

Abstract
Objective
To identify molecular signatures in muscle from patients with amyotrophic lateral sclerosis (ALS) that could provide insight into the disease process and serve as biomarkers.
Methods
RNA sequencing was performed on ALS and control muscle samples to identify Smad family members as potential markers of disease. Validation studies were performed in a cohort of 27 ALS patients and 33 controls. The markers were assessed in the G93A superoxide dismutase (SOD)1 mouse at different stages of disease and in a model of sciatic nerve injury.
Results
Smad8, and to a lesser extent Smad1 and 5, mRNAs were significantly elevated in human ALS muscle samples. The markers displayed a remarkably similar pattern in the G93A SOD1 mouse model of ALS with increases detected at preclinical stages. Expression at the RNA and protein levels as well as protein activation (phosphorylation) significantly increased with disease progression in the mouse. The markers were also elevated to a lesser degree in gastrocnemius muscle following sciatic nerve injury, but then reverted to baseline during the muscle reinnervation phase.
Interpretation
These data indicate that Smad1, 5, 8 mRNA and protein levels, as well as Smad phosphorylation, are elevated in ALS muscle and could potentially serve as markers of disease progression or regression.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating and fatal neurodegenerative disease with no definite etiology identified and no effective treatment. The hallmark of ALS is progressive muscle atrophy and weakness leading to loss of limb and bulbar function.1 While degeneration of motor neurons underlies these clinicopathological changes, there is mounting evidence to support an active role of skeletal muscle in disease progression.2 In animal models of ALS, alterations in muscle, including neuromuscular junction abnormalities and mitochondrial dysfunction, can be detected prior to loss of motor neurons, prompting the hypothesis that motor neuron degeneration is a distal axonopathy.2–4 The participation of skeletal muscle in motor neuron degeneration is further supported by two transgenic mouse models where muscle-restricted expression of the uncoupling protein 1 (UCP1) or superoxide dismutase (SOD) 1 genes led to motor neuron degeneration.2,5–7 In the latter model, another report suggested that muscle-restricted attenuation of mutant SOD1 did not affect the development of disease.8 Skeletal muscle provides an essential role in the health of the motor neuron through production of growth and survival factors such as neurotrophin-4 and bone morphogenetic protein (BMP) 4, and disruption of these factors could potentially modify progression of motor neuron degeneration independent of the underlying cause.9–12 For these reasons, we investigated skeletal muscle gene expression patterns in ALS patients to identify targets that might provide insight into disease-relevant pathways and identify biomarkers. We found a significant upregulation of Smad1, 5, and 8 in ALS patients with Smad8 mRNA showing a disproportionately larger increase. A similar pattern was observed in the G93A mutant SOD1 ALS mouse, and the upregulation paralleled disease progression. In a sciatic nerve injury model, all three Smads increased in muscle during the acute phase and reverted back to baseline in the muscle reinnervation phase. Smad1, 5, and 8 are highly homologous members of the Smad family which is involved in transforming growth factor beta (TGFβ) and BMP signal transduction.13 Referred to as receptor regulated (R) Smads, these proteins become phosphorylated upon activation of BMP receptors (TGFβ for Smad2 and 3), translocate to the nucleus in association with Smad4, and modulate transcription through direct DNA binding or interaction with cofactors.13,14 Our findings suggest that the Smads, particularly Smad8, are elevated and active in ALS muscle and may be useful as markers of disease progression (or regression).
Materials and Methods
Animals
B6.Cg-Tg (SOD1*G93A) 1 Gur/J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Transgenic mice were maintained in the hemizygous state by mating G93A males with C57BL/6J females. Nontransgenic littermates were used as controls. For the sciatic nerve injury experiment, mice were from the C57BL/6J background and bred in-house. All animal procedures were reviewed and approved by the UAB Institutional Animal Care and Use Committee in compliance with the National Research Council Guide for the Care and Use of Laboratory Animals. For sciatic nerve injury, the spared nerve injury (SNI) model was chosen whereby all divisions of the sciatic nerve at the trifurcation, except the sural branch, are transected.15 Under oxygen/isoflurane anesthesia, mice in this experiment received unilateral SNI on the left side. A control group underwent sham surgery in which the sciatic nerve was exposed but not transected.
Behavioral assessment
For the G93A SOD1 mice, clinical progression was evaluated by weight determination and performance on the rotarod (San Diego Instruments, San Diego, CA). End-stage disease was determined when the mouse could not right itself after 30 sec when placed on its side. Rotarod testing was based on previously published methods.16 Briefly, the rod was rotated at a gradually accelerating speed up to 11 rpm over a 2 min interval, and the animal’s latency to fall was recorded. Measurements were collected once a week in a cohort of 20 transgenic SOD1*G93A mice and 20 wild-type (WT) littermate controls. For animals undergoing SNI, pain threshold testing with the Von Frey test and the Basso Mouse Scale for Locomotion (BMS) were used to confirm nerve injury.17,18 For the Von Frey test, the up-down method was used to estimate the 50% withdrawal thresholds using nylon monofilaments (Stoelting Co., Wood Dale, IL). Mice were placed in custom-made Plexiglas cubicles (5 × 8.5 × 6 cm) on a perforated metal sheet and were permitted to habituate for at least 1 h prior to testing. As the sural nerve was spared, the filaments were applied to the lateral aspect of the hindpaw for 1 sec and responses were recorded. Two consecutive measures were taken on both hindpaws at each time point, but only the measures on the ipsilateral paw are shown. All mice (n = 6) were tested at baseline and 1 week post nerve injury (PNI). To determine the extent of motor deficits, mice were assessed 2 days PNI using the BMS. This 10-point scale was assessed by two independent observers blinded to the experimental condition. A score of 0 indicates no ankle movement and 10 is normal locomotion.
