

Abstract
A new series of 4-substituted 2-amino pyrido[3,4-d]pyrimidine derivatives has been designed and synthesized as potential anticancer agents. These compounds were prepared from a common intermediate, 4-chloro-8-methoxy pyrido[3,4-d]pyrimidin-2-amine, followed by palladium catalyzed cross-coupling reactions or nucleophilic aromatic substitutions at the C-4 position. Evaluation of the representative analogs using the US National Cancer Institute’s 60 human cancer cell line (NCI 60) panel identified some of these compounds as exhibiting highly selective activities against breast cancer and renal cancer cell lines. A structure–activity relationship (SAR) study was explored to facilitate further development of this new class of compounds.
Introduction
Cancer is a group of different diseases characterized by uncontrolled cellular growth, tissue damage, invasion and metastases. According to an estimate from the American Cancer Society, a total of 1 638 910 new cancer cases and 577 190 deaths from cancer could occur in the United States this year.1 Although the rate of deaths from cancer has dropped 0.6 percent this year, compared to 2.4 percent fall of deaths from cardiovascular disease,2 the reduction in the overall cancer mortality rate has been disappointing. Currently, cancer is second only to cardiovascular disease as a cause of mortality in the US and is set to become the most common cause in the near future.3 Recent progress in cancer drug discovery has been impeded due to severe side effects, development of resistance to the current drugs and a limited chemical space, which have fueled a high demand for new chemotherapeutic agents with new scaffolds and prompted us to discover new biologically active chemical entities.
Tremendous efforts have been invested in the discovery of new scaffolds in both industry and academia, most of which are heterocyclic structures. Among these scaffolds, the fused pyrimidine derivatives are of considerable pharmacological importance owing to a broad-spectrum of biological activities including antimicrobial,4 anti-inflammatory,5 anti-HIV,6 anti-tubercular,7 anti-malarial,8 etc. Furthermore, fused pyrimidine derivatives have attracted great interest from both medicinal chemists and organic chemists for their well known anticancer activities, and numerous analogs are in development or have been approved for the treatment of cancers. For example, the quinazoline scaffold with fused pyrimidine and benzene rings has been used as a chemical core for a myriad of anticancer agents and four quinazoline analogs, Gefitinib (Iressa), Erlotinib (Tarceva), Lapatinib (Tykerb) and Vandetanib (Caprelsa) (Fig. 1), have been approved by the US FDA for the treatment of cancers. Meanwhile, the fused pyridone scaffolds have also been used as templates for the design of anticancer agents, which resulted in potent anticancer compounds, such as hydroxyfasudil,9 PD 166326,10 NU 1025,11 and Isoquinolone 20 (Fig. 1).12,13 Additionally, the anticancer activities of most recently discovered fused pyrimidine and pyridone derivatives have been attributed to the inhibition of cell signaling transduction pathways which regulate diverse cellular functions, such as proliferation, differentiation, apoptosis, and cell migration. Deregulation of signaling transduction pathways has been considered as a key factor in the development of many cancer types.14,15 Therefore, these fused pyrimidine and pyridone derivatives that specifically target aberrant pathways have represented a new molecularly targeted therapy in cancer treatment with less reliance on non-discriminate killing of tumor and host cells. Recognizing the potential application of both the moieties in drug discovery and development, we designed and explored a new series of pyrido [3,4-d]pyrimidine derivatives bearing a fused six-membered pyridone and pyrimidine ring.
Fig. 1.

Fused pyrimidine and pyridone derivatives as anti-cancer agents.
A variety of synthetic strategies are known for the synthesis of fused pyrimidine systems.16,17 In the present study, the synthesis of the pyrido[4,3-d]pyrimidine heterocyclic system has been accomplished by a thermal cyclocondensation of ethyl 3-amino-2-chloroisonicotinate with chloroformamidine hydrochloride.18,19 Details are described in the following section. Also, the cytotoxicities of products were evaluated using the US National Cancer Institute’s 60 human cancer cell line (NCI 60) screen panel. Herein, we report the synthesis, cytotoxicity profile, and preliminary structure–activity relationship (SAR) of these novel compounds.
Chemistry
The crucial chlorinated intermediate, 4-chloro-8-methoxy pyrido [3,4-d]pyrimidin-2-amine, was prepared by the route depicted in Scheme 1. The nitration of 2-amino-4-picoline with nitric acid in concentrated sulfuric acid gave a mixture of 4-methyl-3-nitropyridin-2-amine (2) and 4-methyl-5-nitropyridin-2-amine (3) and the 3-nitro isomer was obtained by silica gel column chromatography. Then the 2-amino group was converted to a hydroxyl group with sodium nitrite (Sandmeyer condition), and compound 4 was obtained in 96% yield. Following that, chlorination (compound 5 in 78% yield), carboxylation (compound 6 in 81% yield and 7 in 96% yield) and reduction with iron (in 89% yield) were conducted in sequence to provide compound 8, which was condensed with freshly made chloroformamidine hydrochloride to elaborate compound 9 in 93% yield. After replacing the chloro with a methoxy group at the C-8 position and treating with phosphorous oxychloride, the key common intermediate, 4-chloro-8-methoxypyrido[3,4-d]pyrimidin-2-amine (11) was prepared with a satisfactory overall yield.20,21
Scheme 1.

Synthesis of 4-chloro-8-methoxypyrido[3,4-d]pyrimidin-2-amine.
Subsequently, the 4-amino/phenol/thiol analogs were obtained via a direct nucleophilic aromatic substitution (SNAr) reaction, promoted with either acid or base. Meanwhile, the palladium-mediated reactions with boronic acids (Suzuki coupling) or heteroarylamine (Buchwald–Hartwig cross-coupling) afforded the analogs with C–C or C–N connecting substituents (Scheme 2). Finally, the 8-methyl group can be removed by using a 4 M solution of hydrochloric acid in acetonitrile.
