U-shaped curved is described for many drugs and generally is linked to a desensitization effect due to the downregulation of the target. Since the precise target of HIP/PAP is not known, this hypothesis is difficult to test. Another point of interest is that the same dose of HIP/PAP can be protective or with no significant protective effect according to the postinsult time at administration. The reason for this temporal effect is unknown but could be linked to changes of the expression of the target after the insult, changes in blood-brain barrier permeability and/or changes in diffusion properties linked to edema or inflammatory processes. Despite this variability, whatever the dose of HIP/PAP used, no toxic effect was observed. This is in agreement with the lack of toxicity reported in Phase 1 trial in healthy human subjects exposed to escalating doses of HIP/PAP.23
How are the neuroprotective effects of HIP/PAP mediated? In keeping with the involvement of excitotoxic processes both during perinatal brain injury and in the course of neurodegenerative disorders in adulthood, the paradigms we chose to study induced neuronal damage predominantly through an excitotoxic cascade involving the activation of the NMDA-type glutamate receptor. Following the injection of the NMDA agonist ibotenate, the brain of the newborn undergoes a series of cellular and molecular changes, ranging from inflammatory changes such as gliosis and cytokine production within a few hours of the insult, to neuronal loss within a few days, and the subsequent formation of a lesion demarcated by a glial scar and the remodeling of neuronal connections.2,38 In the case of TBI also, there is an initial and rapid excitotoxic reaction mediated by glutamate receptor activation, which gives way to apoptotic degeneration and scar formation over several days.5 Since both these processes (ibotenate-induced excitotoxicity and TBI-induced neuronal death) involve mitochondrial damage and oxidative stress, it appears probable that HIP/PAP acts at least partly at this level to increase neuronal survival and reduce gliosis in these injured brains. Support for this hypothesis comes from our finding that the change in redox status (i.e., the increase in lipid peroxidation) of primary cortical neuronal cultures exposed to NMDA was blocked by the presence of HIP/PAP in the culture medium. This is in keeping with previous findings as to the mode of action of HIP/PAP in vivo in the liver and intestines, where it attenuates free radical damage induced by Fas and dextran sodium sulfate, respectively,39,40 as well as in vitro in primary rat brain neuronal cultures, where it blocks peroxide-induced neurotoxicity.19
In addition to, or perhaps in parallel with, these effects on oxidative damage triggered by neurons themselves, HIP/PAP could also act at the level of inflammatory processes, as indicated by the reduction of reactive gliosis (both astrocytes and microglia) in our animals. Findings from several groups also indicate that HIP/PAP reduces the injury-induced upregulation of inflammatory cytokines, such as TNFα and IL-6, in other tissues,41,42 and blocks the effect of TNFα on macrophage activation and inflammatory function.43 However, we have no direct evidence for an effect of HIP/PAP on glia in our models and any changes may reflect decreased cell death.
HIP/PAP could also affect neuroregeneration or plasticity following injury as indicated by its effects on neurite growth, branching, and fasciculation in this study. The way HIP/PAP promotes neuronal plasticity remains unclear, although this might involve ARPP19. Indeed, HIP/PAP has been shown to induce the overexpression of ARPP19, which is known to promote neurite outgrowth,44 during the course of liver regeneration.24 Regeneration is of particular importance as dying neurons undergo neurite degeneration, and axonopathy is a hallmark of brain damage in preterm infants and those with TBI.35,45,46
This study achieved its aim of describing the neuroprotective effects of HIP/PAP in models of damage to the immature brain. Given the current paucity of neurotherapeutic strategies for excitotoxic injuries in the perinatal and pediatric population, these data suggest that HIP/PAP might have clinical potential to reduce the burden of neurological damage associated with these injuries. Further work to elucidate the precise molecular effectors of HIP/PAP in the brain may strengthen this potential and enhance our ability to effectively use this drug.
Acknowledgments
We thank Laure Jamot and Jérémie Mariaux for their help and support during this study. This study was supported by grants from Inserm, Université Paris Diderot, APHP (Contrat Hospitalier de Recherche Translationnelle to P. Gressens), Fondation PremUP, Fondation Roger de Spoelberch, Fondation Grace de Monaco, Fondation pour le Recherche Médicale, Fondation des Gueules Cassées, Institut pour la Recherche sur la Moëlle Epinière et l’Encéphale, IFCPAR-CEFIPRA (project number 3803-3 to S. Mani and P. Gressens), and the Programme National de recherche en hépato-gastroentérologie Inserm/AFEF, ASE07127LSA. The authors acknowledge financial support from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy’s & St Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust. The supporting bodies played no role in any aspect of study design, analysis, interpretation or decision to publish this data.