Tissue collection
After approval by the UAB Institutional Review Board, the UAB Nerve and Muscle Histopathology Laboratory database was searched for archived biopsy samples (stored at −80°C) representative of ALS, normal, and diseased controls (myopathy and neuropathy). A cohort of 27 patients with ALS was identified (Table 1). At the time of biopsy, 17 patients had clinically probable or definite ALS by revised El Escorial criteria.19 The remaining 10 patients had possible ALS, but developed a definite diagnosis over time. For mouse samples, animals were sacrificed by CO2 inhalation followed by cervical dislocation. Brain, spinal cord, hindlimb, and forelimb muscle tissues were dissected at discrete time points between 40 and 150 days postnatal. Samples were briefly rinsed in phosphate-buffered saline (PBS), and frozen in liquid nitrogen and stored at −80°C prior to biochemical analysis. For immunohistochemical studies, samples were frozen on dry ice in Tissue-Tek compound or fixed for 1 day in 4% paraformaldehyde in 0.1 mol/L sodium phosphate buffer, pH 7.4.
Table 1.
ALS patients (n = 27)
| Age range (years) | 33–82 |
| Mean age (years) | 61 ± 11 |
| Gender | 14 M 13 F |
| Duration (months)1 | 11 ± 7 |
| Spinal onset | 21 |
| Bulbar onset | 6 |
| Median survival (years)2 | 2 |
| MRC grade of muscle (n) | 0–2 (8), 3 (4), 4 (11), 5 (4) |
| Muscle (n) | TA (14); VL (4) |
| Bic (3), Delt (6) | |
| EMG denervation | 25 (2 muscles not tested) |
Bic, biceps; Delt, deltoid; EMG, electromyography; MRC, medical research council; TA, tibialis anterior; VL, vastus lateralis.
Symptom onset to time of biopsy.
From time of diagnosis.
Western blot and immunohistochemistry
2Tissues were homogenized in T-Per (Pierce Endogen, Rockford, IL) and quantitated with a bicinchoninic acid (BCA) protein assay kit. Sixty micrograms of protein were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, blotted, and probed with antibodies to the following targets: p-Smad 1/5/8 (Cell Signaling, Beverly, MA), t-Smad 1/5/8 (Santa Cruz Biotechnology, Paso Robles, CA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell Signaling). Densitometry was done with the VersaDoc Imaging System (Bio-Rad, Hercules, CA) and quantified using Image Lab (Bio-Rad). For immunohistochemistry, 10 micron sections of muscle were fixed in Bouin’s fixative for 15 min and then oxidized in 0.3% H2O2 for 15 min. After blocking, sections were incubated with p-Smad or t-Smad 1/5/8 antibodies (1:10) overnight at 4°C. Slides were incubated for an hour at RT with donkey anti-rabbit secondary antibody conjugated with horseradish peroxidase-labeled polymer (Jackson ImmunoResearch, West Grove, PA). Slides were incubated in TSA Plus Cyanine 3 (PerkinElmer, Waltham, MA) at 1:1500 for 30 min, Wheat Germ Agglutinin (WGA), Alexa Fluor® 488 (Invitrogen, Carlsbad, CA) at 1:200 for 10 min, followed by Hoechst 33342 (Sigma-Aldrich, St. Louis, MO) at 1:20,000 for 10 min. The prepared slides were viewed under an Olympus DP 71 fluorescence microscope (Olympus, Center Valley, PA).
RNA Isolation, next-generation sequencing, and qRT-PCR
RNA was extracted from frozen tissues with Trizol Reagent (Invitrogen) according to the manufacturer’s instructions. Next-Generation sequencing was performed on ALS and disease control muscle RNA samples by the UAB Genomics Core Facility. Transcriptional sequencing methods were performed essentially as described elsewhere.20,21
For qRT-PCR, two micrograms of total RNA were reverse transcribed according to the manufacturer’s specifications (Applied Biosystems, Carlsbad, CA). Multiplex PCR was done using On-Demand Taqman primers (Applied Biosystems) in a ViiA 7 Real Time PCR System (Applied Biosystems). Probes for all target genes were 6-carboxy-fluorescein (FAM) labeled, and the endogenous control GAPDH was labeled with VIC dye/MGB. A standard thermal cycle protocol was used. Data were analyzed with the ViiA 7 Software version v1.1 (Applied Biosystems). The baseline was auto set, and threshold values were adjusted from 0.09 to 0.2 based on the amplification curve of different targets. Quantification of target mRNAs was done by the ∆∆CT method using GAPDH as an internal control.22
Statistical analyses
For qRT-PCR comparisons between ALS group and all other groups, one-way analysis of variance (ANOVA) and pair-wise t tests were conducted on ΔCt values. For Western blot data analysis, we first logarithm-transformed and normalized the data using the GAPDH values for each lane. Protein quantities were modeled using a linear model with genotype, age and their interaction term controlling for blot. For the p-Smad/t-Smad analysis, ratios on the logarithm scale were modeled using a linear model similar to that for the total amount of protein. Paired t tests were used to compare baseline and PNI mechanical sensitivity, and unpaired t tests were used to compare protein or RNA across groups.