Scheme 2.
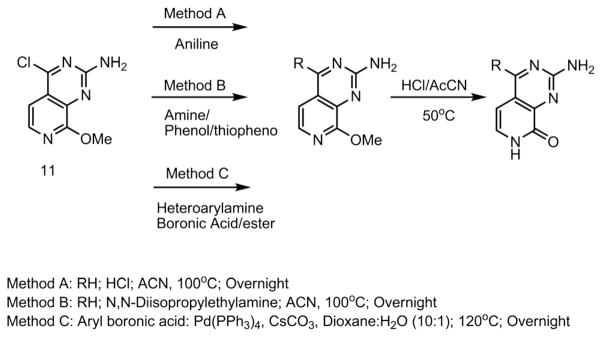
Synthetic scheme for coupling of compound 11.
Results and discussion
The newly synthesized compounds were evaluated for in vitro cytotoxicity and selectivity (differential cytotoxicity) using the NCI 60 human cancer cell line panel, which consists of nine subpanels representing diverse histologies: leukemia, melanoma, and cancers of lung, colon, kidney, ovary, breast, prostate, and the central nervous system (CNS).22 Details of the NCI 60 human tumor cell line screening methodology are described at http://dtp.nci.nih.gov/branches/btb/ivclsp.html. The NCI 60 evaluation has several advantages:23 first, the cellular evaluation would more accurately measure the activity of the test compound in physiological contexts such as the concentration of ATP, substrates and enzymes, etc.;24 second, screening across diverse tumor types can rapidly identify the test compounds with growth-inhibitory or toxic effects on particular tumor types (the disease-oriented concept); third, the patterns of relative drug sensitivity and resistance (differential cytotoxicity profile) would reflect the compounds’ mechanism of action (MAO) and provide guidance for target identification.
A set of sixteen newly synthesized analogs was selected and evaluated using the NCI 60 human tumor cell line screen panel. In general, the majority of cell lines, including leukemia, melanoma, non-small cell lung cancer, colon cancer, ovarian cancer, prostate cancer and CNS cancer, were largely “refractory” to the synthesized compounds without overt cell killing or growth inhibition. Interestingly, with some compounds highly selective growth inhibition activity was seen against UO-31 (renal cancer cell line), MCF-7 (breast cancer cell line) and MDA-MB-468 (breast cancer cell line) compared to the remaining cell lines, which suggested a high tumor-tissue-type (disease-oriented) selectivity. Interestingly, studies have shown that these cell lines frequently over-express various growth factor kinases, including EGFR (epidermal growth factor receptor, ErbB-1), HER2/Neu (ErbB-2, overexpressed in 15–20% of breast cancer), PDGFR (platelet-derived growth factor receptor), VEGFR (vascular endothelial growth factor receptor), etc.25 The growth percentages of test compounds over these three cell lines are shown in Table 1. For the inhibitory activity against the UO31 renal cancer cell line, the compounds (13 and 23) with an amine (–NH–) linker at the C-4 position showed better inhibitory activity than the compounds with thiol (–S–) (16 and 26) or ether (–O–) linkers (17 and 27) and without linker (connected via a C–C bond) (20 and 30), indicating that the amine linker at the C-4 position is essential to enhance growth inhibitory effects. In addition, the analogs with 8-methoxy (fixed lactam form) showed better activity against the UO31 renal cancer cell line compared to those with a hydroxy group (pyridone moiety), such as compounds 13 and 23, 14 and 24, suggesting that a methoxy group was favored at the C-8 position. Compound 13 with a 4-chloro and 14 with a 4-methoxy group showed moderate selectivity against UO31 over MCF-7 and MDA-MB-468, while compound 13 showed better growth inhibition against UO31 compared with 14 and 12 (unsubstituted), suggesting that an electron withdrawing substituent at the para position was favored. For the inhibitory activity against MCF-7 and MDA-MB-468 breast cancer cell lines, compound 21 with 3-fluoro showed the best growth inhibition of 60.77% and 71.42%, respectively, and it also exhibited the highest selectivity compared with the rest of the cell lines in the NCI 60 (Fig. 2) panel. Replacement of the methoxy at the C-8 position by a hydroxy group led to decreased inhibitory activity (32), which also confirmed the importance of the C-8 methoxy moiety. Taken together, among all the analogs synthesized and tested so far, compound 13 showed the best inhibitory activity against the UO31 renal cancer cell line while compound 21 exhibited the highest potency and selectivity against MCF-7 and MDA-MB-468 breast cancer cell lines.
Table 1.
Growth percentage of cell lines in NCI 60

| |||||
|---|---|---|---|---|---|
| Entry | R | R1 | UO31a | MCF7a | MDA-MB-468a |
| 12 | –OMe | –NHPh | 88.35 | 78.45 | 56.49 |
| 13 | –OMe | –NHPh-4-Cl | 42.85 | 62.16 | 59.96 |
| 14 | –OMe | –NHPh-4-OMe | 57.67 | 72.95 | 76.31 |
| 15 | –OMe | –NHCH2Phb | N/A | N/A | N/A |
| 16 | –OMe | –SPh | 75.33 | 91.91 | 112.04 |
| 17 | –OMe | –OPhb | N/A | N/A | N/A |
| 18 | –OMe | –Ph | 93.82 | 96.51 | 98.16 |
| 19 | –OMe | –Ph-4-OMe | 107.88 | 86.28 | 97.88 |
| 20 | –OMe | –Ph-4-Cl | 86.16 | 75.34 | 90.32 |
| 21 | –OMe | –Ph-3-F | 108.82 | 39.23 | 28.58 |
| 22 | –OH | –NHPh | 73.68 | 80.78 | 125.96 |
| 23 | –OH | –NHPh-4-Cl | 54.49 | 80.96 | 134.82 |
| 24 | –OH | –NHPh-4-OMe | 64.56 | 79.74 | 118.38 |
| 25 | –OH | –NHCH2Phb | N/A | N/A | N/A |
| 26 | –OH | –SPh | 86.77 | 91.56 | 119.89 |
| 27 | –OH | –OPhb | N/A | N/A | N/A |
| 28 | –OH | –Ph | 91.94 | 110.76 | 101.12 |
| 29 | –OH | –Ph-4-OMe | 94.64 | 95.69 | 110.12 |
| 30 | –OH | –Ph-4-Cl | 96.90 | 94.14 | 107.87 |
| 31 | –OH | –Ph-3-F | 117.31 | 83.28 | 106.62 |
Cell growth percentage at 10 μM (NCI 60).