Conflict of Interest
Dr. BRECHOT reports In addition, Dr. BRECHOT has a patent WO2010/142800 pending. Dr. AMOUYAL reports In addition, Dr. AMOUYAL has a patent WO2010/142800 pending. Dr. FAIVRE reports In addition, Dr. FAIVRE has a patent WO2010/142800 pending. Dr. Dupuis reports grants from Alfact Innovation, during the conduct of the study; Dr. Amouyal reports In addition, Dr. Amouyal has a patent WO2010/142800 pending. Dr. Gressens reports grants from Alfact Innovation, during the conduct of the study; In addition, Dr. Gressens has a patent WO2010/142800 pending.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Elements taken into account for the quantitative analyses performed on cultured neurons to assess neurite branching.
Effect of rcHIP/PAP on ibotenate-induced (A) and TBI-induced (B) plasticity as determined by immunohistochemistry for pGAP-43. Pups underwent ibotenate injection on P5 or TBI on P7 and were sacrificed 5 days later. PBS or rcHIP/PAP (0.75 μg) was injected i.p. immediately after the ibotenate injection or the TBI. Values represent mean ± SEM densitometry of immunostaining as determined with the ImageJ software (NIH). Asterisks indicate statistically significant difference from the PBS-treated animals; *P < 0.05, **P < 0.01, in Mann–Whitney test.
References
- Mesples B, Plaisant F, Fontaine RH, Gressens P. Pathophysiology of neonatal brain lesions: lessons from animal models of excitotoxicity. Acta Paediatr. 2005;94:185–190. doi: 10.1111/j.1651-2227.2005.tb01888.x. [DOI] [PubMed] [Google Scholar]
- Marret S, Mukendi R, Gadisseux JF, et al. Effect of ibotenate on brain development: an excitotoxic mouse model of microgyria and posthypoxic-like lesions. J Neuropathol Exp Neurol. 1995;54:358–370. doi: 10.1097/00005072-199505000-00009. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399(6738 suppl):A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Wahlestedt C, Golanov E, Yamamoto S, et al. Antisense oligodeoxynucleotides to NMDA-R1 receptor channel protect cortical neurons from excitotoxicity and reduce focal ischaemic infarctions. Nature. 1993;363:260–263. doi: 10.1038/363260a0. [DOI] [PubMed] [Google Scholar]
- Palmer AM, Marion DW, Botscheller ML, et al. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J Neurochem. 1993;61:2015–2024. doi: 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- Guo Q, Sebastian L, Sopher BL, et al. Neurotrophic factors [activity-dependent neurotrophic factor (ADNF) and basic fibroblast growth factor (bFGF)] interrupt excitotoxic neurodegenerative cascades promoted by a PS1 mutation. Proc Natl Acad Sci USA. 1999;96:4125–4130. doi: 10.1073/pnas.96.7.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Olney JW, Labruyere J, Wang G, et al. NMDA antagonist neurotoxicity: mechanism and prevention. Science. 1991;254:1515–1518. doi: 10.1126/science.1835799. [DOI] [PubMed] [Google Scholar]
- Olney JW, Labruyere J, Price MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science. 1989;244:1360–1362. doi: 10.1126/science.2660263. [DOI] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Hartman RE, Izumi Y, et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–882. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Manning SM, Talos DM, Zhou C, et al. NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J Neurosci. 2008;28:6670–6678. doi: 10.1523/JNEUROSCI.1702-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M, Griesmaier E, Auer M, et al. Dextromethorphan is protective against sensitized N-methyl-d-aspartate receptor-mediated excitotoxic brain damage in the developing mouse brain. Eur J Neurosci. 2008;27:874–883. doi: 10.1111/j.1460-9568.2008.06062.x. [DOI] [PubMed] [Google Scholar]
- Namikawa K, Fukushima M, Murakami K, et al. Expression of Reg/PAP family members during motor nerve regeneration in rat. Biochem Biophys Res Commun. 2005;24:126–134. doi: 10.1016/j.bbrc.2005.04.105. [DOI] [PubMed] [Google Scholar]