Results
Smad1, 5, and 8 mRNAs are elevated in muscle biopsies of patients with ALS
We performed next-generation sequencing of RNA isolated from muscle biopsies of patients with ALS, diseased controls, and normal controls. We observed a significant (four- to eightfold) elevation of Smad8 mRNA in ALS muscle samples over controls (data not shown). To validate this finding, we tested a large cohort of ALS and control muscle samples by qRT-PCR (Tables 1, 2). The ALS cohort (n = 27) reflected demographics that matched our overall ALS population, including a mean age of 61 years at diagnosis, and a slight male predominance.23 Medical Research Council (MRC) grades were available for all muscles and ranged from 0 to 5. Electromyography (EMG) testing of the biopsied muscle was available for 25 patients and all showed evidence of active and/or chronic denervation. Our control population consisted of histologically normal patients (referred for nonspecific complaints, usually myalgias or cramps), and diseased controls (histologically proven myopathy or neuropathy-related denervation). We assessed Smad8 mRNA levels by qRT-PCR in these samples, using GAPDH as an internal control (Fig. 1A). We found a significant upregulation of Smad8 in ALS patients at ~19-fold compared to normals, threefold to neuropathy and 5.6-fold to myopathy controls (P < 0.0001 for all comparisons). We looked at the closely related family members, Smad1 and 5, and found significant increases, but to a much smaller degree (~threefold over controls and ~1.5-fold over diseased controls, P < 0.05). Correlation between all Smads in the ALS samples was highly significant (Fig. 1B), with Smad1 and 5 being the strongest (Pearson coefficient of 0.88, P < 0.0001). Smad mRNA levels and MRC grade showed a positive correlation trend which was not statistically significant at the 0.05 level (P values 0.09 to 0.23; data not shown).
Table 2.
Control patients
| Normal (n = 13) | Myopathy (n = 11) | Neuropathy (n = 9) | |
|---|---|---|---|
| Age range (years) | 24–67 | 38–74 | 39–88 |
| Mean age (years) | 53 ± 11 | 61 ± 14 | 56 ± 18 |
| Diagnosis | – | Inflammatory (7) | Axonal (2) |
| Mitochondrial (4) | Plexopathy (2) | ||
| CIDP (2) | |||
| GBS (1) | |||
| Muscle (all) | Nonspecific (2) | ||
| TA (5); | |||
| VL (10); | |||
| Bic (11); | |||
| Delt (7) |
Bic, biceps; CIDP, Chronic inflammatory demyelinating polyneuropathy; Delt, deltoid; GBS, Guillain Barre Syndrome; TA, tibialis anterior; VL, vastus lateralis.
Figure 1.
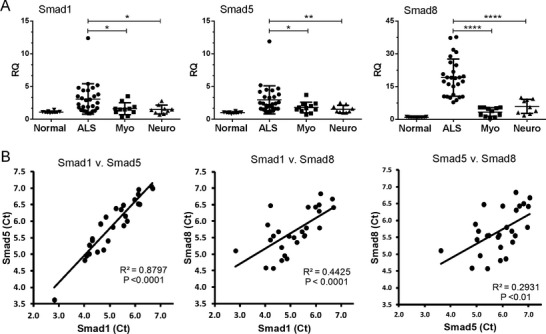
Smad1, 5, and 8 mRNA expression is upregulated in muscle samples from amyotrophic lateral sclerosis (ALS) patients. (A) Total RNA from muscle biopsy samples was analyzed by qRT-PCR for Smad1, 5, and 8 mRNA expression in patients with ALS (n = 27), myopathy (n = 11), neuropathy (n = 9), or no neuromuscular disease (n = 13). RQ, relative quantity. *P < 0.05; ****P < 0.0001. (B) Correlation plots between the Smad Ct values from the ALS muscle samples.
Smad1, 5, and 8 mRNAs are elevated in muscle from G93A SOD1 mice
As a separate validation, we next examined expression of Smad mRNA in muscle samples from the G93A SOD1 mouse. This model recapitulates many of the pathological and clinical features of ALS, including progressive weakness, motor neuron loss, and muscle denervation.24 Our mouse model is on the C57/BL6 background and displays a later onset of clinical symptoms and longer survival than the original BL6/SJL mouse (Fig. 2A).25 In this model, subtle differences in motor function between controls can be detected 45 days (postnatal), well before overt clinical manifestations which usually occur at 90–100 days.26 By weeks 7–8, there is a significant decrease in the weight of hindlimb muscles accompanied by increased expression of proteins associated with stress response, oxidative metabolism, and autophagy.26–28 We sampled the gastrocnemius muscle at different clinical stages, including three preclinical stages (40, 60, and 105 days), one just after onset of motor deterioration as measured by rotarod testing (125 days) and one at end stage (150 days). WT littermates served as a control. RNA was extracted and assessed by qRT-PCR. We observed a pattern of upregulation very similar to the human samples, with Smad8 showing a substantially greater fold-change over WT mice (up to 17-fold at end stage) compared to Smad1 and 5 (up to fivefold at end stage). There was a clinical stage-dependent increase in all Smads, beginning in presymptomatic stages of the disease (60 and 105 days). At day 40, however, there was no difference between ALS and control samples. We assessed these markers in forelimb muscles which have a slower onset of denervation, atrophy, and functional impairment.24,27–29 We observed no elevation in any of the Smad mRNAs at day 60 but a significant increase at day 125 (Fig. S1). The increase was less than the hindlimb muscles by two- to fourfold.
Figure 2.
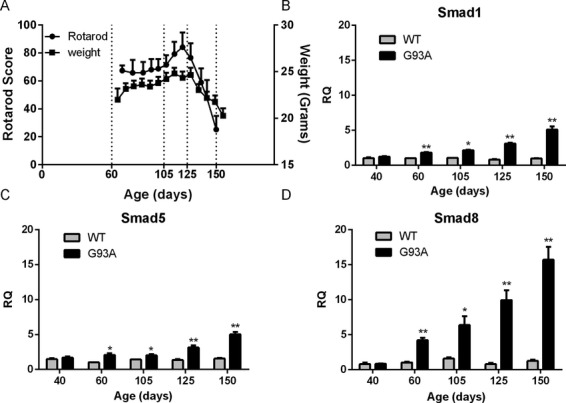
Smad1, 5, and 8 mRNAs are upregulated in G93A SOD1 mouse muscle and increase with disease progression. (A) Rotarod testing and weight measurement of G93A SOD1 mice over the course of the disease. Gastrocnemius muscle samples were obtained at the ages shown and also at day 40. Control muscle samples were obtained from age-matched nontransgenic littermates. (B) Smad1, (C) Smad5, (D) Smad8 mRNA levels were quantitated using qRT-PCR and normalized to an internal housekeeping (GAPDH) control. All data points represent the mean ± SEM of 3–8 independent mice. *P < 0.01; **P < 0.001.