Compounds were synthesized but not selected using NCI 60.
Fig. 2.

Cytotoxicity profile of compound 21 against NCI 60.
Conclusions
In summary, we have designed and synthesized a new series of 4-substituted-2-amino-pyrido[3,4-d]pyrimidine analogs as potential anticancer agents. A convenient and efficient synthetic strategy has been developed which can rapidly generate analogs with diverse substituents at the C-4 position of the core scaffold. Screening of the representative analogs using the NCI 60 cancer cell line panel has shown highly selective inhibitory effects against the growth of the UO-31 renal cancer cell line, MDA-MB-468 and MCF-7 breast cancer cell lines. Compounds 13 and 21 represent promising starting points for the development of this new compound class as molecularly targeted chemotherapeutic agents. The mechanism of action (MOA) studies to determine their cellular targets are underway. The results of our study suggest that this compound class (in particular compounds 13 and 21) has merits for further development towards potential candidates for clinical trials in the treatment of cancers.
Experimental section
Methodology of NCI 60 human cancer cell line screening
The human tumor cell lines of the cancer screening panel are grown in an RPMI 1640 medium containing 5% fetal bovine serum and 2 mM L-glutamine. For a typical screening experiment, cells are inoculated into 96 well microtiter plates in 100 μl at plating densities ranging from 5000 to 40 000 cells per well depending on the doubling time of individual cell lines. After cell inoculation, the microtiter plates are incubated at 37 °C, 5% CO2, 95% air and 100% relative humidity for 24 h prior to addition of experimental drugs. After 24 hours, two plates of each cell line are fixed in situ with TCA to represent a measurement of the cell population for each cell line at the time of drug addition (Tz). All test compounds are solubilized in dimethyl sulfoxide at a single dose of 10 μM and incubated with the cells for an additional 48 hours at 37 °C, 5% CO2, 95% air, and 100% relative humidity. For adherent cells, the assay is terminated by the addition of cold TCA. Cells are fixed in situ by the gentle addition of 50 μl of cold 50% (w/v) TCA (final concentration, 10% TCA) and incubated for 60 minutes at 4 °C. The supernatant is discarded, and the plates are washed five times with tap water and air dried. Sulforhodamine B (SRB) solution (100 μl) at 0.4% (w/v) in 1% acetic acid is added to each well, and plates are incubated for 10 minutes at room temperature. After staining, the unbound dye is removed by washing five times with 1% acetic acid and the plates are air dried. Bound stain is subsequently solubilized with 10 mM trizma base, and the absorbance is read on an automated plate reader at a wavelength of 515 nm. For suspension cells, the methodology is the same except that the assay is terminated by fixing settled cells at the bottom of the wells by gently adding 50 μl of 80% TCA (final concentration, 16% TCA). Using the seven absorbance measurements [time zero, (Tz), control growth, (C), and test growth in the presence of drug at the five concentration levels (Ti)], the percentage growth is calculated at each of the drug concentration levels. The percentage growth inhibition is calculated as:
Three dose response parameters are calculated for each experimental agent. Growth inhibition of 50% (GI50) is calculated from [(Ti − Tz)/(C − Tz)] × 100 = 50, which is the drug concentration resulting in a 50% reduction in the net protein increase (as measured by SRB staining) in control cells during the drug incubation. The drug concentration resulting in total growth inhibition (TGI) is calculated from Ti = Tz. The LC50 (concentration of drug resulting in a 50% reduction in the measured protein at the end of the drug treatment as compared to that at the beginning) indicating a net loss of cells following treatment is calculated from [(Ti − Tz)/Tz] × 100 = −50. Values are calculated for each of these three parameters if the level of activity is reached; however, if the effect is not reached or is exceeded, the value for that parameter is expressed as greater or less than the maximum or minimum concentration tested.
Chemistry
Melting points were measured with an Electrothermal Mel-Temp melting apparatus without correction. 1H and 13C NMR spectra were measured on a 400 MHz Varian Gemini 2000 spectrometer using TMS as an internal standard. The solvent used was DMSO unless indicated. Mass spectra were measured on an Agilent LC-MS 2010 instrument. Thin-layer chromatography (TLC) and preparative TLC were performed on precoated silica gel GF plates purchased from Merck, Inc. Isco Rf Companion systems were used for flash chromatography. The SCX (–SO3) ion exchange column was purchased from Phenomenex (S-210-45). All other chemicals were obtained from Aldrich, Inc. All final compounds are >95% pure on LC-Mass analysis.
Synthesis of 4-methyl-3-nitropyridin-2-amine (2) and 4-methyl-5-nitropyridin-2-amine (3)
To 50 g of 2-amino-4-picoline (1) was slowly added 200 ml of conc. sulfuric acid in an ice-bath. The reaction was vigorously stirred and the reaction temperature was carefully maintained under 20 °C. Subsequently, the reaction mixture was cooled to 10 °C and 70 ml of a mixture (1 : 1) of conc. sulfuric acid and nitric acid was added. After addition, the reaction mixture was slowly warmed to 35–40 °C (must be cautious, the temperature must be slowly increased, the reaction is exothermic) overnight to complete the reaction. Then the mixture was carefully poured over ice and neutralized with aq. ammonia to give the mixture of 4-methyl-3-nitropyridin-2-amine (2) (yield: 38%) and 4-methyl-5-nitropyridin-2-amine (3) (yield: 26%). ESI MS m/z 154 (M + H)+.