- Ampo K, Suzuki A, Konishi H, Kiyama H. Induction of pancreatitis-associated protein (PAP) family members in neurons after traumatic brain injury. J Neurotrauma. 2009;26:1683–1693. doi: 10.1089/neu.2008.0847. [DOI] [PubMed] [Google Scholar]
- Marz-Weiss P, Kunz D, Bimmler D, et al. Expression of pancreatitis-associated protein after traumatic brain injury: a mechanism potentially contributing to neuroprotection in human brain. Cell Mol Neurobiol. 2011;31:1141–1149. doi: 10.1007/s10571-011-9715-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averill S, Davis DR, Shortland PJ, et al. Dynamic pattern of reg-2 expression in rat sensory neurons after peripheral nerve injury. J Neurosci. 2002;22:7493–7501. doi: 10.1523/JNEUROSCI.22-17-07493.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey FJ, O’Brien JA, Li M, et al. A Schwann cell mitogen accompanying regeneration of motor neurons. Nature. 1997;390:614–618. doi: 10.1038/37615. [DOI] [PubMed] [Google Scholar]
- Nishimune H, Vasseur S, Wiese S, et al. Reg-2 is a motoneuron neurotrophic factor and a signalling intermediate in the CNTF survival pathway. Nat Cell Biol. 2000;2:906–914. doi: 10.1038/35046558. [DOI] [PubMed] [Google Scholar]
- Moniaux N, Song H, Darnaud M, et al. Human hepatocarcinoma-intestine-pancreas/pancreatitis-associated protein cures fas-induced acute liver failure in mice by attenuating free-radical damage in injured livers. Hepatology. 2011;53:618–627. doi: 10.1002/hep.24087. [DOI] [PubMed] [Google Scholar]
- Lieu HT, Demauger F, Simon M, et al. HIP/PAP stimulates liver regeneration by increasing growth associated protein-43 expression through the PKA-dependent phosphorylation of cAMP regulated phosphoprotein-19. 41st Annual Meeting of the European-Association for the Study of the Liver, Austria. J Hepatol. 2006;44:S136–S136. [Google Scholar]
- Irwin N, Chao S, Goritchenko L, et al. Nerve growth factor controls GAP-43 mRNA stability via the phosphoprotein ARPP-19. Proc Natl Acad Sci USA. 2002;99:12427–12431. doi: 10.1073/pnas.152457399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosevitsky MI. Nerve ending “signal” proteins GAP-43, MARCKS, and BASP1. Int Rev Cytol. 2005;245:245–325. doi: 10.1016/S0074-7696(05)45007-X. [DOI] [PubMed] [Google Scholar]
- Gupta SK, Mishra R, Kusum S, et al. GAP-43 is essential for the neurotrophic effects of BDNF and positive AMPA receptor modulator S18986. Cell Death Differ. 2009;16:624–637. doi: 10.1038/cdd.2008.188. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Qin Y, Labruyere J, et al. Prevention of trauma-induced neurodegeneration in infant rat brain. Pediatr Res. 1996;39:1020–1027. doi: 10.1203/00006450-199606000-00015. [DOI] [PubMed] [Google Scholar]
- Haldipur P, Bharti U, Govindan S, et al. Expression of Sonic hedgehog during cell proliferation in the human cerebellum. Stem Cells Dev. 2012;21:1059–1068. doi: 10.1089/scd.2011.0206. [DOI] [PubMed] [Google Scholar]
- Favrais G, van de Looij Y, Fleiss B, et al. Systemic inflammation disrupts the developmental program of white matter. Ann Neurol. 2011;70:550–565. doi: 10.1002/ana.22489. [DOI] [PubMed] [Google Scholar]
- Carlsson Y, Schwendimann L, Vontell R, et al. Genetic inhibition of caspase-2 reduces hypoxic-ischemic and excitotoxic neonatal brain injury. Ann Neurol. 2011;70:781–789. doi: 10.1002/ana.22431. [DOI] [PubMed] [Google Scholar]
- Degos V, Charpentier TL, Chhor V, et al. Neuroprotective effects of dexmedetomidine against glutamate agonist-induced neuronal cell death are related to increased astrocyte brain-derived neurotrophic factor expression. Anesthesiology. 2013;118:1123–1132. doi: 10.1097/ALN.0b013e318286cf36. [DOI] [PubMed] [Google Scholar]