To determine whether Smad upregulation was specific to muscle or a more global phenomenon, we assessed mouse brain and spinal cord tissues at presymptomatic (60 days) and symptomatic (125 days) stages. No differences were observed between WT and mutant mice (Fig. 3). These findings indicate that Smad1, 5, and 8 upregulation is an early and specific event in the muscle of ALS mice, and that the gradual rise in gene expression, particularly Smad8, parallels disease progression.
Figure 3.
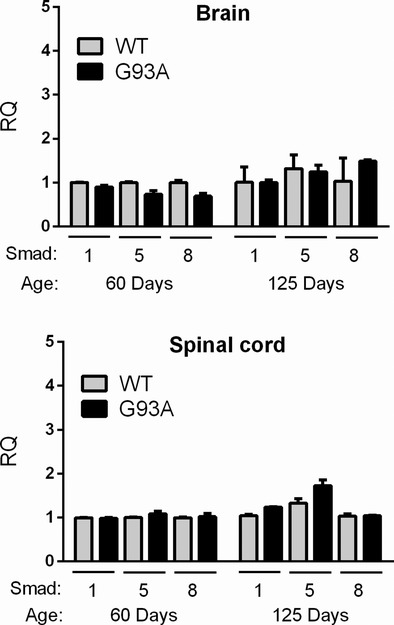
Smad1, 5, and 8 mRNAs are not elevated in G93A mouse spinal cord or brain tissues. Spinal cord and brain tissues from G93A SOD1 and nontransgenic littermates (Wild-type [WT]) were assessed by qRT-PCR for Smad1, 5, and 8 expression at the ages indicated. Values were expressed relative to WT at day 60 which was set at 1. Data represent the mean ± SEM of 3 mice.
Smad1, 5, 8 protein is elevated in muscle from G93A SOD1 mice and Human ALS patients
Since the data indicated a significant upregulation of Smad1, 5, and 8 mRNA levels, we next determined whether Smad protein was increased and/or activated. We performed Western blot analysis of mouse muscle samples using an antibody which recognizes the phosphorylated (activated) form of all three proteins. We observed an increase in activated Smad at each of the stages, most prominently at 150 days (representative blot shown in Fig. 4A). Reprobing the blot with an antibody that recognizes total Smad1, 5, and 8, however, showed an increase in total levels consistent with the mRNA expression patterns. To quantify changes both in total Smad levels and p-Smad/t-Smad ratios, we assessed three to six independent mutant and WT littermates per clinical stage. We observed a significant increase in total Smad1, 5, and 8 protein over WT control over the course of the disease (P < 0.05; Fig. 4B). The fold-difference between mutant and WT mice progressively increased, and at end stage it was nearly threefold. Despite the increase in total Smad protein, however, the p-Smad/t-Smad ratio was still higher than WT at each clinical stage (Fig. 4C). At end stage, the p-smad/t-smad ratio was nearly 15-fold greater than WT (P < 0.0001) and significantly higher than earlier stages of the ALS mice (P < 0.05). We also assessed forelimb muscles for p-Smad and observed no differences at day 60, but a clear increase at day 125, consistent with a delayed onset in forelimb muscles (Fig. S2). In summary, smad1, 5, and 8 protein increased with disease progression in parallel with Smad activation.
Figure 4.
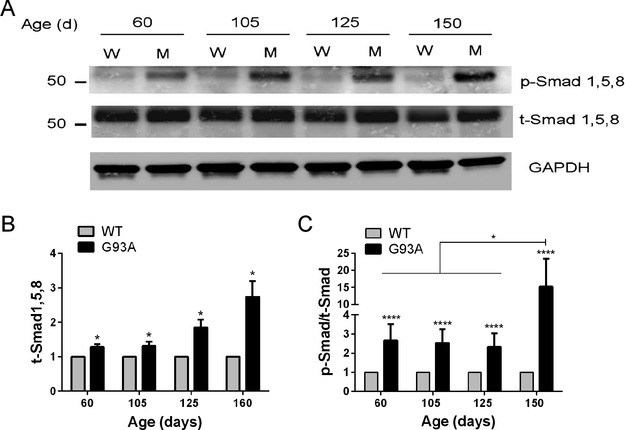
Smad1, 5, and 8 protein expression and activation increase with disease progression in the G93A SOD1 mouse. (A) Representative Western blot of gastrocnemius samples from G93A SOD1 mice (M) and littermate controls (W) at different ages. Antibodies are shown to the right of the blot. (B) total Smad1, 5, and 8 was quantified by densitometry, normalized to GAPDH, and compared to nontransgenic littermate controls (set at 1). (C) p-Smad/t-Smad ratios were calculated in G93A mice and compared to littermate controls (set at 1). Data points represent the mean ± SEM of 4–6 mice in each group. *P < 0.05; ****P < 0.0001.