Synthesis of 4-methyl-3-nitropyridin-2-ol (4)
To 10 g of compound 2 in 50 ml of 30% sulfuric acid solution was added aq. NaNO2 solution, the precipitate was filtered and collected. The crude was dried over vacuum and purified via chromatography using 0–5% MeOH in DCM. Yield: 96%; M.P. = 234–235 °C. ESI MS m/z 155 (M + H)+.
1H NMR (DMSO-d6): 11.0 (br, 1H), 8.02 (d, 1H), 7.46 (s, 1H), 7.03 (d, 1H), 2.40 (s, 3H).
13C NMR (DMSO-d6): 149.6, 141.9, 125.0, 17.2.
Synthesis of 2-chloro-4-methyl-3-nitropyridine (5)
5 g of compound 4 was added to 15 ml of phosphorus oxychloride and the reaction mixture was heated at 120 °C for 5 hours. The excess phosphorus oxychloride was removed under vacuum. The residue was poured over ice with vigorous stirring; the solid was filtered and collected to give 2-chloro-4-methyl-3-nitropyridine (5). Yield: 78%, M.P. = 52–53 °C. ESI MS m/z 173 (M + H)+.
1H NMR (CDCl3): 8.37 (d, 1H), 7.24 (d, 1H), 2.39 (s, 3H).
13C NMR (CDCl3): 149.8, 142.0, 125.1, 17.1.
Synthesis of 2-chloro-3-nitroisonicotinic acid (6)
To a mixture of compound 5 (10.3 g, 60 mmol) in 60 ml conc. sulfuric acid was added Na2Cr2O7 (1.2 eq., 72 mmol), and during the addition the reaction temperature was maintained between 35 and 40 °C. The reaction mixture was stirred overnight and poured over ice. The white solid was filtered and collected. The solid was dissolved in EtOAc and washed with brine three times. The organic phase was dried over anhydrous Na2SO4. The solvent was removed to give a white solid. Yield: 81%; M.P. = 159–160 °C; ESI MS m/z 203 (M + H)+.
1H NMR (DMSO-d6): 13.5 (br, 1H), 9.10 (d, J = 4.95 Hz, 1H), 8.00 (d, J = 4.96 Hz, 1H).
13C NMR (DMSO-d6): 164.1, 155.3, 146.5, 142.3, 139.5, 124.0.
Synthesis of ethyl 2-chloro-3-nitroisonicotinate (7)
To a mixture of 5.0 g of compound 6 in 32 ml thionyl chloride was added 2 ml DMF and refluxed for 2 h. The solution was concentrated and 20 ml anhydrous EtOH was added to the residue. The mixture was stirred at room temperature for 1 hour. Removal of EtOH in vacuo gave the product as a white solid. Yield: 96%; M.P. = 102–103 °C; ESI MS m/z 231 (M + H)+.
1H NMR (CDCl3):8.68 (d, J = 4.98 Hz, 1H),7.88(d, J =4.96 Hz, 1H), 4.43 (q, J = 5.89 Hz, J = 13.0 Hz, 2H), 1.38 (t, J = 7.14, 3H).
13C NMR (CDCl3): 160.5, 151.1, 143.1, 132.8, 123.1, 63.6, 13.6.
Synthesis of ethyl 3-amino-2-chloroisonicotinate (8)
To a solution of iron (10 g), EtOH (40 ml), water (10 ml) and conc. hydrochloric acid (0.5 ml) was added compound 7 (10 g, 50 mmol) and the reaction mixture was refluxed for 2 hours, cooled to 40 °C and filtered through Celite. The residue was washed with EtOH three times (10 ml × 3), the filtrate was combined, evaporated and purified by chromatography using hexane–EtOAC (10 : 1) to give the product as a white solid. Yield: 89%; M.P. = 92–93 °C. ESI MS m/z 201.0 (M + H)+.
1H NMR (CDCl3):8.67 (d, J = 4.96 Hz, 1H),7.86(d, J =4.95 Hz, 1H), 4.43 (q, J = 5.86, J = 13.0 Hz, 2H), 1.38 (t, J = 7.10 Hz, 3H).
13C NMR (CDCl3): 160.5, 151.0, 143.1, 132.7, 123.1, 63.6, 13.6.
Synthesis of 2-amino-8-chloropyrido[3,4-d]pyrimidin-4-ol (9)
Chloroformamidine hydrochloride was prepared by bubbling hydrochloric acid gas through a solution of cyanamide in ethyl ester. The precipitate (white power) was collected, dried and directly used for the condensation. A mixture of compound 8 (500 mg, 2.5 mmol), chloroformamidine hydrochloride and dimethyl sulfone (1.5 g) was heated at 140 °C for 4 h. The mixture was cooled to 70 °C and 7.5 ml 2 M ammonium in MeOH was added to neutralize the suspension (pH = 8). The product was collected by filtration, washed with EtOH and purified by chromatography (0–20% MeOH–DCM) to give the product as a pale yellow powder. Yield: 93%. M.P. = 225–253 °C. ESI MS m/z 197 (M + H)+.
1H NMR (DMSO): 8.00 (d, J = 8 Hz, 1H), 7.71 (d, J = 4 Hz, 1H), 7.02 (br, 2H).
13C NMR (DMSO): 162.0, 161.9, 146.9, 145.2, 136.3, 116.5, 115.8.