- Medja F, Lelievre V, Fontaine RH, et al. Thiorphan, a neutral endopeptidase inhibitor used for diarrhoea, is neuroprotective in newborn mice. Brain. 2006;129(Pt 12):3209–3223. doi: 10.1093/brain/awl239. [DOI] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Pohl D, et al. Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol. 1999;45:724–735. doi: 10.1002/1531-8249(199906)45:6<724::aid-ana6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Kaindl AM, Zabel C, Stefovska V, et al. Subacute proteome changes following traumatic injury of the developing brain: implications for a dysregulation of neuronal migration and neurite arborization. Proteomics Clin Appl. 2007;1:640–649. doi: 10.1002/prca.200600696. [DOI] [PubMed] [Google Scholar]
- Gupta SK, Meiri KF, Mahfooz K, et al. Coordination between extrinsic extracellular matrix cues and intrinsic responses to orient the centrosome in polarizing cerebellar granule neurons. J Neurosci. 2010;30:2755–2766. doi: 10.1523/JNEUROSCI.4218-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husson I, Mesples B, Bac P, et al. Melatoninergic neuroprotection of the murine periventricular white matter against neonatal excitotoxic challenge. Ann Neurol. 2002;51:82–92. doi: 10.1002/ana.10072. [DOI] [PubMed] [Google Scholar]
- Redecker C, Hagemann G, Witte OW, et al. Long-term evolution of excitotoxic cortical dysgenesis induced in the developing rat brain. Brain Res Dev Brain Res. 1998;109:109–113. doi: 10.1016/s0165-3806(98)00065-0. [DOI] [PubMed] [Google Scholar]
- Lieu HT, Simon MT, Nguyen-Khoa T, et al. Reg2 inactivation increases sensitivity to Fas hepatotoxicity and delays liver regeneration post-hepatectomy in mice. Hepatology. 2006;44:1452–1464. doi: 10.1002/hep.21434. [DOI] [PubMed] [Google Scholar]
- Lv Y, Yang X, Huo Y, et al. Adenovirus-mediated hepatocarcinoma-intestine-pancreas/pancreatitis-associated protein suppresses dextran sulfate sodium-induced acute ulcerative colitis in rats. Inflamm Bowel Dis. 2012;18:1950–1960. doi: 10.1002/ibd.22887. [DOI] [PubMed] [Google Scholar]
- Yang X, Jin H, Liu K, et al. A novel peptide derived from human pancreatitis-associated protein inhibits inflammation in vivo and in vitro and blocks NF-kappa B signaling pathway. PLoS ONE. 2011;6:e29155. doi: 10.1371/journal.pone.0029155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viterbo D, Bluth MH, Lin YY, et al. Pancreatitis-associated protein 2 modulates inflammatory responses in macrophages. J Immunol. 2008;181:1948–1958. doi: 10.4049/jimmunol.181.3.1948. [DOI] [PubMed] [Google Scholar]
- Vasseur S, Hoffmeister A, Garcia-Montero A, et al. Mice with targeted disruption of p8 gene show increased sensitivity to lipopolysaccharide and DNA microarray analysis of livers reveals an aberrant gene expression response. BMC Gastroenterol. 2003;3:25. doi: 10.1186/1471-230X-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RE, Giffard RG. MicroRNA-320 induces neurite outgrowth by targeting ARPP-19. NeuroReport. 2012;23:590–595. doi: 10.1097/WNR.0b013e3283540394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klementiev B, Novikova T, Korshunova I, et al. The NCAM-derived P2 peptide facilitates recovery of cognitive and motor function and ameliorates neuropathology following traumatic brain injury. Eur J Neurosci. 2008;27:2885–2896. doi: 10.1111/j.1460-9568.2008.06245.x. [DOI] [PubMed] [Google Scholar]
- Delcour M, Olivier P, Chambon C, et al. Neuroanatomical, sensorimotor and cognitive deficits in adult rats with white matter injury following prenatal ischemia. Brain Pathol. 2012;22:1–16. doi: 10.1111/j.1750-3639.2011.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elements taken into account for the quantitative analyses performed on cultured neurons to assess neurite branching.
Effect of rcHIP/PAP on ibotenate-induced (A) and TBI-induced (B) plasticity as determined by immunohistochemistry for pGAP-43. Pups underwent ibotenate injection on P5 or TBI on P7 and were sacrificed 5 days later. PBS or rcHIP/PAP (0.75 μg) was injected i.p. immediately after the ibotenate injection or the TBI. Values represent mean ± SEM densitometry of immunostaining as determined with the ImageJ software (NIH). Asterisks indicate statistically significant difference from the PBS-treated animals; *P < 0.05, **P < 0.01, in Mann–Whitney test.