p-Smad1, 5, 8 protein localizes to nuclei and cell membrane in muscle from G93A SOD1 mice and human ALS
We assessed localization of p-Smad1, 5, and 8 by immunofluorescence in mouse at a preclinical stage (60 days) and detected staining in multiple myofibers, but no staining in an age-matched WT mouse (Fig. 5). There were both punctate and more diffuse staining observed in the periphery of the cells. Higher power views showed overlap of the punctate signal with Hoechst-stained myonuclei, indicating nuclear localization. Additionally, we saw immunoreactivity along the periphery of the myocytes which overlapped with WGA, a lectin stain which identifies myofiber membrane boundaries. Although the data suggest localization to the cell membrane, we cannot exclude that this may be related to muscle atrophy and membrane leakiness. In a biopsy sample from a patient with ALS at an early stage, there was also specific localization of p-Smad to the myonuclei (Fig. 5). We then assessed a muscle sample from an older mutant mouse (131 days) and found a similar pattern but with more extensive staining (Fig. 6). This finding is consistent with Western blot results shown in Figure 4. A muscle sample from an ALS patient at end stage also showed extensive p-Smad staining of myonuclei and myofiber borders suggesting a similar pattern of Smad1, 5, and 8 accumulation with disease progression.
Figure 5.
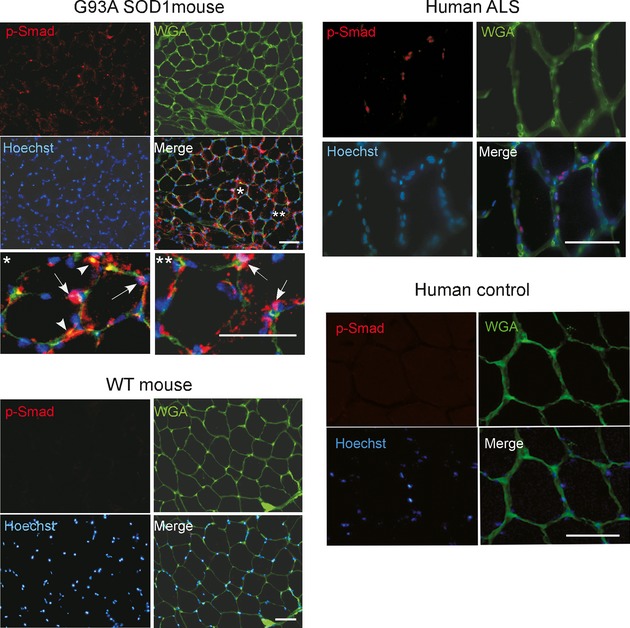
Smad1, 5, and 8 is activated in mouse and human amyotrophic lateral sclerosis (ALS) muscle samples. Muscle sections from G93A SOD1 mouse (day 60) and a biopsy from a patient with ALS were stained with p-Smad1, 5, and 8 antibody, Wheat Germ Agglutinin (WGA) lectin, and Hoechst as indicated. For the G93A SOD1 mouse, the bottom panels represent higher power views of areas highlighted by asterisks. The arrows indicate colocalization of p-Smad1, 5, and 8 with Hoechst (nuclear) signal, and arrowheads show colocalization with WGA. An age-matched nontransgenic littermate and a normal human muscle biopsy sample were used as controls. Scale bars, 50 microns.
Figure 6.
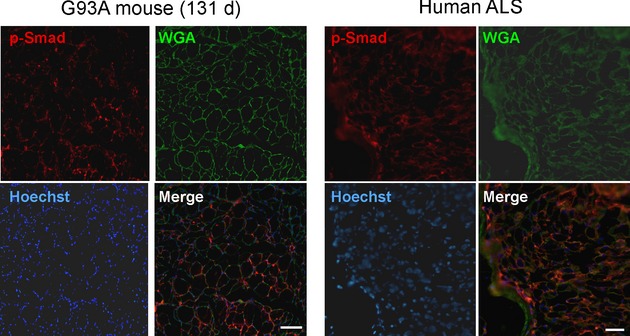
p-Smad1, 5, and 8 accumulates as the disease progresses. Increased p-Smad1, 5, and 8 staining at a later stage of disease in amyotrophic lateral sclerosis (ALS) mouse muscle (131 days) and in a patient with end-stage ALS. Scale bars, 50 microns.
Smads are elevated in a peripheral nerve injury model
To assess Smad induction in non-ALS muscle denervation, we used a model of sciatic nerve injury. Mice underwent sciatic nerve transection or sham surgery and Smad profiles were assessed at 2.5 days, 1 and 4 weeks PNI. Nerve injury was confirmed by motor defects using the BMS and the development of allodynia using the Von Frey withdrawal test (Fig. 7A). Injured mice showed significant locomotion defects (day 2 PNI) and mechanical allodynia (day 7 PNI). We assessed Smad mRNA expression in the gastrocnemius muscle in the injured limb. All comparisons were made to a control group which underwent a sham procedure where the sciatic nerve was exposed but not transected. We observed a progressive increase in Smad1 and 5 mRNAs to ~fivefold over control at 1 week PNI which then returned to baseline at 4 weeks, a time point in which a majority of muscle fibers are reinnervated (Fig. 7B).30 Smad8 increased to a lesser extent (~fourfold) and also reverted back toward baseline at 4 weeks. Assessment of protein expression by Western blot revealed an increase in total Smad1, 5, and 8 at 1 week post injury (P < 0.05) which then declined at 4 weeks to that of control (Fig. 7C and D). Phosphorylated Smad1, 5, and 8 was significantly elevated at 1 week and remained elevated at 4 weeks, whereas the p-smad/t-smad ratio was only increased at 4 weeks. In summary, there was increased expression of the Smads to a similar degree following nerve injury, which reversed over time. Smad activation persisted in the muscle reinnervation phase out of proportion to total Smad expression.
Figure 7.