Synthesis of 2-amino-8-methoxypyrido[3,4-d]pyrimidin-4-ol (10)
10 ml dry MeOH was added to sodium (400 mg) in an ice bath and the mixture was stirred until no solid was left. The solution was added to compound 9 (3 mmol, 570 mg) and was stirred at reflux for 1 day. 1 ml acetic acid was added to neutralize the reaction mixture and solid was collected and washed with EtOAc and water. The solid was purified by chromatography using 0–20% MeOH–DCM to give the product as a light yellow solid: yield: 83%. M.P. = 260–261 °C. ESI MS m/z 193 (M + H)+.
1H NMR (DMSO): 7.61 (d, J = 8 Hz, 1H), 7.40 (d, J = 4 Hz, 1H), 6.26 (br, 2H), 3.89 (s, 3H).
13C NMR (DMSO): 162.0, 161.9, 158.6, 139.7, 133.9, 114.5, 110.2, 53.4.
Synthesis of 4-chloro-8-methoxypyrido[3,4-d]pyrimidin-2-amine (11)
A mixture of compound 10 (1.1 g, 5.7 mmol) in phosphorus oxychloride (10.5 ml, 115 mmol) was adjusted to reflux (105 °C) overnight. The phosphorus oxychloride was removed by vacuum (with toluene). The residue was added to aqua ammonia to neutralize the excess acid. The crude was further purified by chromatography using 0–100% EtOAc–hexane to give the light yellow solid: yield: 40%; M.P. = 241–242 °C. ESI MS m/z 211 (M + H)+.
1H NMR (DMSO): 8.00 (d, J = 4 Hz, 1H), 7.68 (d, J = 8 Hz, 1H), 7.35 (br, 2H), 4.07 (s, 3H).
13C NMR (DMSO): 167.3, 161.6, 147.1, 146.1, 138.1, 116.9, 116.5, 55.1.
Method A: general procedure for coupling of compound 11 and aniline
To a solution of compound 11 (0.5 mmol) and aniline (1 mmol) in 1.5 ml acetonitrile was added 1–2 drops of conc. HCl. The reaction mixture was heated at 100 °C in a sealed tube for 3 h. After cooling to room temperature, the reaction was neutralized with aqua ammonia, then the solvent was evaporated in vacuo. The crude product was further purified by chromatography using 0–100% EtOAc–hexane to afford compounds 12–14.
Method B: general procedure for coupling of compound 11 and amine/phenol/thiophenol
A solution of compound 11 (0.5 mmol), N,N-diisopropylethylamine (1.5 mmol) and amine/phenol/thiophenol (1.5 mmol) in 1.5 ml acetonitrile in a sealed tube was heated at 100 °C overnight. After cooling to room temperature, the reaction was treated with water and extracted by EtOAc. The crude product was further purified by chromatography using 0–100% EtOAc–hexane to afford compounds 15–17.
Method C: general procedure for coupling of compound 11 and aryl boronic acid
To a mixture of compound 11 (1 mmol), aryl boronic acid (1.2 mmol), cesium carbonate (1.5 mmol) and (tri-phenylphosphine)palladium(0) (Pd(PPh3)4) (0.1 mmol) in a sealed tube was added 1.5 ml nitrogen-debubbled mixture of 10% water in dioxane under the atmosphere of nitrogen. The mixture was heated at 120 °C overnight. After cooling to room temperature, the reaction mixture was loaded onto a SCX cartridge (–SO3) and washed with methanol. Then the cartridge was flushed with 2 M ammonium in MeOH, the ammonium eluting was collected and evaporated in vacuo. The crude product was further purified by chromatography using 0–100% EtOAc–hexane to afford compounds 18–21.
General procedure for removal of methyl group to produce cyclic amide
A solution of methylated compound (0.5 mmol) in 1 ml 4 M HCl–acetonitrile was heated to 50 °C in a sealed tube for 5 hours. After cooling to room temperature, 1 ml DCM was added to the mixture and stirred for 30 min. The solid was collected and dried in vacuo to afford pure cyclic amide products: 22–31.
The following compounds were prepared using a similar procedure described in method A
8-Methoxy-N4-phenylpyrido[3,4-d]pyrimidine-2,4-diamine (12)
Light yellow solid: yield: 75%; M.P. = 236–237 °C. ESI MS m/z 268 (M + H)+.
1H NMR (DMSO): 8.74 (s, 1H), 7.91 (d, J = 4 Hz, 2H), 7.78 (d, J = 4 Hz, 1H), 7.33 (t, 2H), 7.01 (m, 4H), 4.05 (s, 3H).
13C NMR (DMSO): 167.2, 160.4, 150.0, 140.6, 139.1, 136.8, 129.2 (2C), 121.9, 118.7 (2C), 113.0, 106.0, 54.7.
N4-(4-Chlorophenyl)-8-methoxypyrido[3,4-d]pyrimidine-2,4-diamine (13)
Off-white solid: yield: 87%; M.P. = 243–244 °C. ESI MS m/z 302 (M + H)+.
1H NMR (DMSO): 8.87 (s, 1H), 7.98 (d, J = 4 Hz, 2H), 7.79 (d, J = 4 Hz, 1H), 7.37 (d, J = 4 Hz, 2H), 7.05 (d, J = 4 Hz, 1H), 7.01 (s, 2H), 4.05 (s, 3H).
13C NMR (DMSO): 167.2, 160.4, 149.8, 139.7, 139.1, 136.7, 129.0 (2C), 125.1, 120.3 (2C), 113.1, 106.5, 54.7.
8-Methoxy-N4-(4-methoxyphenyl)pyrido[3,4-d]pyrimidine-2,4-diamine (14)
White solid: yield: 45%; M.P. = 225–226 °C. ESI MS m/z 298 (M + H)+.
1H NMR (DMSO): 8.59 (s, 1H), 7.81 (d, J = 8 Hz, 2H), 7.73 (d, J = 8 Hz, 1H), 6.97 (s, 2H), 6.95 (d, J = 8 Hz, 1H), 6.93 (d, J = 8 Hz, 2H), 4.04 (s, 3H), 3.73 (s, 3H).
13C NMR (DMSO): 167.2, 160.4, 154.6, 150.2, 139.0, 137.0, 134.0, 120.5 (2C), 114.4 (2C), 112.8, 105.3, 55.6, 54.7.
The following compounds were prepared using a similar procedure described in method B
N4-Benzyl-8-methoxypyrido[3,4-d]pyrimidine-2,4-diamine (15)
Light yellow solid: yield: 35%; M.P. = 130–131 °C. ESI MS m/z 282 (M + H)+.
1H NMR (DMSO): 7.51 (d, J = 4 Hz, 1H), 7.33 (m, 3H), 7.22 (m, 3H), 6.38 (s, 2H), 4.64 (s, 2H), 4.26 (s, 1H), 4.06 (s, 3H).
13C NMR (DMSO): 167.3, 162.7, 161.9, 155.8, 145.1, 139.9, 129.0 (2C), 127.1 (2C), 127.0, 104.3, 112.4, 54.2; 43.5.
8-Methoxy-4-(phenylthio)pyrido[3,4-d]pyrimidin-2-amine (16)
White solid: yield: 56%; M.P. = 136–137 °C. ESI MS m/z 285 (M + H)+.
1H NMR (DMSO): 7.88 (d, J = 4 Hz, 1H), 7.57 (m, 2H), 7.46 (m, 3H), 7.36 (d, J = 4 Hz, 1H), 7.20 (b, 2H), 4.06 (s, 3H).
13C NMR (DMSO): 167.1, 160.7, 156.9, 145.8, 139.1, 136.0 (2C), 130.3, 129.5 (2C), 129.2, 113.3, 112.4, 54.8.
8-Methoxy-4-phenylpyrido[3,4-d]pyrimidin-2-amine (17)
Light yellow solid: yield: 98%; M.P. = 178–179 °C. ESI MS m/z 253 (M + H)+.
1H NMR (DMSO): 8.34 (d, J = 4 Hz, 1H), 8.18 (d, J = 8 Hz, 2H), 7.67 (d, J = 4 Hz, 1H), 7.43 (m, 3H), 6.97 (s, 2H), 4.08 (s, 3H).
13C NMR (DMSO): 167.5, 160.6, 154.1, 146.6, 139.2, 139.0, 130.8 (2C), 128.6, 127.3 (2C), 116.0, 115.4, 54.7.
The following compounds were prepared using a similar procedure described in method C
8-Methoxy-4-(4-methoxyphenyl)pyrido[3,4-d]pyrimidin-2-amine (18)
Light yellow solid: yield: 88%; M.P. = 126–127 °C. ESI MS m/z 283 (M + H)+.
1H NMR (DMSO): 8.33 (d, J = 4 Hz, 1H), 8.26 (d, J = 8 Hz, 2H), 7.64 (d, J = 4 Hz, 1H), 7.11 (b, 2H), 7.04 (d, J = 8 Hz, 2H), 4.09 (s, 3H), 3.84 (s, 3H).
13C NMR (DMSO): 167.6, 160.3, 160.1, 153.0, 145.7, 139.1, 132.2 (2C), 130.8, 116.2, 114.9, 113.5 (2C), 55.6, 54.9.
4-(4-Chlorophenyl)-8-methoxypyrido[3,4-d]pyrimidin-2-amine (19)
White solid: yield: 95%; M.P. = 208–209 °C. ESI MS m/z 287 (M + H)+.
1H NMR (DMSO): 8.33 (d, J = 4 Hz, 1H), 8.29 (d, J = 8 Hz, 2H), 7.68 (d, J = 4 Hz, 1H), 7.51 (d, J = 8 Hz, 2H), 7.02 (s, 2H), 4.07 (s, 3H).
13C NMR (DMSO): 167.5, 160.7, 152.4, 146.6, 139.2, 137.6, 133.6, 132.5 (2C), 128.0 (2C), 116.2, 115.8, 54.8.
4-(3-Fluorophenyl)-8-methoxypyrido[3,4-d]pyrimidin-2-amine (20)
White solid: yield: 92%; M.P. = 202–203 °C. ESI MS m/z 271 (M + H)+.
1H NMR (DMSO): 8.35 (d, J = 8 Hz, 1H), 8.11 (d, J = 8 Hz, 2H), 7.70 (d, J = 8 Hz, 1H), 7.47 (q, 1H), 7.25 (m, 1H), 7.07 (s, 2H), 4.08 (s, 3H).
13C NMR (DMSO): 167.5, 163.2, 160.8, 151.9, 146.6, 141.1, 139.2, 129.8, 126.7, 117.5, 116.3, 116.1, 115.6, 54.8.
8-Methoxy-4-phenoxypyrido[3,4-d]pyrimidin-2-amine (21)
White solid: yield: 76%; M.P. = 68–69 °C. ESI MS m/z 269 (M + H)+.
1H NMR (DMSO): 7.48 (d, J = 4 Hz, 1H), 7.25 (m, 2H), 7.23 (m, 3H), 7.19 (d, J = 4 Hz, 1H), 6.99 (b, 2H), 4.05 (s, 3H).
13C NMR (DMSO): 167.4, 165.1, 162.9, 158.9, 139.1, 129.5 (2C), 124.6.3, 122.3 (2C), 129.2, 105.0, 98.6, 54.2.
The following compounds were prepared using the general procedure for removal of methyl group
2-Amino-4-(phenylamino)pyrido[3,4-d]pyrimidin-8-ol (22)
White solid; yield: 99%; M.P. = 273–274 °C. ESI MS m/z 254 (M + H)+.