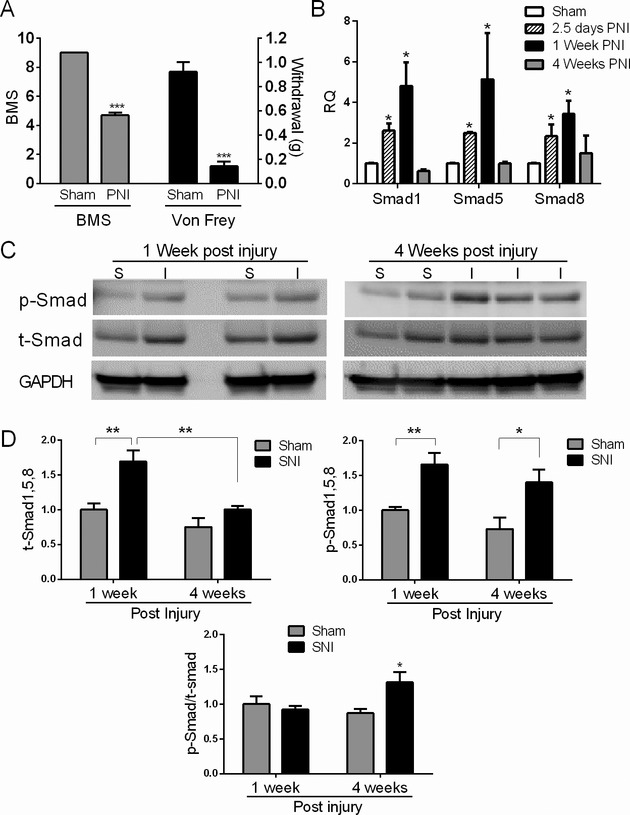
Muscle Smads are induced and activated in a peripheral nerve injury model. (A) Mice were subjected to sciatic nerve injury (SNI) as described in the Materials and Methods. To determine efficacy of the injury, mice were assessed behaviorally with the Basso mouse scale for locomotion (BMS) 2 days post nerve injury (PNI) and the Von Frey mechanical sensitivity test 1 week PNI (except for the 2.5 day group). There was a significant reduction in locomotion and mechanical threshold to withdraw indicative of nerve injury. ***P < 0.001. (B) Smad mRNA levels were assessed in the gastrocnemius muscle by qRT-PCR at 2.5 days, 1 and 4 weeks post nerve injury (PNI) and compared to a control group which underwent sham surgery. All RNA values were expressed as a fold-change over the 1 week control group. Data are from 3–6 mice in each group. (C) Representative Western blot of muscle extracts from injured (I) or sham controls (S). Antibodies are shown to the left of the blots. (D) Quantitative densitometry of three Western blots representative of 3–6 mice in each test group. For t- and p-Smad quantitation, band densities were adjusted to the loading control (GAPDH) and then expressed as a fold-change over the sham control at 1 week PNI. P-Smad/t-Smad ratios were calculated and expressed as a fold-change over the control group at week 1 PNI. *P < 0.05; **P < 0.005.
Discussion
We have identified Smad1, 5, and 8 as potential muscle biomarkers of disease progression in ALS at the transcriptional (mRNA), translational (protein) and posttranslational (phosphorylation) levels. Smad8, first identified by RNA sequencing of human ALS muscle biopsy samples, showed the highest specificity for ALS patients at the mRNA level (Fig. 1). The markers were validated in a SOD1 mouse model of ALS where there was a gradual accumulation of Smad protein and mRNA, starting in preclinical stages to end stage. Concomitantly, there was activation of these Smad members which enhanced significantly as the disease progressed.
The impact of our findings is at the clinical and basic science levels of ALS. A challenge facing investigations of novel therapeutic compounds in ALS is the ability to monitor clinical responses in a timely manner to determine efficacy. Standard clinical testing, including the ALS Functional Rating Score, is relatively insensitive.31,32 Based on our findings, two features of Smads correlated with disease progression in the ALS mouse: gene upregulation (mRNA and protein) and protein activation (phosphorylation). These molecular changes also occurred before overt clinical manifestations, suggesting that the markers are sensitive to more subtle loss of motor neurons. Furthermore, the hindlimb muscles showed earlier expression and activation of Smads compared to forelimb muscles providing further support that these markers may track disease progression.24,27–29
The sciatic nerve injury findings (Fig. 7) indicate that mRNA upregulation of these Smads is not specific to ALS. The disproportionate increase in Smad8, however, in both human ALS and mutant mouse muscle, was not observed in the sciatic nerve injury model or human neuropathy controls, suggesting a level of specificity. Whether this signature is unique to motor neuron disease or a nonspecific consequence of muscle denervation remains to be determined. The sciatic nerve injury data also indicate that Smad mRNA and protein levels can revert to baseline upon reinnervation, supporting a potential role for Smads in tracking disease regression. The second finding in this report, phosphorylation of Smads in ALS muscle, has been reported in other nerve injury models.33,34 In a study by Winbanks et al., activation of Smad1, 5, and 8 was detected in the G93A SOD1 mouse (in the Bl6/SJL background) at 60 and 90 days to equal degrees.33 Interestingly, total Smad was assessed by a Smad5 antibody which showed lower levels in the ALS mouse, in contrast to our findings of an increase in total Smad1, 5, and 8 (Fig. 4). Later stages were not assessed in that study to determine if activation tracked disease progression. Interestingly, in our report, Smad phosphorylation persisted in the muscle reinnervation phase of sciatic nerve injury when RNA and protein levels returned to baseline. This finding suggests that gene induction may be a better metric than protein activation for detecting early reinnervation. Activation or upregulation of other Smad members has been reported in motor neurons of ALS patients. In one study, activation of Smad2 and 3, as reflected by nuclear translocation of phosphorylated forms, was observed in motor neurons of sporadic and familial ALS patients.35 Smad members were identified in hyaline and skein-like inclusions in motor neurons of ALS patients where they colocalized with TDP-43.35,36 The investigators postulated that there may be aberrant TGF-β signal transduction in neurons with these inclusions.