1H NMR (DMSO): 11.38 (b, 1H), 8.51 (s, 1H), 7.85 (d, J = 8 Hz, 2H), 7.80 (d, J = 4 Hz, 1H), 7.32 (t, 2H), 7.08 (d, J = 4 Hz, 1H), 6.96 (t, 1H), 6.70 (s, 2H).
13C NMR (DMSO): 161.7, 152.9, 150.2, 140.8, 137.7, 136.1, 129.2 (2C), 121.7, 120.0, 118.6 (2C), 108.5.
2-Amino-4-(4-chlorophenylamino)pyrido[3,4-d]pyrimidin-8-ol (23)
White solid; yield: 99%; M.P. = 289–290 °C. ESI MS m/z 288 (M + H)+.
1H NMR (DMSO): 11.38 (b, 1H), 8.61 (s, 1H), 7.91 (d, J = 8 Hz, 2H), 7.81 (d, J = 4 Hz, 1H), 7.36 (d, J = 8 Hz, 2H), 7.11 (d, J = 4 Hz, 1H), 6.67 (b, 2H).
13C NMR (DMSO): 161.6, 152.9, 150.0, 139.8, 137.7, 136.2, 129.0 (2C), 124.9, 120.2, 120.0 (2C), 109.0.
2-Amino-4-(4-methoxyphenylamino)pyrido[3,4-d]pyrimidin-8-ol (24)
White solid; yield: 98%; M.P. = 263–264 °C. ESI MS m/z 284 (M + H)+.
1H NMR (DMSO): 11.34 (b, 1H), 8.34 (s, 1H), 7.73 (m, 3H), 7.02 (d, J = 8 Hz, 1H), 6.92 (d, J = 8 Hz, 2H), 6.65 (b, 2H), 3.73 (s, 3H).
13C NMR (DMSO): 161.7, 154.5, 152.8, 150.5, 137.9, 135.9, 134.1, 120.3 (2C), 119.8, 114.4 (2C), 107.8, 55.6.
2-Amino-4-(benzylamino)pyrido[3,4-d]pyrimidin-8-ol (25)
White solid; yield: 99%; M.P. = 135–136 °C. ESI MS m/z 268 (M + H)+.
1H NMR (DMSO): 11.43 (b, 1H), 7.52 (d, J = 4 Hz, 1H), 7.29 (m, 3H), 7.18 (m, 3H), 6.42 (b, 2H), 4.42 (s, 2H), 4.24 (b, 1H).
13C NMR (DMSO): 162.1, 161.0, 158.8, 156.5, 146.2, 139.5, 129.1 (2C), 127.0 (2C), 126.8, 111.3, 115.2, 43.6.
2-Amino-4-(phenylthio)pyrido[3,4-d]pyrimidin-8-ol (26)
White solid; yield: 98%; M.P. = 156–157 °C. ESI MS m/z 271 (M + H)+.
1H NMR (DMSO): 11.37 (b, 1H), 7.88 (d, J = 4 Hz, 1H), 7.53 (m, 2H), 7.43 (m, 3H), 7.40 (d, J = 4 Hz, 1H), 6.83 (b, 2H).
13C NMR (DMSO): 165.1, 161.5, 153.2, 143.9, 140.2, 136.0 (2C), 130.6, 129.5 (2C), 129.1, 120.5, 115.0.
2-Amino-4-phenylpyrido[3,4-d]pyrimidin-8-ol (27)
White solid; yield: 99%; M.P. = 200–201 °C. ESI MS m/z 239 (M + H)+.
1H NMR (DMSO): 11.48 (s, 1H); 8.33 (d, J = 4 Hz, 1H), 8.06 (d, J = 4 Hz, 2H), 7.72 (d, J = 4 Hz, 1H), 7.41 (m, 3H), 6.68 (b, 2H).
13C NMR (DMSO): 161.8, 153.9, 152.6, 140.5, 139.2, 139.1, 130.6 (2C), 128.4, 127.9 (2C), 123.5, 118.1.
2-Amino-4-(4-methoxyphenyl)pyrido[3,4-d]pyrimidin-8-ol (28)
White solid; yield: 99%; M.P. = 197–198 °C. ESI MS m/z 269 (M + H)+.
1H NMR (DMSO): 11.39 (s, 1H); 8.30 (d, J = 4 Hz, 1H), 8.16 (d, J = 8 Hz, 2H), 7.66 (d, J = 4 Hz, 1H), 6.98 (d, J = 8 Hz, 2H), 6.59 (b, 2H).
13C NMR (DMSO): 161.9, 159.7, 153.3, 152.4, 144.5, 140.4, 132.1 (2C), 123.4, 117.3, 114.3, 113.2 (2C), 55.6.
2-Amino-4-(4-chlorophenyl)pyrido[3,4-d]pyrimidin-8-ol (29)
White solid; yield: 98%; M.P. = 251–252 °C. ESI MS m/z 273 (M + H)+.
1H NMR (DMSO): 11.40 (b, 1H); 8.34 (d, J = 4 Hz, 1H), 8.16 (d, J = 8 Hz, 2H), 7.74 (d, J = 4 Hz, 1H), 7.49 (d, J = 8 Hz, 2H), 6.66 (b, 2H).
13C NMR (DMSO): 161.8, 152.7, 152.0, 140.5, 137.7, 133.3, 132.4 (2C), 131.2, 1287.9 (2C), 123.7, 118.5.
2-Amino-4-(3-fluorophenyl)pyrido[3,4-d]pyrimidin-8-ol (30)
White solid; yield: 99%; M.P. = 214–215 °C. ESI MS m/z 257 (M + H)+.
1H NMR (DMSO): 11.42 (s, 1H); 8.34 (d, J = 4 Hz, 1H), 8.00 (d, J = 8 Hz, 2H), 7.75 (d, J = 4 Hz, 1H), 7.47 (q, 1H), 7.22 (t, 1H), 6.67 (b, 2H).