Several fundamental questions emerge from this report. First, what is driving Smad upregulation and activation, particularly Smad8? Although Smad1, 5, and 8 activation is classically associated with BMP ligands, there is clear evidence that they can be activated by TGF-β ligands in a wide range of cell types.37,38 Our RNA sequencing data have provided clues that both TGFβ and BMP ligands may be involved in Smad activation and gene upregulation in ALS muscle (not shown). Work is underway to further delineate the relative contributions of these pathways. The second question is whether the Smad molecular response is a physiological one related to progressive loss of muscle innervation, or a pathological one that contributes to disease progression. For the SOD1 mouse, we did not observe Smad upregulation in the spinal cord or brain of ALS mice, or in cultured astrocytes and cortical neurons expressing G93A SOD1 (Fig. 3 and data not shown), suggesting that the response was specific to muscle. Activation of Smad1, 5, and 8 via BMP signaling, has been shown to be a major hypertrophic signaling pathway in muscle, and inhibition leads to an accentuation of atrophy following experimentally induced nerve injury.33,34 Likewise, augmenting this signaling pathway lessened denervation-induced atrophy. These findings suggest that Smad upregulation in muscle is a physiological response, possibly to offset muscle atrophy from motor neuron loss. The disproportionate increase in Smad8 mRNA expression in ALS muscle is of unclear biological significance. Smad1, 5, and 8 are highly homologous, and a Smad8-specific antibody is not yet available to determine the relative contribution of this member to the overall increase in total Smad protein noted in this report (Figs. 4, 5, 6). Although these Smads have functional overlap in certain biological contexts, Smad8 plays a nonredundant role in miRNA processing (a noncanonical function previously linked to Smads).13,39–43 Mutations of Smad8 in patients with heritable pulmonary arterial hypertension, for example, lead to aberrant miRNA processing that can be reversed with restored Smad8 function.43 Since altered miRNA patterns have been identified in ALS skeletal muscle, it is possible that this noncanonical function may be relevant.30,44
In summary, we have identified Smads as potential muscle biomarkers of disease progression in ALS. These markers may have utility in detecting arrest or reversal of muscle denervation prior to changes in motor function, which would be advantageous when assessing efficacy of experimental therapies. The applicability of these markers (and muscle markers in general) to clinical assessments in ALS remains to be seen in light of the asymmetrical nature of denervation (and progression) of the disease and the potentially broad distribution of muscles affected. Although all of our human ALS muscles samples had elevated Smads, longitudinal studies will be required to determine whether there is correlation with disease progression as in the G93A mouse. Since the Smads are intracellular and not typically secreted, they are less likely to be detected in blood. The sensitivity of qRT-PCR for measuring mRNA levels, however, would require only small amounts of tissue that could be obtained from a needle biopsy, thereby facilitating multiple samplings. Moreover, it may be possible in the future to develop an in vivo tracer for activated p-Smads such that a global assessment of disease could be determined at any one time.
Acknowledgments
This work was supported by National Institute of Neurological Disorders NS064133, R21NS081743, R21NS085497 (P. H. K.), NS057664 (L. L.), and a Merit Review award, BX001148, from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Program (P. H. K.). We wish to thank Terry Lewis, Ph.D. and the UAB Neuroscience Molecular Detection Core Facility (P30 NS47466) for assistance with muscle immunohistochemistry, and Dr. Michael Crowley and the UAB Genomic Core Facility for performing the next-generation sequencing.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Elevation of Smad mRNA is delayed in the forelimb muscles of ALS mice. Forelimb muscles from G93A and WT mice were assessed by qRT-PCR for Smad mRNA expression. All data points represent the mean ± SEM of 3 mice. *P < 0.05.
{kind=link}
Upregulation of p-Smad is delayed in the forelimb muscles of ALS mice. Western blot analysis of forelimb muscle tissue using p- and total-Smad1, 5, and 8 antibodies at the ages shown. This blot is representative of two blots done on two independent G93A and WT mice with similar results.