13C NMR (DMSO): 163.2, 161.8, 160.8, 152.7, 151.8, 144.9, 140.5, 129.7, 126.6, 123.8, 118.7, 117.4, 115.3.
2-Amino-4-phenoxypyrido[3,4-d]pyrimidin-8-ol (31)
White solid; yield: 98%; M.P. = 78–79 °C. ESI MS m/z 255 (M + H)+.
1H NMR (DMSO): 11.53 (s, 1H), 7.48 (d, J = 4 Hz, 1H), 7.22 (m, 2H), 7.20 (m, 3H), 7.16 (d, J = 4 Hz, 1H), 6.96 (b, 2H).
13C NMR (DMSO): 164.4, 162.2, 161.5, 156.8, 140.2, 129.2 (2C), 124.3, 122.5 (2C), 119.2, 115.0, 101.6.
Acknowledgments
The authors would like to thank the NCI Developmental Therapeutics Program for the 60 cell line screen of compounds described in this paper. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
References
- 1.Siegel R, Naishadham D, Jemal A. Ca-Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society. Cancer Facts and Figures 2012. Atlanta, GA: 2012. [Google Scholar]
- 3.Boyle P, Levin B. World Cancer Report 2008. IARC; 2008. p. 512. [Google Scholar]
- 4.Desai K, Patel R, Chikhalia K. Indian J Chem, Sect A: Inorg, Bioinorg, Phys, Theor Anal Chem. 2006;45:773–778. [Google Scholar]
- 5.Amr EA, Nermien MS, Abdulla MM. Monatsh Chem. 2007;138:699–707. [Google Scholar]
- 6.Fujiwara N, Nakajima T, Ueda Y, Fujita H, Kawakami H. Bioorg Med Chem. 2008;16:9804–9816. doi: 10.1016/j.bmc.2008.09.059. [DOI] [PubMed] [Google Scholar]
- 7.Ballell L, Field RA, Chung GA, Young RJ. Bioorg Med Chem Lett. 2007;17:1736–1740. doi: 10.1016/j.bmcl.2006.12.066. [DOI] [PubMed] [Google Scholar]
- 8.Gorlitzer K, Herbig S, Walter RD. Pharmazie. 1997;52:670–672. [Google Scholar]
- 9.Shibuya M, Suzuki Y, Sugita K, Saito I, Sasaki T, Takakura K, Nagata I, Kikuchi H, Takemae T, Hidaka H, et al. J Neurosurg. 1992;76:571–577. doi: 10.3171/jns.1992.76.4.0571. [DOI] [PubMed] [Google Scholar]
- 10.Huron DR, Gorre ME, Kraker AJ, Sawyers CL, Rosen N, Moasser MM. Clin Cancer Res. 2003;9:1267–1273. [PubMed] [Google Scholar]
- 11.Griffin RJ, Srinivasan S, Bowman K, Calvert AH, Curtin NJ, Newell DR, Pemberton LC, Golding BT. J Med Chem. 1998;41:5247–5256. doi: 10.1021/jm980273t. [DOI] [PubMed] [Google Scholar]
- 12.Snow RJ, Cardozo MG, Morwick TM, Busacca CA, Dong Y, Eckner RJ, Jacober S, Jakes S, Kapadia S, Lukas S, Panzenbeck M, Peet GW, Peterson JD, Prokopowicz AS, 3rd, Sellati R, Tolbert RM, Tschantz MA, Moss N. J Med Chem. 2002;45:3394–3405. doi: 10.1021/jm020113o. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg DR, Butz T, Cardozo MG, Eckner RJ, Hammach A, Huang J, Jakes S, Kapadia S, Kashem M, Lukas S, Morwick TM, Panzenbeck M, Patel U, Pav S, Peet GW, Peterson JD, Prokopowicz AS, 3rd, Snow RJ, Sellati R, Takahashi H, Tan J, Tschantz MA, Wang XJ, Wang Y, Wolak J, Xiong P, Moss N. J Med Chem. 2003;46:1337–1349. doi: 10.1021/jm020446l. [DOI] [PubMed] [Google Scholar]
- 14.Dalton WS, Friend SH. Science. 2006;312:1165–1168. doi: 10.1126/science.1125948. [DOI] [PubMed] [Google Scholar]
- 15.Lapenna S, Giordano A. Nat Rev Drug Discovery. 2009;8:547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- 16.Katritzky AR, Rees CW. Comprehensive Heterocyclic Chemistry. Pergamon; Oxford: 1984. pp. 1–8. [Google Scholar]
- 17.Eicher T, Hauptmann S. The Chemistry of Heterocycles. 1995 [Google Scholar]
- 18.Colbry NL, Elslager EF, Werbel LM. J Heterocycl Chem. 1984;21:1521–1525. [Google Scholar]
- 19.Colbry NL, Elslager EF, Werbel LM. J Med Chem. 1985;28:248–252. doi: 10.1021/jm00380a018. [DOI] [PubMed] [Google Scholar]
- 20.Collins I. J Chem Soc, Perkin Trans 1. 2000:2845–2861. [Google Scholar]
- 21.Collins I. J Chem Soc, Perkin Trans 1. 2002:1921–1940. [Google Scholar]
- 22.Shoemaker RH. Nat Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 23.Warmuth M, Kim S, Gu XJ, Xia G, Adrian F. Curr Opin Oncol. 2007;19:55–60. doi: 10.1097/CCO.0b013e328011a25f. [DOI] [PubMed] [Google Scholar]
- 24.Knight ZA, Shokat KM. Chem Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 25.Janne PA, Gray N, Settleman J. Nat Rev Drug Discovery. 2009;8:709–723. doi: 10.1038/nrd2871. [DOI] [PubMed] [Google Scholar]