References
- Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:639–649. doi: 10.1038/nrneurol.2011.153. [DOI] [PubMed] [Google Scholar]
- Musaro A. State of the art and the dark side of amyotrophic lateral sclerosis. World J Biol Chem. 2010;1:62–68. doi: 10.4331/wjbc.v1.i5.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L, Loeffler J-P. Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic models. Curr Opin Pharmacol. 2009;9:341–346. doi: 10.1016/j.coph.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, et al. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L, Gonzalez de Aguilar JL, Echaniz-Laguna A, et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS One. 2009;4:e5390. doi: 10.1371/journal.pone.0005390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Martin LJ. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum Mol Genet. 2010;19:2284–2302. doi: 10.1093/hmg/ddq106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolny G, Aucello M, Rizzuto E, et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008;8:425–436. doi: 10.1016/j.cmet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Miller TM, Kim SH, Yamanaka K, et al. Gene transfer demonstrates that muscle is not a primary target for non-cell-autonomous toxicity in familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2006;103:19546–19551. doi: 10.1073/pnas.0609411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieshammer U, Lewandoski M, Prevette D, et al. Muscle-specific cell ablation conditional upon Cre-mediated DNA recombination in transgenic mice leads to massive spinal and cranial motoneuron loss. Dev Biol. 1998;197:234–247. doi: 10.1006/dbio.1997.8859. [DOI] [PubMed] [Google Scholar]
- Chou H-J, Lai D-M, Huang C-W, et al. BMP4 is a peripherally-derived factor for motor neurons and attenuates glutamate-induced excitotoxicity in vitro. PLoS One. 2013;8:e58441. doi: 10.1371/journal.pone.0058441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funakoshi H, Belluardo N, Arenas E, et al. Muscle-derived neurotrophin-4 as an activity-dependent trophic signal for adult motor neurons. Science. 1995;268:1495–1499. doi: 10.1126/science.7770776. [DOI] [PubMed] [Google Scholar]
- Lowrie MB, Vrbova G. Dependence of postnatal motoneurones on their targets: review and hypothesis. Trends Neurosci. 1992;15:80–84. doi: 10.1016/0166-2236(92)90014-y. [DOI] [PubMed] [Google Scholar]
- Katagiri T, Tsukamoto S. The unique activity of bone morphogenetic proteins in bone: a critical role of the Smad signaling pathway. Biol Chem. 2013;394:703–714. doi: 10.1515/hsz-2012-0310. [DOI] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Shields SD, Eckert WA, III, Basbaum AI. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain. 2003;4:465–470. doi: 10.1067/s1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- Kaspar BK, Llado J, Sherkat N, et al. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, et al. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, et al. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Miller RG, Swash M, Munsat TL World Federation of Neurology Research Group on Motor Neuron D. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BAA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Kazamel M, Cutter G, Claussen G, et al. Epidemiological features of amyotrophic lateral sclerosis in a large clinic-based African American population. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:334–337. doi: 10.3109/21678421.2013.770030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Heiman-Patterson TD, Deitch JS, Blankenhorn EP, et al. Background and gender effects on survival in the TgN(SOD1-G93A)1Gur mouse model of ALS. J Neurol Sci. 2005;236:1–7. doi: 10.1016/j.jns.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Hayworth CR, Gonzalez-Lima F. Pre-symptomatic detection of chronic motor deficits and genotype prediction in congenic B6.SOD1(G93A) ALS mouse model. Neuroscience. 2009;164:975–985. doi: 10.1016/j.neuroscience.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capitanio D, Vasso M, Ratti A, et al. Molecular signatures of amyotrophic lateral sclerosis disease progression in hind and forelimb muscles of an SOD1(G93A) mouse model. Antioxid Redox Signal. 2012;17:1333–1350. doi: 10.1089/ars.2012.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu AY, Zhai P, Dal Canto MC, et al. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6:349–362. doi: 10.1006/mcne.1995.1027. [DOI] [PubMed] [Google Scholar]
- Azzouz M, Leclerc N, Gurney M, et al. Progressive motor neuron impairment in an animal model of familial amyotrophic lateral sclerosis. Muscle Nerve. 1997;20:45–51. doi: 10.1002/(sici)1097-4598(199701)20:1<45::aid-mus6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Williams AH, Valdez G, Moresi V, et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326:1549–1554. doi: 10.1126/science.1181046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumer D, Talbot K, Turner MR. Advances in motor neurone disease. J R Soc Med. 2014;107:14–21. doi: 10.1177/0141076813511451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowser R, Turner MR, Shefner J. Biomarkers in amyotrophic lateral sclerosis: opportunities and limitations. Nat Rev Neurol. 2011;7:631–638. doi: 10.1038/nrneurol.2011.151. [DOI] [PubMed] [Google Scholar]
- Winbanks CE, Chen JL, Qian H, et al. The bone morphogenetic protein axis is a positive regulator of skeletal muscle mass. J Cell Biol. 2013;203:345–357. doi: 10.1083/jcb.201211134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori R, Schirwis E, Blaauw B, et al. BMP signaling controls muscle mass. Nat Genet. 2013;45:1309–1318. doi: 10.1038/ng.2772. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Ito H, Wate R, et al. Phosphorylated Smad2/3 immunoreactivity in sporadic and familial amyotrophic lateral sclerosis and its mouse model. Acta Neuropathol. 2008;115:327–334. doi: 10.1007/s00401-007-0337-z. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Banno H, et al. Transforming growth factor-beta signaling in motor neuron diseases. Curr Mol Med. 2011;11:48–56. doi: 10.2174/156652411794474356. [DOI] [PubMed] [Google Scholar]
- Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- Daly AC, Randall RA, Hill CS. Transforming growth factor β-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol Cell Biol. 2008;28:6889–6902. doi: 10.1128/MCB.01192-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CY, Wang HP, Zhu ZY, Sun YH. Transcriptional factors smad1 and smad9 act redundantly to mediate zebrafish ventral specification downstream of smad5. J Biol Chem. 2014;289:6604–6618. doi: 10.1074/jbc.M114.549758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvis GD, Jamin SP, Kwan KM, et al. Functional redundancy of TGF-beta family type I receptors and receptor-Smads in mediating anti-Mullerian hormone-induced Mullerian duct regression in the mouse. Biol Reprod. 2008;78:994–1001. doi: 10.1095/biolreprod.107.066605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BN, Hilyard AC, Nguyen PH, et al. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell. 2010;39:373–384. doi: 10.1016/j.molcel.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake KM, Zygmunt D, Mavrakis L, et al. Altered MicroRNA processing in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184:1400–1408. doi: 10.1164/rccm.201106-1130OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivonen JM, Manzano R, Oliván S, et al. MicroRNA-206: a potential circulating biomarker candidate for amyotrophic lateral sclerosis. PLoS One. 2014;9:e89065. doi: 10.1371/journal.pone.0089065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elevation of Smad mRNA is delayed in the forelimb muscles of ALS mice. Forelimb muscles from G93A and WT mice were assessed by qRT-PCR for Smad mRNA expression. All data points represent the mean ± SEM of 3 mice. *P < 0.05.
Upregulation of p-Smad is delayed in the forelimb muscles of ALS mice. Western blot analysis of forelimb muscle tissue using p- and total-Smad1, 5, and 8 antibodies at the ages shown. This blot is representative of two blots done on two independent G93A and WT mice with similar results.